⭐⭐⭐C5aR1 inhibition reprograms tumor associated macrophages and reverses PARP inhibitor resistance in breast cancer
摘要¶
尽管聚腺苷二磷酸核糖聚合酶(PARP)抑制剂(PARPi)已被批准用于多种疾病,包括 BRCA1/2 突变的乳腺癌,但其反应通常是短暂的,需要使用组合疗法以获得最佳疗效。因此,我们在此研究了在雌性小鼠中使用两种内在对 PARPi 敏感(T22)和耐药(T127)的同基因小鼠乳腺癌模型,探索对 PARPi 的敏感性和耐药性的机制。我们发现肿瘤相关巨噬细胞(TAM)可能对 PARPi 的不同敏感性起到重要作用。通过单细胞 RNA 测序,我们识别出一个表达与抗炎活性相关基因的 TAM_C3 亚群,该亚群在 PARPi 耐药的 T127 肿瘤中富集,而在 T22 肿瘤中显著减少。Rps19/C5aR1 信号在 TAM_C3 中选择性升高。C5aR1 的抑制或转移 C5aR1 高表达细胞分别增加和降低了对 PARPi 的敏感性。在人类乳腺癌中,高 C5aR1 水平与对免疫检查点阻断疗法的差反应相关。因此,靶向 C5aR1 可能选择性地清除促肿瘤巨噬细胞,并增强对 PARPi 及其他疗法的敏感性。1
引言¶
基于与 BRCA1/2 和其他同源重组(HR)途径缺陷引起的合成致死性,聚腺苷二磷酸核糖聚合酶(PARP)抑制剂(PARPi)在实验室和临床中得到了广泛探索。尽管 PARPi 与化疗联合用于管理 BRCA 相关的转移性三阴性乳腺癌(TNBC)已获批准,其益处并不持久,几乎普遍出现复发。PARPi 耐药机制已被广泛研究,包括 BRCA1/2 逆转突变、低功能 BRCA1/2 等位基因、BRCA1/2 高甲基化的丧失、盾构体复合物的丧失以及 MAPK 和 PI3K 等促存活通路的激活。然而,综上所述,这些机制只能解释部分患者的 PARPi 耐药性。为了扩大 PARPi 在更广泛的 TNBC 患者中的适用性,多个临床研究表明,PARPi 联合疗法对没有 BRCA1/2 突变或 HR 途径异常的患者有效,从而为那些肿瘤没有 BRCA1/2 异常的患者提供了机会。因此,理解在有无 BRCA1/2 突变或 HR 途径异常肿瘤中的 PARPi 耐药机制,并确定和实施在突变亚型中有效且能预防或消除 PARPi 耐药的新组合疗法,是一个优先事项。
巨噬细胞,源自髓系的先天免疫细胞,其表观基因组和转录组的可塑性在一定程度上受到微环境变化的驱动。肿瘤相关巨噬细胞(TAMs)可以通过其极化为抗肿瘤/促炎或促肿瘤效应的巨噬细胞,对肿瘤清除或保护产生贡献。促炎和促肿瘤的 TAMs 通过产生相互排斥的细胞因子集来介导其促和抗肿瘤活动。促炎 TAMs 表达诱导 TH1 反应的细胞因子,如 IL1α、IL1β、IL12 和 TNFα,而促肿瘤 TAMs 表达促进肿瘤的 IL10、CCL8 和 CCL22。然而,单细胞 RNA 测序(scRNA-seq)研究表明,抗肿瘤/促炎或促肿瘤巨噬细胞极化的二分法不足以充分描述巨噬细胞状态的多样性和可塑性。不同的 TAMs 亚型经常与混合表达与血管生成、吞噬、促炎、增殖、转移、检查点抑制和代谢活动相关的基因,而不是仅仅一种功能基因表达群。
我们和其他人已证明,PARPi 可以引发功能性 STING 反应,导致干扰素(IFNs)产生和免疫参与,并进一步增强对免疫检查点阻断(ICB)的敏感性,无论是在 BRCA1/2 突变还是野生型细胞中。此外,STING 诱导的免疫激活似乎有助于 PARPi 与 ICB 联合疗法在临床试验中的活性,包括在没有 BRCA1/2 异常的患者中。有趣的是,STING 激动剂可以通过在 BRCA1 缺陷乳腺癌模型中将 TAMs 重新编程为主导的促炎表型来促进 PARPi 敏感性。然而,尽管在临床前模型中表现出强劲活性,并有证据表明 STING 反应和免疫系统参与,但 PARPi 与 ICB 的组合在临床试验中普遍令人失望,可能是由于对基础机制缺乏了解和缺乏预测性生物标志物。
在此,我们使用两种由转基因小鼠乳腺上皮表达溶血磷脂酸 1(LPA1)受体的同种移植(MDST)模型,即内在对 PARPi 敏感的 LPA1-T22(T22)和耐药的 LPA1-T127(T127),以探索内在 PARPi 敏感性和耐药性的机制。这两个模型是 BRCA1/2 野生型和 HR 功能正常的,表明这些研究结果可能适用于超出 BRCA1/2 突变 HR 缺陷(HRD)肿瘤的情况。单细胞 RNA 分析识别了 T22 和 T127 模型中的四种肿瘤相关巨噬细胞(TAM)亚型:TAM_C0、TAM_C1、TAM_C3 和 TAM_C4。TAM_C0 在 T22 内在 PARPi 肿瘤中呈现出与促炎和抗炎状态相关基因的混合表达。TAM_C3 主要表达与抗炎/促肿瘤状态相关的基因,在 T22 中水平较低,但在 PARPi 耐药的 T127 中水平较高,并且在 T22 中显著减少。与 PARPi 耐药相关的 TAM_C3 可以进一步细分为 C5aR1 高和 C5aR1 低亚群。C5aR1 高表达的 TAM_C3 亚群表达与抗炎活性相关的基因,而 C5aR1 低亚群则不表达与抗炎或促炎活性相关的显著水平基因。为了模拟转移状态,我们将 T22 和 T127 分别植入相对的乳腺脂肪垫中。结果是内在对 PARPi 敏感的 T22 变得对 PARPi 耐药。C5aR1 阳性巨噬细胞足以使 T22 对 PARPi 耐药。显著的是,靶向 C5aR1 在 PARPi 耐药模型中减少了 CD206 高表达巨噬细胞,同时保留了 MHCII 高表达巨噬细胞,使两个模型对 PARPi 敏感。
结果¶
共同移植 PARPi 耐药的 LPA1-T127 与 PARPi 敏感的 LPA1-T22 使 LPA1-T22 对 PARPi 耐药¶
LPA1-T22(T22)和 LPA1-T127(T127)是内在对 PARPi 敏感和耐药的小鼠同种移植(MDST)肿瘤。T22 对抗 PDL1 有温和反应,但在免疫功能正常的小鼠中,对 PARPi(奥拉帕尼)和抗 PDL1 组合有显著反应。相反,T127 对 PARPi 和抗 PDL1 耐药,并且对组合疗法也无反应。由于 PARPi 耐药是乳腺癌和其他癌症中一个主要的新兴挑战,我们探索了在 T127 和 T22 中介导内在 PARPi 耐药和敏感性的机制。此外,为了模拟在不同远处部位(或肿瘤不同位置)中肿瘤亚克隆对 PARPi 耐药的潜在影响,我们通过将 T22 和 T127 肿瘤分别移植到同一小鼠的左侧和右侧乳腺脂肪垫中(共同移植或 co),探讨 PARPi 耐药是否可以系统地从 T127 转移到 T22。由于 T22 比 T127 建立得更慢,因此 T22 在 T127 前两周移植。作为对照,T22 和 T127 在共同移植模型(共同移植或 co)中同时双侧移植(单一或 ss)。双侧移植到同一小鼠中的 T22_ss(T22 单一模型)对奥拉帕尼保持敏感,而 T127_ss(T127 单一模型)对奥拉帕尼保持耐药。相反,当 T22 和 T127 共同移植到同一小鼠时,T22 对奥拉帕尼变得耐药。由于未知原因,T127 在与 T22 共同移植时生长更快,无论它们是否接受奥拉帕尼或载体处理。
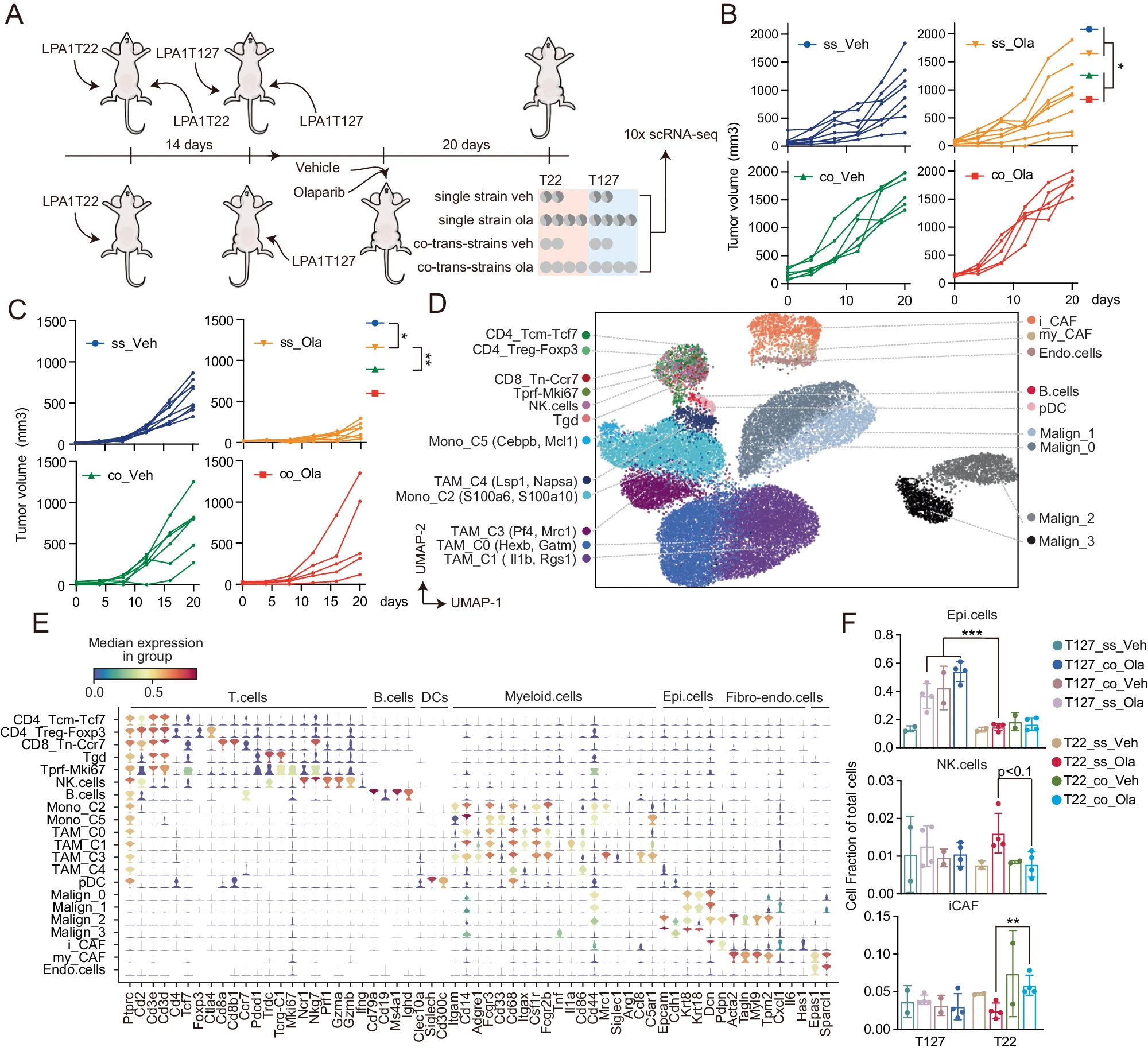
A 研究设计:T127 和 T22 单株(上)和共移植 T22 和 T127(下)MDST 模型接受奥拉帕尼治疗 20 天。单只小鼠的双侧肿瘤分别用深灰色和浅灰色表示。B T127 模型接受奥拉帕尼治疗 20 天的生长曲线。单株移植模型的肿瘤大小表示单株小鼠两侧的平均值。ss:单株移植模型。co:共移植模型中的 T127 侧。单株模型,n=8,接受载体治疗的共移植,n=6,接受奥拉帕尼治疗的共移植,n=5。C T22 模型接受奥拉帕尼治疗 20 天的生长曲线。单株移植模型的肿瘤大小表示单株小鼠两侧的平均值。ss:单株移植模型。co:共移植模型中的 T22 侧。接受载体治疗的单株模型,n=8,接受奥拉帕尼治疗的单株模型,n=7;接受载体治疗的共移植,n=6,接受奥拉帕尼治疗的共移植,n=5。D 从单株和共移植肿瘤中分层和细胞类型识别的 T22 和 T127 肿瘤。T127(Malign_0,Malign_1)和 T22(Malign_2,Malign_3)的恶性细胞显示出不同的细胞簇。Tcm:中央记忆 T 细胞,Treg:调节性 T 细胞,Tn:初始 T 细胞,Tgd:γδT 细胞,Tprf:增殖性 T 细胞,my_CAF:肌成纤维细胞相关癌细胞,i_CAF:炎症相关癌细胞,pDC:浆细胞样树突细胞。接受载体治疗的,n=2,接受奥拉帕尼治疗的,n=4。E 小提琴图显示了图 1D 中代表每个细胞谱系的标记物。F 柱状图显示了每个模型中定义为 E 所列细胞谱系标记的上皮细胞(高 CNV 细胞)、NK 细胞和 i_CAF 的相对数量(以百分比表示)。统计显著性仅在接受奥拉帕尼治疗的组间进行评估。所有数据均以均值 ± SD 表示。p 值来自单因素方差分析(one-way ANOVA)。* p < 0.05,** p < 0.01,*** p < 0.001。源数据和精确 p 值提供在源数据文件中。源数据作为源数据文件提供。Veh:载体。Ola:奥拉帕尼。
随后,我们对模型进行了单细胞 RNA 测序(scRNA-seq)分析,共获得了超过 30,000 个细胞(个体样本列表见补充图 1D)。通过使用 CaSpER 进行 CNV 调用鉴定出恶性细胞(补充图 1C)。根据图 1E 中列出的标记进行细胞谱系分配23。在单一模型和共移植模型中捕获到两个不同的 T22 恶性细胞簇(恶性 2 和 3)和两个不同的 T127 恶性细胞簇(恶性 0 和恶性 1)(图 1D、E 和补充图 1D、E,见图 1E 用于定义细胞亚型的标记)。T22 中的恶性 2 和恶性 3 的 Epcam 和 Cldn1 水平高于 T127 中的恶性 0 和恶性 1。恶性 2 表达与成纤维细胞谱系相关的多个标记,表明这种肿瘤亚型已经历上皮间质转化(EMT),而恶性 3 表达 Krt8 和 Krt18 水平升高,与上皮分化一致。奥拉帕尼治疗对 T22 和 T127 中的恶性细胞簇分布没有显著影响(补充图 1D、E)。如生长曲线所示,T127 模型中的上皮肿瘤细胞比例因奥拉帕尼和与 T22 的共移植而增加(图 1F)。T22 模型中的恶性上皮细胞比例未因治疗显著变化(注意归一化至 1 部分消除了治疗对肿瘤大小的影响)(图 1F)。
补充图 1
(A) 代表性免疫荧光图像展示了在指定的MDST肿瘤中RAD51焦点的形成。Veh:载体。Ola:奥拉帕利。数据代表了三次重复实验的结果。
(B) T127单株移植和共同移植肿瘤在使用奥拉帕利治疗20天后的肿瘤生长曲线。单株移植模型的肿瘤体积是双侧平均值。ss:单株移植模型。co:共同移植模型。T22_co:共同移植模型的T22部位。Veh:载体。Ola:奥拉帕利。样本数量在图1B和C中有标注。所有数据均以均值 +/- 标准差表示。p值来自单因素方差分析。* p < 0.05,** P < 0.01,* p < 0.001,** p < 0.0001。源数据和精确的p值提供在源数据文件中。
(C) 通过CaSpER对T22和T127肿瘤进行的CNV分析。
(D) 通过图1E中的标记确定的不同细胞在指定处理下T22和T127肿瘤中的细胞分数(上皮细胞、成纤维细胞、内皮细胞、NK细胞、T细胞、髓系细胞和DC细胞)。
(E) 图1D中UMAP展示的T127和T22肿瘤中的细胞分布。ss:单株移植模型。co:共同移植模型。T127_co_Ola_1:1089个细胞,T127_co_Ola_2:890个细胞,T127_co_Ola_3:1180个细胞,T127_co_Ola_4:1170个细胞,T127_co_Veh_1:508个细胞,T127_co_Veh_2:801个细胞,T127_ss_Ola_1:1639个细胞,T127_ss_Ola_2:1408个细胞,T127_ss_Ola_3:534个细胞,T127_ss_Ola_4:676个细胞,T127_ss_Veh_1:597个细胞,T127_ss_Veh_2:454个细胞,T22_co_Ola_1:1892个细胞,T22_co_Ola_2:989个细胞,T22_co_Ola_3:415个细胞,T22_co_Ola_4:613个细胞,T22_co_Veh_1:801个细胞,T22_co_Veh_2:856个细胞,T22_ss_Ola_1:2481个细胞,T22_ss_Ola_2:2108个细胞,T22_ss_Ola_3:1755个细胞,T22_ss_Ola_4:1665个细胞,T22_ss_Veh_1:435个细胞,T22_ss_Veh_2:709个细胞。载体处理的样本数为n = 2,奥拉帕利处理的样本数为n = 4。
(F) UMAP中每个髓系细胞亚群的分布。
(G) 热图展示了每个髓系细胞亚群的前20个差异表达基因。
(H) 点图展示了每个TAM亚群的代表性路径标记。
(I) 点图总结了每个TAM亚群的每条路径的表达水平。用于功能路径评分的基因列表列在(H)中。
(J) 从泛癌浸润髓系细胞研究转移到MDST数据集的指定巨噬细胞亚群的预测评分。
(K) 通过scGPT在每个人类髓系细胞群中标注的MDST髓系细胞群的细胞分数。源数据提供在源数据文件中。
补充图 1D 所示,在共移植模型中,T22(恶性 2 和恶性 3)中几乎没有发现 T127(恶性 0 和恶性 1)的恶性细胞,反之亦然,这消除了共移植 T22 中 PARPi 耐药是由于 T127 转移性细胞在 T22 肿瘤中过度生长的可能性。重要的是,与模型特异性的恶性细胞相比,T22 和 T127 肿瘤中的血液细胞、成纤维细胞和内皮细胞在有无奥拉帕尼治疗的情况下共同聚类(图 1D,补充图 1D、E)。单一和共移植模型及处理和未处理的样本之间 T 细胞或 B 细胞亚型没有统计学显著变化(图 1D 和补充图 1E)(见图 1E 用于定义每个簇的标记)。奥拉帕尼在 T22_ss 模型中诱导 NK 细胞增加,而在 T22_co(共移植模型的 T22 侧)中没有(图 1F)。相比之下,奥拉帕尼在 T22_co 模型中轻微但显著地增加了癌症相关成纤维细胞(CAF),而在 T22_ss 模型中没有。这些变化对 NK 细胞和 CAF 的影响尚待阐明。
T127 与 T22 的共移植改变了 PARPi 治疗后 T22 中的髓系亚型¶
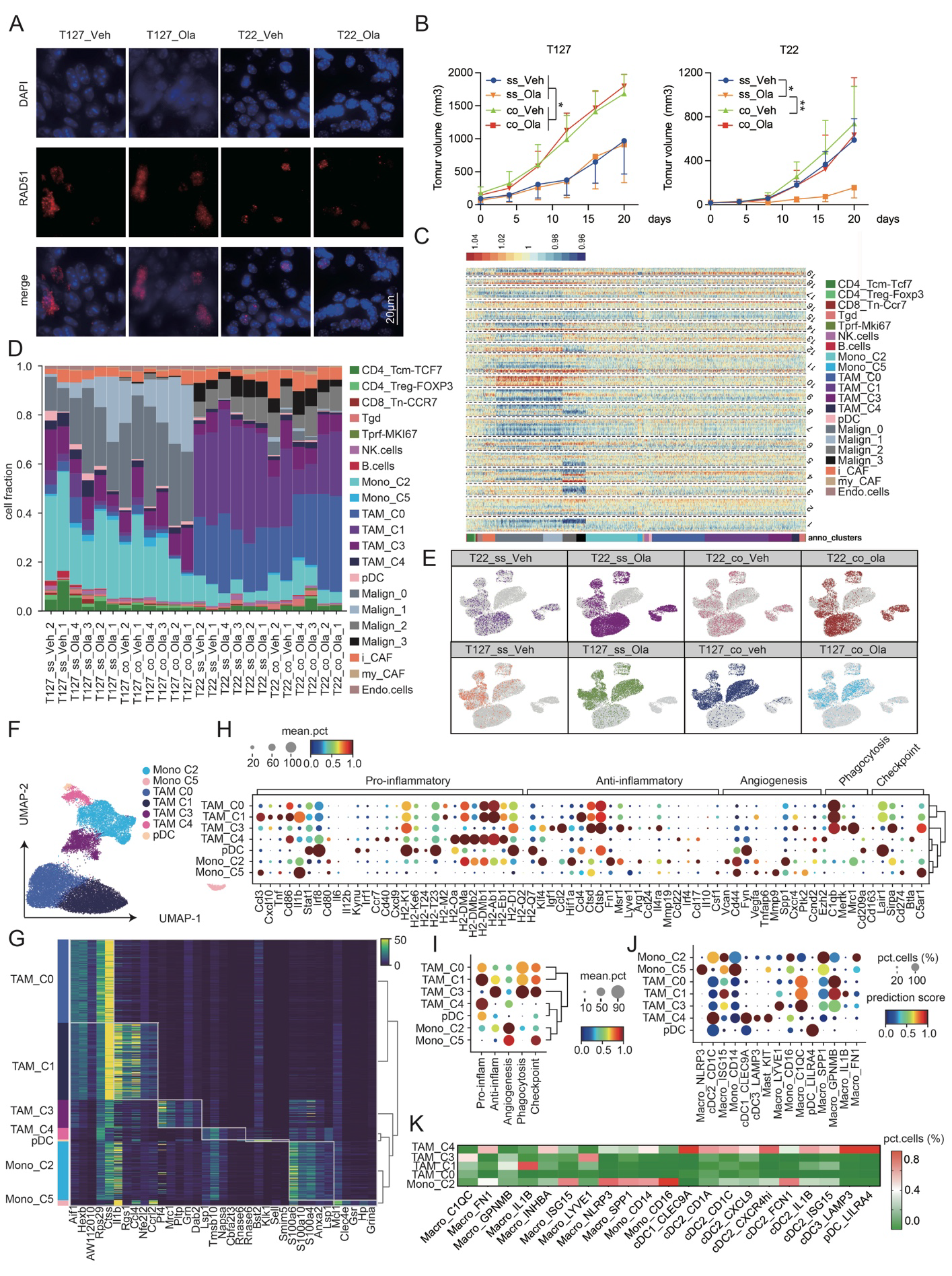
两种模型之间免疫细胞构成最显著的差异在于基于重新聚类最初簇中鉴定的单核细胞和巨噬细胞群的不同 TAM 亚簇的比例。对单核细胞和巨噬细胞群进行无监督重新聚类(补充图 1F)鉴定了两个单核细胞群(Mono_C2 和 Mono_C5,Cd14 表达较高)和 4 个巨噬细胞群(TAM_C0、TAM_C1、TAM_C3 和 TAM_C4,Cd68 表达较高),它们的特征是表达与单核细胞和巨噬细胞功能相关的多个标记(补充图 1F、G,见补充数据 1 中每个群体表达的基因列表)。这些单核细胞和 TAM 簇被注释为无监督 Leiden 簇,投射到图 1D 上,然后根据每个 Leiden 簇的顶级差异表达基因(DEGs)进行命名。随后评估这些单核细胞和 TAM 群体的特征单核细胞和巨噬细胞标记的表达,这些标记在先前的出版物中有所定义。TAM_C0,其特征为 Hexb 和 Gatm DEGs,表达与促炎、抗炎、吞噬和检查点活性相关的混合基因(补充图 1H、I)。TAM_C1,其特征为 Il1b 和 Rgs1 DEGs,与 TAM_C0 聚类接近,富含与促炎活性相关的一系列标记,并有 KEGG 坏死相关通路和 GO 干扰素 γ 相关通路活性(补充图 1H、I 和补充图 2A)。TAM_C3,其特征为 Mrc1 和 Pf4 DEGs,富含与促肿瘤活动相关的表达基因,包括抗炎、吞噬和检查点活性,以及 KEGG 脂质相关通路和 GO I 型干扰素相关通路活性(补充图 1H、I 和补充图 2A)。TAM_C4,其特征为 Lsp 和 Napsa DEGs,像 TAM_C1 一样,与促炎标记最为相关,其相对低水平的 Adgre1 提示其可能与巨噬细胞祖细胞有关(补充图 1H、I)。Mono_C2,其特征为 S100a6 和 S100a10 DEGs,和 Mono_C5,其特征为 Cebpb 和 Mcl1 DEGs,均表达与血管生成相关的基因,而 Mono_C5 还表达与检查点活性相关的基因(补充图 1H、I)。
然后使用标签转移进一步表征 TAM 亚型(见方法),并与先前在泛癌浸润髓系细胞大规模分析中识别的亚型进行比较。TAM_C1 得分预测与 Macro_IL1B 和 Macro_ISG15 亚型相关,这些亚型据报道与促炎活动有关,并且还具有相对较高的 Macro_GPNMB 预测得分,Macro_GPNMB 之前被报道为抗炎和代谢相关亚型(补充图 1J)。TAM_C3 在 Macro_LYVE1 和 Macro_C1QC 巨噬细胞亚型中得分最高,据报道这些亚型与抗炎亚型相关(补充图 1J)。TAM_C0 与 TAM_C1 相似,也与 Macro_GPNMB 表型相关,但 Macro_IL1B 和 Macro_ISG15 特征低于 TAM_C1。TAM_C4 得分与 cDC 细胞得分相关,需要进一步探讨。为了进一步澄清巨噬细胞表型,使用 MDST 数据集作为训练模型和大型人类髓系数据集作为测试数据进行了反向注释(见方法)。类似于标签转移结果,大多数人类促炎 Macro_IL1B 巨噬细胞被注释为 TAM_C1,而超过一半的抗炎 Macro_LYVE1 和 Macro_C1QC 巨噬细胞被注释为 TAM_C3(补充图 1K)。如其他研究中所示,不同的 TAM 亚群可能会表达与促炎和抗炎亚型相关的混合基因表达,并且在我们的研究中也发现了 TAM_C0、TAM_C1 和 TAM_C4 包含了促炎和抗炎表型的混合体。然而,TAM_C1 与促炎活动相关基因的表达以及 TAM_C3 与促肿瘤/抗炎活动相关基因的表达的强烈关联表明,它们可能是 T22 和 T127 对奥拉帕尼相对反应性的贡献者。
由于不同的 TAM 亚型与癌症患者的预后相关,我们探讨了在我们的研究中鉴定的 TAM 亚型与 TCGA 基底型乳腺癌队列预后的关系。为此,我们计算了每个细胞谱系的细胞特征排名,然后进行了多变量生存分析(见方法,补充图 2B)。与上述基因表达模式一致,较高的 TAM_C3 特征显著与较差的预后相关。较高的 NK 细胞评分显著与较好的预后相关。TCGA 基底型乳腺癌中没有其他特征评分与预后相关。
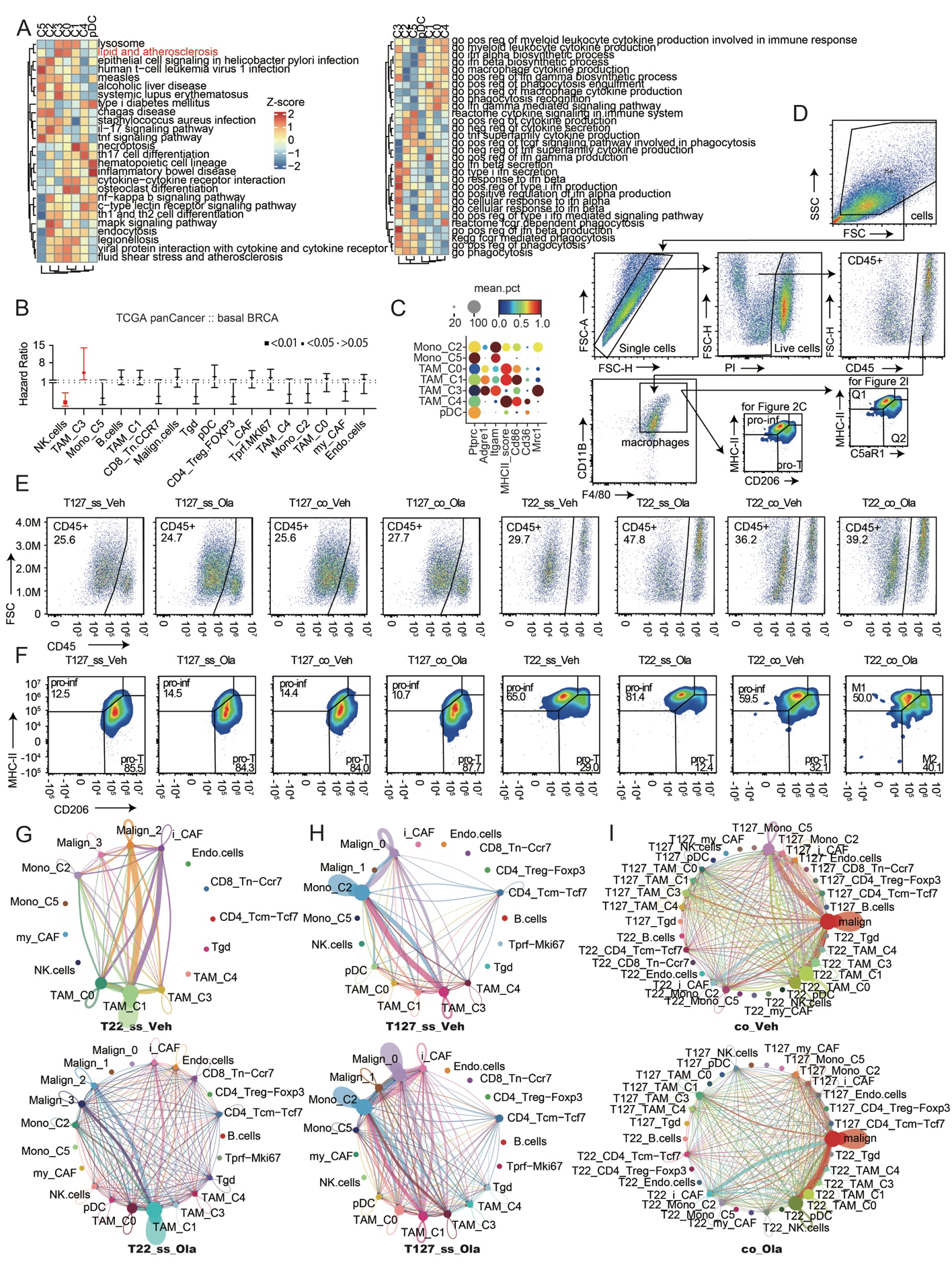
SupFig 2 (A) 热图显示了指定TAM亚群的不同表达路径的GSVA富集分数(左),以及指定TAM亚群的免疫相关路径的GSVA富集分数(右)。(B) 点图显示了在TCGA基底型乳腺癌数据库中指定细胞特征等级的风险比。细胞特征等级的截止值由‘survival’包的surv_cutpoint函数设置。p值通过点的大小表示。n=171。(C) 指定TAM亚群的亚型相关标记的点图。(D) 图2C和2J中淋巴细胞面板流式细胞术分析的工作流程。通过用CD11B和F4/80染色将细胞门控为巨噬细胞,对于图2C,通过MHCII和CD206染色将巨噬细胞亚型区分为MHC-IIhiCD206lo或MHCIIloCD206hi,对于图2J,通过MHCII和C5aR1染色将巨噬细胞亚型区分为MHC-IIhiC5aR1lo或MHCIIloC5aR1hi。(E) 指定组中CD45+细胞的分布模式。n=5。(F) 指定组中MHC-IIhiCD206lo或MHCIIloCD206hi TAM的分布模式。n=5。(G-I) 每个治疗组中各成分间的CellChat细胞-细胞通讯的圆形图。ss:单株移植模型。co:共同移植模型。Veh:载体。Ola:奥拉帕利。源数据提供在源数据文件中。
与奥拉帕尼治疗的 PARPi 敏感 T22 单一模型相比,奥拉帕尼治疗的 PARPi 耐药 T127(两种模型)和 T22 共移植模型显示出 TAM 和单核细胞群体的显著差异(图 2A、B)。在 T22 单一模型中,奥拉帕尼显著减少了 TAM_C3,但这种减少在 T22 共移植模型中被抑制,这表明共移植模型中的 PARPi 耐药与 TAM_C3 对奥拉帕尼作用的变化有关,TAM_C3 主要与抗炎活动相关基因的表达相关,并与 TCGA 基底型乳腺癌中的较差预后相关(补充图 1H、I 和补充图 2A、B)。相反,奥拉帕尼在任何 T127 模型中都没有减少 TAM_C3。与奥拉帕尼治疗的 T22 单一模型相比,奥拉帕尼治疗的 T22 共移植模型显示 TAM_C1 减少,TAM_C0 增加(图 2A、B)。有趣的是,在 T22 单一模型中增加的 TAM_C1 在共移植模型的 T22 肿瘤中减少,并进一步减少了奥拉帕尼治疗。TAM_C0 在 T22 单一和 T22 共移植模型中均高于 T127 模型,且在奥拉帕尼存在下维持或略有增加。Mono_C2 在 T127 单一和 T127 共移植模型中均高于 T22 模型(图 2B)。有趣的是,奥拉帕尼在所有模型中略微减少了 Mono_C2。Mono_C5 在两种 T127 模型中均因奥拉帕尼增加,但在两种 T22 模型中未发生变化或略有减少(图 2B)。综上所述,基于 TAM_C1 和 TAM_C3 相关的基因表达模式,T22 共移植模型中 TAM_C1 减少和 TAM_C3 增加可能是 T127 对 T22 共移植后 PARPi 敏感性降低的主要 TAM 群体。4
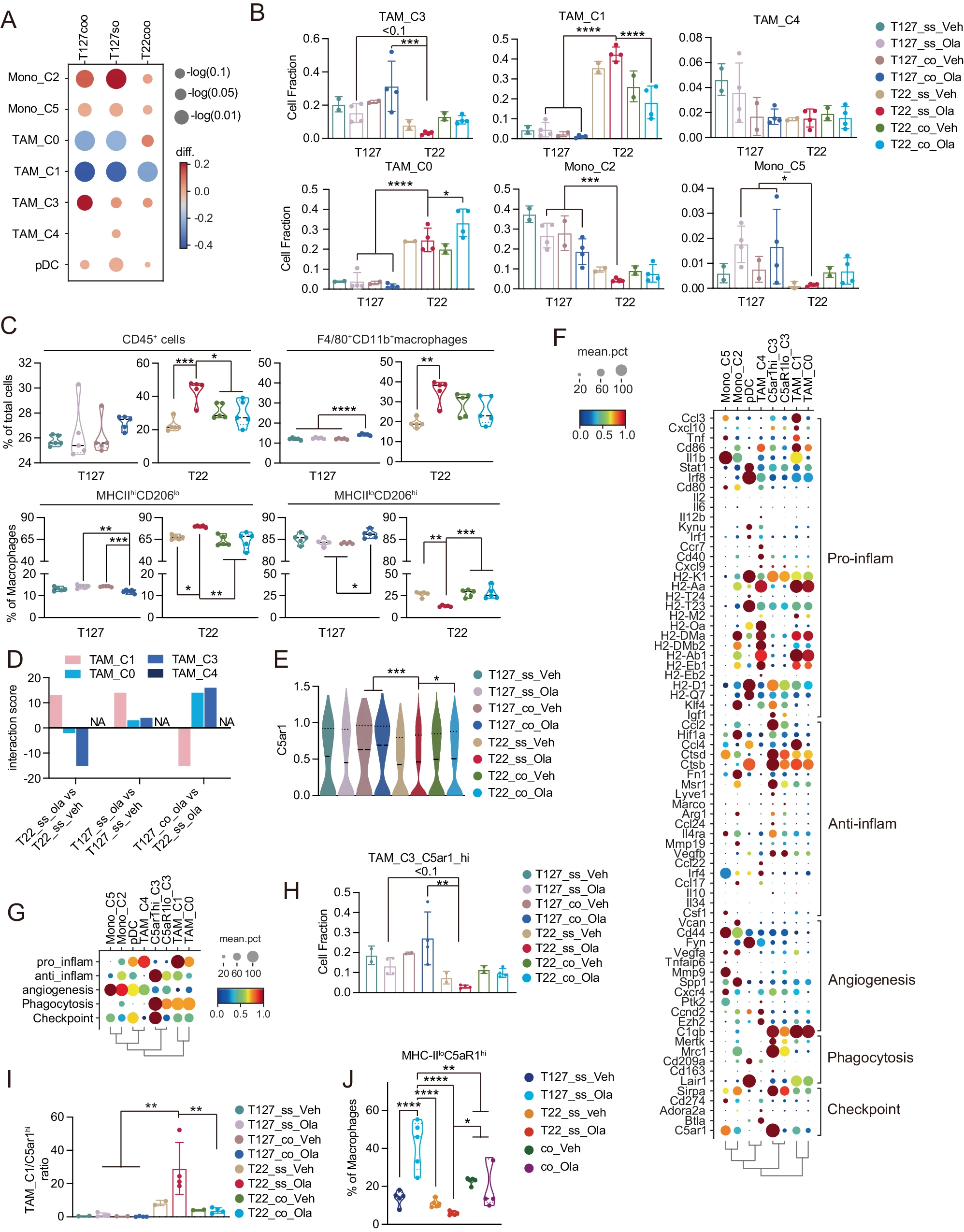
A 点图显示了每个模型中指示的细胞类型相对于在图 1 中的 T22 单移植模型中的相对差异。n=4。B 每个模型中指示的细胞类型在图 1 中的 scRNAseq 数据集中的细胞比例。统计显著性仅在奥拉帕尼处理组中进行评估。C 每个模型中通过流式细胞术评估的 CD45+ 细胞、总巨噬细胞(CD45+F4/80+CD11b+)、促炎性(CD45+F4/80+CD11b+MHCII 高 CD206 低)、促肿瘤(CD45+F4/80+CD11b+MHCII 低 CD206 高)TAM 的细胞比例。n=5。D 条形图显示在指示的比较对中,恶性细胞与指示的 TAM 细胞之间的配体 - 受体对相互作用计数估计的减少。E 小提琴图显示了指示组中 TAM 细胞中 C5ar1 的相对表达水平。F 点图显示了每个指示的 TAM 簇的代表每个通路的标志物。G 点图总结了每个指示的 TAM 簇的每个通路的表达水平。H 条形图显示了每个模型中 TAM_C3_C5ar1 高细胞在图 1 的 scRNAseq 数据集中的比例。统计显著性仅在奥拉帕尼处理组中进行评估。I 条形图显示了每个模型中 TAM_C1 细胞和 TAM_C3_C5ar1 高细胞在图 1 的 scRNAseq 数据集中的比例。统计显著性仅在奥拉帕尼处理组中进行评估。J 每个指示治疗组中外周血中总巨噬细胞中 MHC-II 低 C5aR1 高细胞的比例(见补充图 2 的门控策略),通过流式细胞术评估。n=5。统计显著性仅在奥拉帕尼处理组中进行评估。对于 B–E 和 H–I,所有数据均以平均值 ± 标准差表示,载体处理,n=2,奥拉帕尼处理,n=4。对于所有数据,p 值来自单向 ANOVA。* p < 0.05,** P < 0.01,*** p < 0.001,**** p < 0.0001。源数据和精确的 p 值作为源数据文件提供。
Mrc1 编码 CD206 在 TAM_C3 中高度表达,而在 TAM_C0、TAM_C1 和 TAM_C4 中非常低(补充图 2C)。相反,抗肿瘤 T 细胞反应所需的 MHCII 在 TAM_C0、TAM_C1 和 TAM_C4 中也高度表达,但在 TAM_C3 中非常低(补充图 2C)。在流式细胞术中,(F4/80+MHCII 低/CD206 高) 巨噬细胞代表 TAM_C3 巨噬细胞,而 (F4/80+MHCII 高/CD206 低) 代表 TAM_C0 和 TAM_C1 巨噬细胞。流式细胞术显示,与 T127 相比,T22 单一和 T22 共移植模型中的总 CD45+ 造血细胞以及 CD45、F4/80 和 CD11b 阳性巨噬细胞增加(图 2C 和补充图 2D–F)。奥拉帕尼在 T22 单一模型中诱导 CD45+ 造血细胞和 F4/80+CD11b+ 巨噬细胞增加,但在 T22 共移植模型中则被抑制。显著的是,T22 中 CD45+、F4/80+CD11b+、MHCII 高/CD206 低的巨噬细胞显著增加,代表 TAM_C0、TAM_C1 和 TAM_C4 巨噬细胞,这些细胞在 T22 单一模型中被奥拉帕尼增加,但在 T22 共移植模型中则被抑制。相反,CD45+、F4/80+CD11b+、MHCII 低/CD206 高的巨噬细胞在 T127 模型中水平显著更高。奥拉帕尼在 T22 单一模型中进一步减少了 CD45+、F4/80+CD11b+、MHCII 低/CD206 高的巨噬细胞,而在 T22 共移植模型中则被抑制(图 2C)。
这一结果再次支持了在共移植模型中 T127 对 T22 奥拉帕尼敏感性的影响可能依赖于 T22 单一和 T22 共移植模型中 TAM_C3 与 TAM_C0、TAM_C1 和 TAM_C4 巨噬细胞群体的相对水平,并且这些影响在奥拉帕尼存在下更加显著。
因此,我们探讨了可能导致 TAM 群体之间显著差异以及奥拉帕尼对 T127 和 T22 模型中 TAM 群体不同影响的机制,包括共移植肿瘤中的影响。CellChat,用于推断 scRNA-seq 数据集内细胞状态特异性信号通讯,已被用来可视化肿瘤细胞和 TAM 之间的配体 - 受体相互作用(补充图 2G–I,补充图 3A–C)。CellChat 捕捉到了巨噬细胞和肿瘤之间已知的相互作用,如 Lgals9-Cd45、Csf1-Csf1r、Cx3cl1-Cx3Cr1、Ccl7-Ccr2。然后,我们寻找在奥拉帕尼耐药模型中上调但在奥拉帕尼敏感模型中未上调的肿瘤细胞和 TAM 之间的配体 - 受体对(补充图 3D)。我们然后通过汇总在奥拉帕尼存在下与 T22 单一模型相比在 T22 共移植模型中增加的相互作用,并减去那些减少的相互作用,创建一个相互作用计数估计(见方法,补充图 3E)。基于共移植模型中的相互作用计数估计,只有 TAM_C3 和恶性细胞之间的 Rps19-C5ar1 相互作用在奥拉帕尼耐药模型中表现为上调,而在奥拉帕尼敏感模型中表现为下调(图 2D)。在受体转录本中,C5ar1 在 T127 和 T22 共移植模型中的 TAMs 中被奥拉帕尼显著上调,但在 T22 单一模型中则没有(图 2E)。基于此,我们探讨了 C5ar1 作为 T127 共移植使 T22 对奥拉帕尼耐药的潜在介导因子。因此,TAM_C3 根据其 C5ar1 表达重新分组。
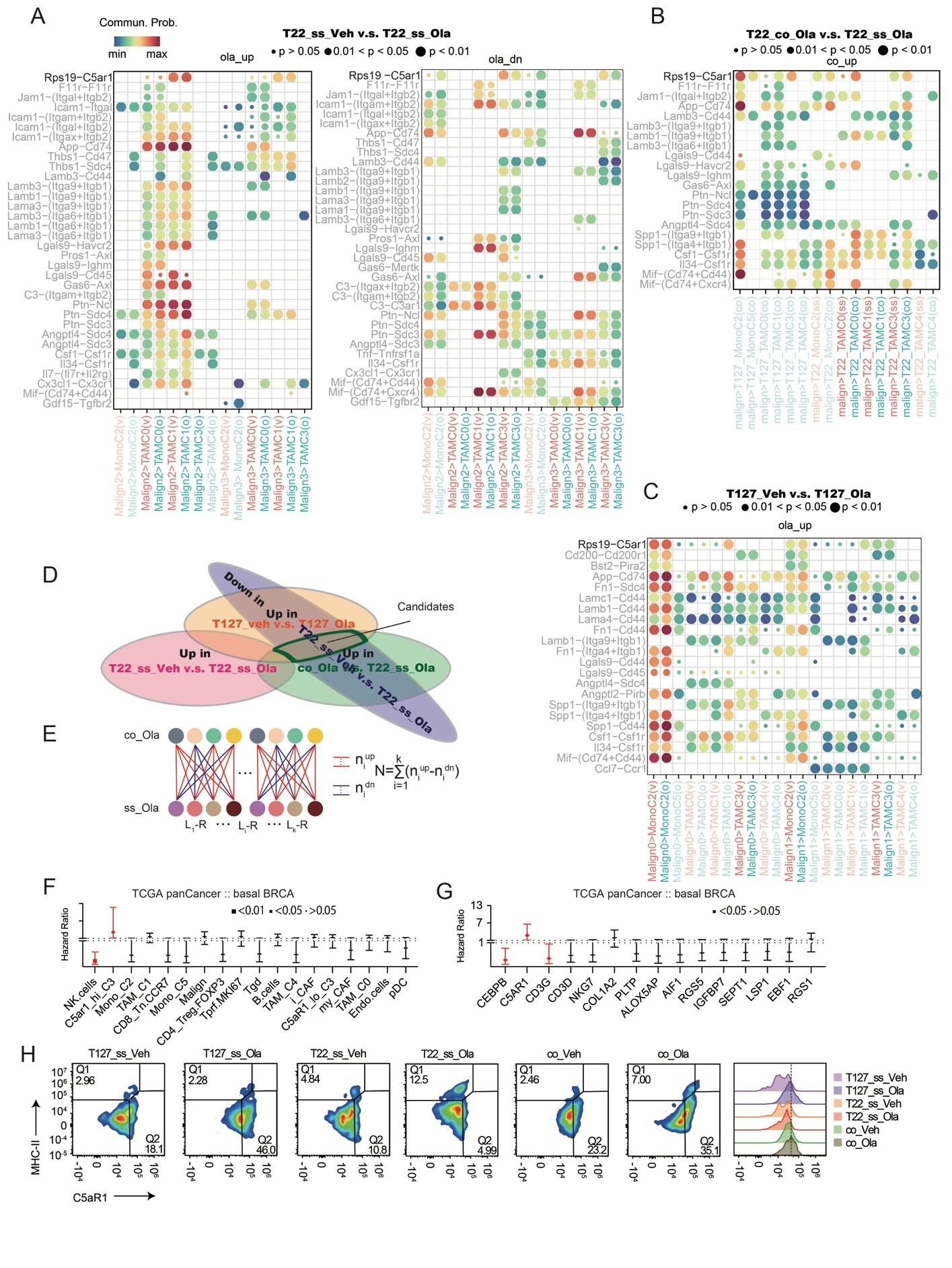
(A) 单株移植的 T22 肿瘤在有和没有奥拉帕利治疗情况下的配体-受体对变化。 (B) 奥拉帕利治疗下共同移植的 T22 肿瘤与单株移植的 T22 肿瘤相比的配体-受体对变化。ss: 单株移植模型。co: 共同移植模型。Veh: 载体。Ola: 奥拉帕利。图 1G 中的配体-受体对交互作用被突出显示。 (C) 单株移植的 T127 肿瘤在有和没有奥拉帕利治疗情况下的配体-受体对变化。 (D) 配体-受体候选选择策略的设计。候选配体-受体对表示在 T127 单株中介导肿瘤细胞和巨噬细胞之间通信并且被奥拉帕利在 T127 中上调(对奥拉帕利耐药),在 T22 单株中不存在或弱存在(对奥拉帕利敏感),并且在奥拉帕利治疗的共同移植 T22 模型中上调(对奥拉帕利耐药)的那些。 (E) 基于 CellChat 输出的交互次数估计设计,比较奥拉帕利治疗的单株 T22 肿瘤与共同移植的 T22 肿瘤。交互次数估计是增加的每个比较对的交互总和,并减去每个比较对中减少的交互(见方法)。 (F) TCGA 基底型乳腺癌数据库中指定细胞特征排名的危险比点图。细胞特征排名的截止值由‘survival’包的 surv_cutpoint 函数设定。p 值通过点大小显示。n=171。 (G) TCGA 基底型乳腺癌数据库中指定基因的危险比点图。每个基因的截止值由‘survival’包的 surv_cutpoint 函数设定。p 值通过点大小显示。n=171。 (H) 奥拉帕利治疗和未治疗情况下,来自 T22、T127 和共同移植模型的血液中巨噬细胞的代表性细胞分布模式。来源数据作为来源数据文件提供。
将 TAM_C3 重新分组为 C5ar1 高和 C5ar1 低群体后,与抗炎活性相关的基因表达仅在 C5ar1 高表达的 C3 群体中,而 C5ar1 高表达的亚群缺乏与促炎活性相关的基因表达(图 2F、G)。相反,C5ar1 低表达的 C3 亚群未表达显著水平的促炎活性或抗炎活性相关的基因。与 TAM_C3 一致,C5ar1 高表达的 C3 群体在 TCGA 基底型乳腺癌中主要与较差的预后相关(补充图 3F),并且在通过多变量分析评估簇顶级 DEG 对 TCGA 基底型乳腺癌预后的贡献时,C5AR1 独立与较差的预后相关(补充图 3G)。在奥拉帕尼敏感模型(T22 单一)中,C5aR1 高表达的 C3 巨噬细胞减少,但在耐药模型(T127 和 T22 共移植模型)中没有(图 2H)。当绘制 TAM_C1 与 C5ar1 高表达的 C3 巨噬细胞的比例时,差异效果最为明显(图 2I)。
在 T22 共移植模型中 TAM 数量和表型变化的一个潜在解释是 TAMs 从 T127 向 T22 的转移。支持这一观点的是,外周血细胞的流式细胞术显示,与 T22 单一模型相比,T127 模型和 T22 共移植模型中的血液中 MHC-II 低 C5aR1 高的巨噬细胞水平更高(图 2J 和补充图 3H)。在奥拉帕尼治疗下,T127 单一和 T127 共移植模型中的血液中 MHC-II 低 C5aR1 高的巨噬细胞数量进一步增加,而 T22 单一模型中略有减少。
总的来说,在 T127 单一和 T127 共移植模型中的血液中发现 MHC-II 低 C5aR1 高的巨噬细胞水平升高,并且在奥拉帕尼治疗下进一步增加,这与 MHC-II 低 C5aR1 高的巨噬细胞从 T127 向 T22 转移一致,使通常对奥拉帕尼敏感的 T22 模型对奥拉帕尼耐药。
奥拉帕尼在 T22 单一模型中诱导巨噬细胞亚群和通讯的快速变化¶
为了捕捉奥拉帕尼在较早时间点对肿瘤微环境的影响,我们在治疗奥拉帕尼 4 天和 8 天后评估了 T22 和 T127(图 3A)。为了减少批次效应,所有肿瘤在同一时间收获,通过在 8 天后移植“4 天肿瘤”来实现(图 3A)。肿瘤建立 14 天后,使用奥拉帕尼治疗所有肿瘤,并在同一天收获所有肿瘤进行 scRNA-seq 分析(图 3A)。在严格细胞选择后的 scRNA-seq 数据中,有超过 20,000 个细胞可供进一步分析。即使短期治疗 T22 而非 T127 也足以减少肿瘤生长和肿瘤重量(图 3B、C)。
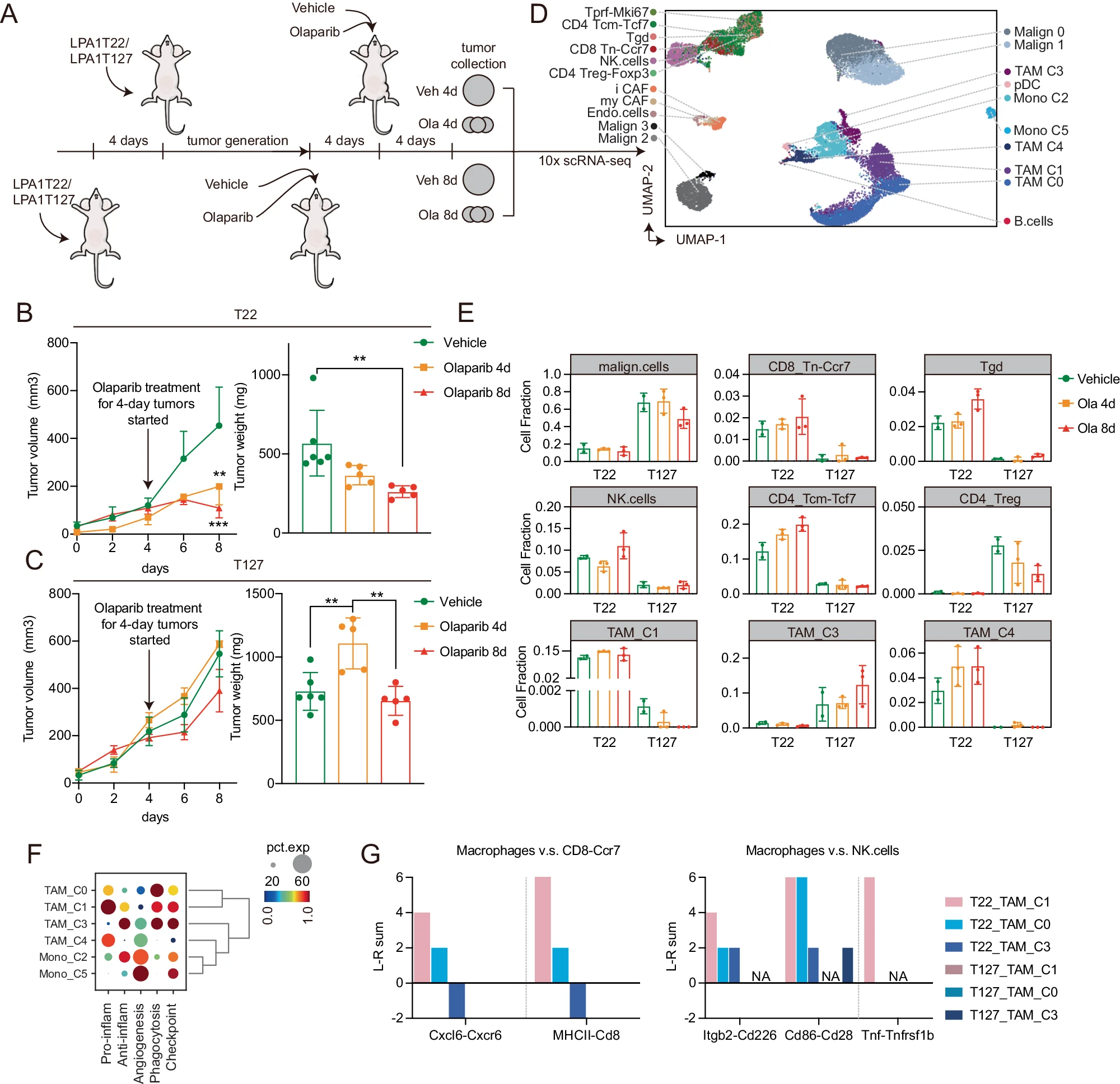
A T22 和 T127 模型在奥拉帕尼治疗 4 天或 8 天的研究设计。4d:4 天,8d:8 天。B T22 模型在奥拉帕尼(50 mg/kg,po,每日)治疗 4 天和 8 天后的生长曲线(左)和研究结束时的肿瘤重量(右)。载体处理组,n=6,奥拉帕尼处理组,n=5。数据以平均值 ± 标准差表示。p 值来自单向 ANOVA。** P < 0.01。C T127 模型在奥拉帕尼治疗 4 天或 8 天后的生长曲线(左)和研究结束时的肿瘤重量(右)。载体处理组,n=6,奥拉帕尼处理组,n=5。数据以平均值 ± 标准差表示。p 值来自单向 ANOVA。** P < 0.01。D 从 A 中指示的小鼠模型中分层和细胞类型鉴定的 T22 和 T127 肿瘤。T127(Malign_0,Malign_1)和 T22(Malign_2,Malign_3)的恶性细胞显示出不同的细胞簇。Tcm:中央记忆 T 细胞,Treg:调节性 T 细胞,Tn:初始 T 细胞,Tgd:γδT 细胞,Tprf:增殖性 T 细胞,my_CAF:肌成纤维细胞相关癌细胞,i_CAF:炎症相关癌细胞,pDC:浆细胞样树突细胞。载体处理组,n=2,奥拉帕尼处理组,n=3。E 条形图显示了在 D 中的 scRNAseq 数据集中定义的每个细胞谱系标记(图 1E)中上皮细胞(高 CNV 细胞)、NK 细胞和 i_CAF 的相对数量(百分比)。数据以平均值 ± 标准差表示。由于每组样本量较小,统计显著性不适用。F 点图显示了每个指示的 TAM 簇的每个通路的总结表达水平。用于功能通路得分的基因列表列在图 2F 中。G CellChat 估计的每个 TAM 簇与 CD8 初始 T 细胞(左)或 NK 细胞(右,定义见图 1E)之间的配体 - 受体相互作用计数在奥拉帕尼治疗后的变化。由于 T127 中 T 细胞数量较少,因此 T127 中未记录到相互作用。
使用图 1 和图 2 中的细胞谱系定义方法,恶性 T127(Malign_0,Malign_1)和 T22(Malign_2,Malign_3)再次形成了独立的簇,而两种肿瘤模型的非恶性细胞类型则融合在一起(图 3D 和补充图 4A)。与晚期时间点(见图 1)相比,早期(4 天和 8 天,图 3D)的肿瘤中免疫细胞占更大比例。NK 细胞和 T 细胞几乎完全存在于 T22 肿瘤中(图 3E 和补充图 4B、C)。CD4 Treg 细胞仅在 T127 肿瘤中存在,但在奥拉帕尼治疗后没有富集(图 3E)。T22 或 T127 肿瘤的奥拉帕尼处理和未处理样本之间的 B 细胞或 CAF 亚型没有统计学显著变化。
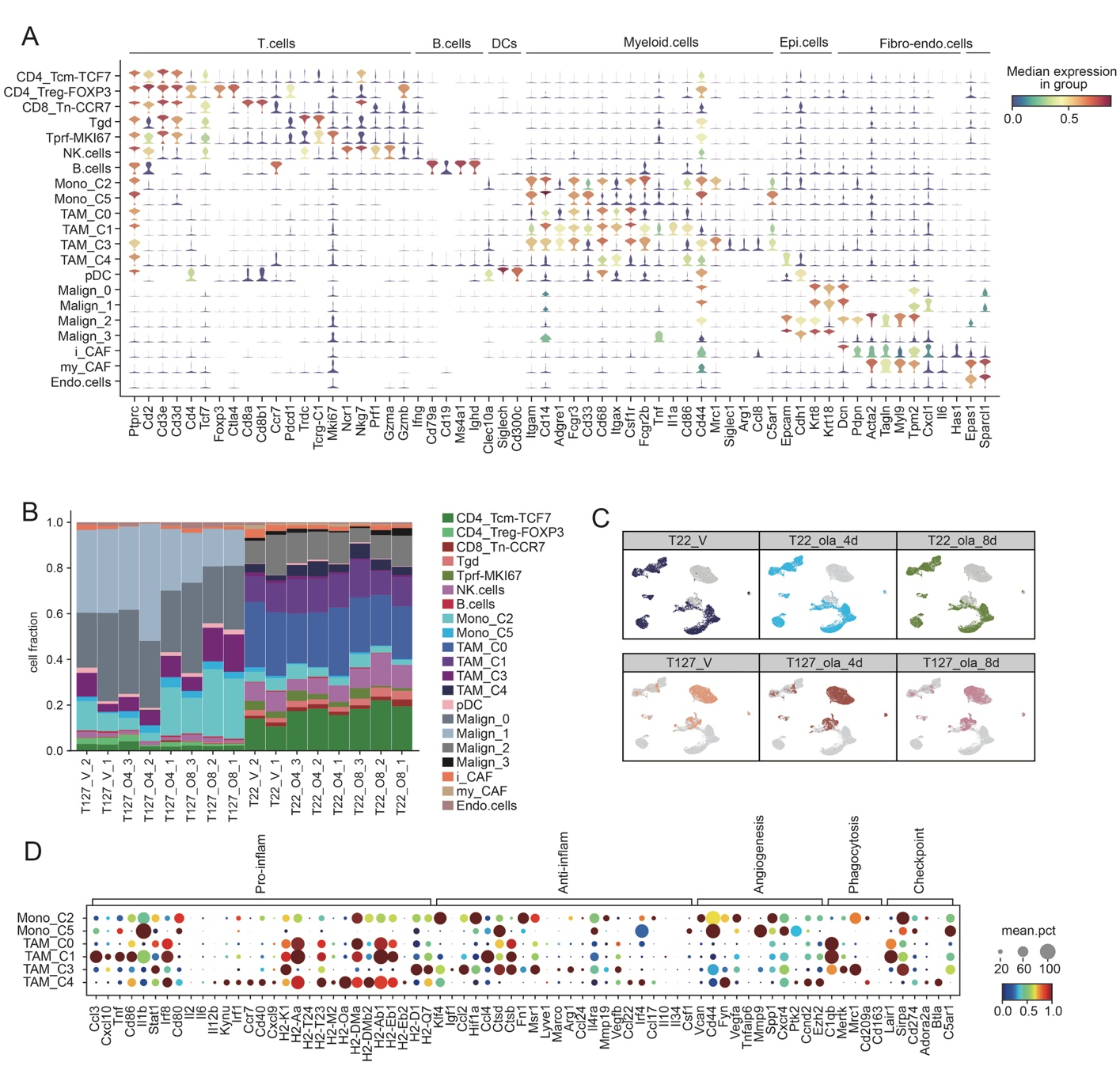
(A) 小提琴图显示用于验证 T 亚型和巨噬细胞亚型注释的标记,以及用于上皮细胞、成纤维细胞、内皮细胞、树突细胞和 NK 细胞亚型划分的标记。 (B) T22 和 T127 肿瘤中每种细胞类型的细胞分数。V:载体,O4:奥拉帕利处理 4 天,O8:奥拉帕利处理 8 天。 (C) UMAP 图(图 3D)中每只小鼠的 T22 和 T127 肿瘤簇的分布。V:载体,O4:奥拉帕利处理 4 天,O8:奥拉帕利处理 8 天。T22_O4_1:2152 个细胞,T22_O4_2:2482 个细胞,T22_O4_3:1694 个细胞,T22_O8_1:1701 个细胞,T22_O8_2:1094 个细胞,T22_O8_3:1807 个细胞,T22_V_1:1436 个细胞,T22_V_2:2114 个细胞,T127_O4_1:1147 个细胞,T127_O4_2:771 个细胞,T127_O4_3:1136 个细胞,T127_O8_1:1113 个细胞,T127_O8_2:1203 个细胞,T127_O8_3:832 个细胞,T127_V_1:1180 个细胞,T127_V_2:695 个细胞。载体处理,n = 2;奥拉帕利处理,n = 3。 (D) 点图显示表示每个 TAM 簇通路的标记。
与长期治疗一致,在 T22 中,奥拉帕尼在第 4 天增加了 TAM_C1 细胞,这一现象持续到第 8 天,同时 TAM_C3 细胞在第 4 天有所减少,这一现象也持续到第 8 天(图 3E,补充图 4B、C)。此外,短期模型中的 TAM_C1 和长期模型一样,表达与促炎表型相关的基因,而 TAM_C3 再次类似于长期模型,表达与抗炎、吞噬和检查点抑制特征相关的基因(图 3F 和补充图 4D)。根据 CellChat,奥拉帕尼显著增加了 T22 模型中恶性细胞、TAM_C1、NK 细胞、i_CAF、CD8_Tn.CCR7 和 CD4_Tcm.Tcf7 细胞之间的细胞 - 细胞通讯,但在 T127 模型中没有显著增加(补充图 5A、B)。在 T127 模型中,恶性细胞和 TAM_C3 之间的通讯显著,并且在整个治疗期间被奥拉帕尼增加(补充图 5A、B)。
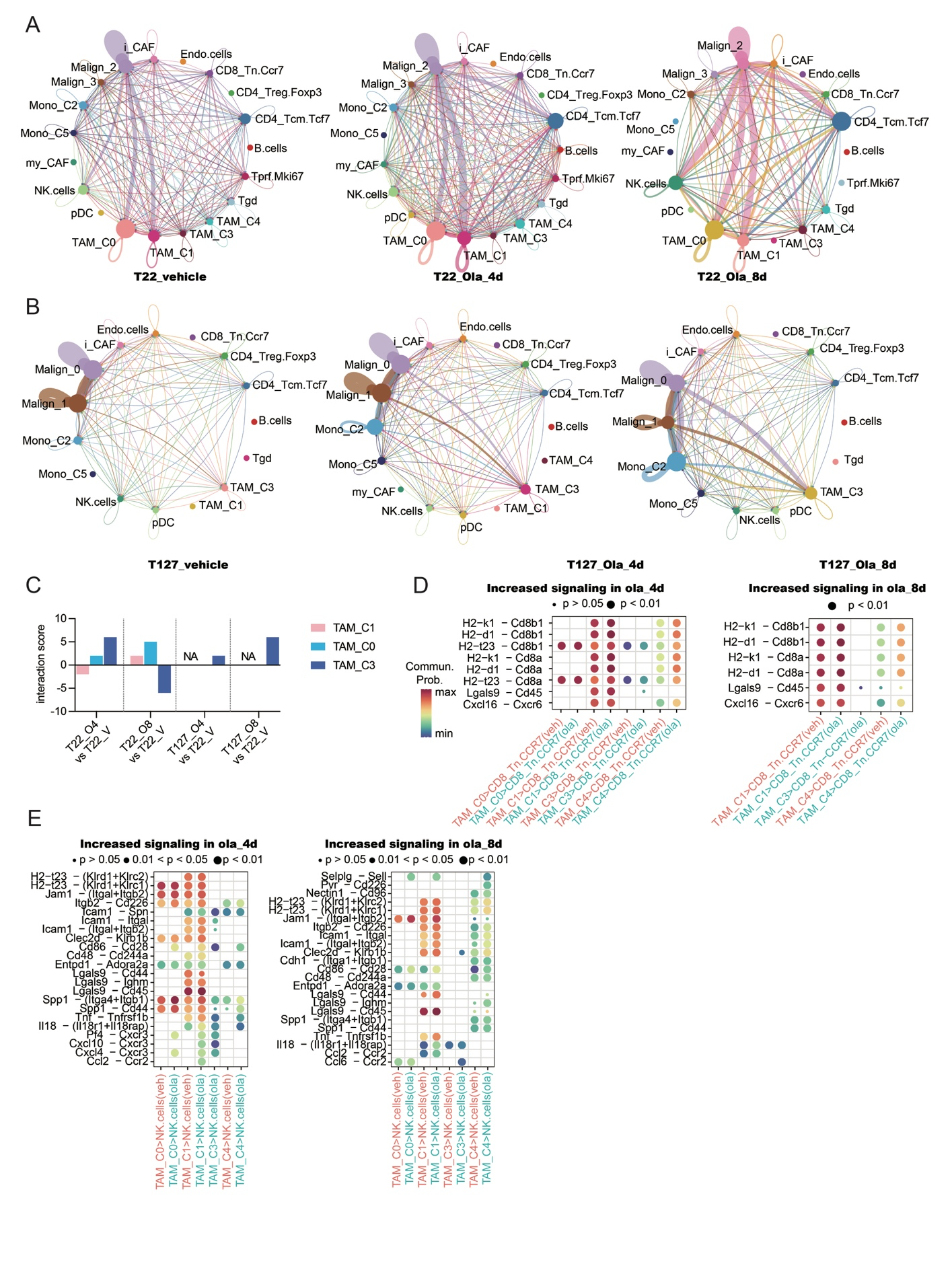
(A) 圆形图显示了用载体或奥拉帕利处理4天或8天的T22肿瘤中每种细胞类型之间的细胞间通信。 (B) 圆形图显示了用载体或奥拉帕利处理4天或8天的T127肿瘤中每种细胞类型之间的细胞间通信。 (C) 条形图显示奥拉帕利处理后的T22或T127肿瘤中,恶性细胞与每个TAM簇之间的配体-受体对交互作用次数估计值,与载体处理后的T22或T127肿瘤相比。 (D) 显著上调的配体-受体通信在载体或奥拉帕利处理4天或8天的T22中,巨噬细胞和初始CD8 T细胞之间。 (E) 显著上调的配体-受体通信在载体或奥拉帕利处理4天或8天的T22中,巨噬细胞和NK细胞之间。来源数据作为来源数据文件提供。
为了量化奥拉帕尼对细胞类型之间相互作用的影响,相对于载体,视觉化奥拉帕尼诱导的配体 - 受体相互作用计数估计的变化。恶性细胞和 TAM_C3 之间的 Rps19-C5ar1 相互作用在 T22 模型中被奥拉帕尼治疗 4 天后短暂增加,但在第 8 天减少。相反,在 T127 模型中,恶性细胞和 TAM_C3 之间的 Rps19-C5ar1 相互作用在所有时间点都被奥拉帕尼增加(补充图 5C)。在 T22 中,奥拉帕尼增加了 TAM_C1 和 CD8_Tn-Ccr7 细胞之间的 Cxcl16-Cxcr6、MHCII-Cd8 相互作用计数估计,但没有增加 TAM_C3 和 CD8_Tn-Ccr7 细胞之间的相互作用(图 3G 和补充图 5D)。奥拉帕尼还诱导了 NK 细胞和 TAM_C1 之间的 Itgb2-Cd226、Cd86-Cd28 和 Tnf-Tnfrsf1b 相互作用增加,但在 T127 模型中没有(图 3G 和补充图 5E)。
这些数据一致表明,奥拉帕尼在 T22 中诱导了与促炎特征相关基因表达的迅速增加,以及 TAM_C1 巨噬细胞与 T22 中的初始 T 细胞和 NK 细胞之间的通讯增加,而在 T127 中没有。
C5aR1 抑制使共移植的 T22 和内在 PARPi 耐药的 T127 对奥拉帕尼敏感¶
我们的数据表明,C5aR1 在 TAM_C3 巨噬细胞中高水平表达,CellChat 表明它参与了 TAM_C3 巨噬细胞和肿瘤细胞之间的通讯。为了确定 C5aR1 是否可能导致内在 T127 的 PARPi 耐药性和共移植模型中 T22 的 PARPi 耐药性,我们在共移植的 T22 和 T127 中在有无奥拉帕尼的情况下抑制了 C5aR1。在小鼠模型中,使用 PMX53(IC50 为 20nM 的合成多肽活性补体 C5a 受体拮抗剂)抑制 C5aR1。显著的是,奥拉帕尼和 PMX53 单药治疗活性有限,但奥拉帕尼和 PMX53 的联合治疗不仅在共移植模型中重新使 T22 对奥拉帕尼敏感,还控制了内在奥拉帕尼耐药的 T127 的生长曲线和肿瘤重量,且对体重没有明显变化(图 4A、B,补充图 6A–C)。

Fig4 A T127 的生长曲线(左)和代表性 T127 肿瘤的图像(右),来自接受对照、奥拉帕尼、PMX53 或联合治疗 14 天的共移植 T22 和 T127 肿瘤模型。n=5。B T22 的生长曲线(左)和代表性 T22 肿瘤的图像(右),来自接受对照、奥拉帕尼、PMX53 或联合治疗 14 天的共移植 T22 和 T127 肿瘤模型。n=5。C 每个指示治疗组中总巨噬细胞(F4/80+CD11b+)中促炎性(MHCII 高 CD206 低)TAM(左)、促肿瘤(MHCII 低 CD206 高)TAM(中,见补充图 2B-D 中的门控策略)和促炎性与促肿瘤 TAM 的比例(右),通过流式细胞术评估。n=5。D 每个指示组中 CD3+F4/80− 细胞(左上)、CD3−F4/80− 细胞(右上)、CD3+CD8+ 细胞(右上)和 CD3−CD335+ 细胞(右下)的比例,通过流式细胞术评估。NK 细胞门控为 CD3−F4/80−CD335+ 群体。n=5。E 接受对照、抗 CD8(5 mg/kg,i.p.,隔日一次),PMX53(3 mg/kg,s.c.,隔日一次)联合抗 CD8 或抗 CD8、PMX53 和奥拉帕尼(50 mg/kg,o.g.,每日一次)联合治疗 14 天的共移植 T22 和 T127 肿瘤模型的 T127(左)和 T22(右)肿瘤生长曲线。aCD8:抗 CD8。P + O:PMX53 联合奥拉帕尼。n=5。F 接受(5×10^5)C5aR1 高细胞转移后,载体或奥拉帕尼处理 8 天的小鼠中 T22 的肿瘤生长曲线。治疗在 C5aR1 高细胞转移后的第二天开始。n=5。G 每个治疗组中 MHC-II 低 C5aR1 高、CD3+F4/80− 细胞、CD3−F4/80− 细胞、CD3+CD8+ 细胞和 CD3−CD335+ 细胞的比例,通过流式细胞术评估。n=5。所有数据均以平均值 ± 标准差表示。p 值来自单向 ANOVA。* p < 0.05,** P < 0.01,*** p < 0.001,**** p < 0.0001。
为了将我们的发现扩展到另一个同种模型和 PARP1 选择性抑制剂 AZD5305,我们用 PMX53、奥拉帕尼或 AZD5305 以及 PMX53 和 PARPi 的组合治疗 KPCA 卵巢移植模型。与乳腺癌模型相似,组合治疗显示显著的生长控制,而单药治疗无显著效果(补充图 6D)。
在 T22 共移植模型中,PMX53 增加了 MHCII 高 CD206 低的巨噬细胞数量(代表 TAM_C0、TAM_C1 和 TAM_C4),这种增加在 PMX53 和奥拉帕尼的组合中进一步增加(图 4C)。同时,PMX53 减少了 MHCII 低 CD206 高的巨噬细胞数量(代表 TAM_C3),组合治疗在 T22 共移植模型中有轻微的额外效果。PMX53 显著且统计显著地增加了 MHCII 高 CD206 低/MHCII 低 CD206 高的比率,这一比率在组合治疗中进一步增加(图 4C)。在 T127 共移植模型中,奥拉帕尼减少了 MHCII 高 CD206 低的巨噬细胞数量(如前所示,图 4C,补充图 6E)。单独使用 PMX53 对 T127 中的 MHCII 高 CD206 低的巨噬细胞影响有限,但 PMX53 逆转了奥拉帕尼单独诱导的 MHCII 高 CD206 低巨噬细胞的减少,实际上增加了 MHCII 高 CD206 低的巨噬细胞数量。在 T127 中,对 MHCII 低 CD206 高的巨噬细胞的影响与对 MHCII 高 CD206 低的巨噬细胞的影响相反(图 4C,补充图 6E)。虽然奥拉帕尼增加了 MHCII 高 CD206 低的巨噬细胞数量,而 PMX53 单独效果不大,但组合治疗显著减少了 MHCII 低 CD206 高的巨噬细胞数量。对 MHCII 高 CD206 低和 MHCII 低 CD206 高巨噬细胞的组合效应导致 T127 中 MHCII 高 CD206 低/MHCII 低 CD206 高的比率显著增加,这可能促进了对 PMX53 和奥拉帕尼组合的响应。

SupFig 6 (A) 共同移植的T22和T127肿瘤模型分别在控制组(载体 + PMX-53C)、奥拉帕利、PMX-53或组合治疗14天后的T22(左)和T127(右)肿瘤重量。n=5。 (B) 根据最后一天的肿瘤体积计算的肿瘤生长抑制率。PO = PMX53 + 奥拉帕利。n=5。 (C) 小鼠在接受控制组、奥拉帕利、PMX-53或组合治疗前后的体重变化。n=5。 (D) KPCA小鼠模型在接受控制组、奥拉帕利、AZD5305、PMX53、PMX53和奥拉帕利组合或PMX53和AZD5305组合治疗28天后的肿瘤生长曲线。第29天的体积是通过解剖肿瘤测量的,比在小鼠体内测量的更准确。载体处理组和AZD5305+PMX53处理组,n=7;其他组,n=8。 (E) 图4C中每种治疗的MHCIIhiCD206lo和MHCIIloCD206hi细胞分布模式。n=5。 (F) 图4D中淋巴细胞面板的流式细胞术分析工作流程。n=5。 (G) 磁柱选择后通过流式细胞术评估的C5aR1hi细胞纯度。 (H) 接受C5aR1hi细胞的小鼠在接受控制组、奥拉帕利、PMX53或组合治疗8天后的T22肿瘤重量。n=5。 (I) 接受和未接受奥拉帕利治疗的接受C5aR1hi细胞的小鼠在治疗前后的体重变化。n=5。 (J) 图5中CD8 T面板的流式细胞术分析工作流程。n=3。 (K) 共培养实验工作流程:从小鼠中纯化T127肿瘤细胞,然后在培养中用载体或AZD5305(5nM)处理4天。从肿瘤未感染小鼠中分离的CD11b+C5aR1-/+巨噬细胞在第4天添加。1×10^6 个T127肿瘤细胞在培养中用或不用AZD5305(5nM)处理4天。然后与从肿瘤未感染小鼠CD11b+阳性脾细胞中分离的5×10^5 个CD11b+C5aR1-/+巨噬细胞共培养1天,用或不用PMX53(40nM)。在第5天,分离CD11b+C5aR1-/+巨噬细胞,然后与从肿瘤未感染小鼠中分离的5×10^5个CD8 T细胞共培养。通过流式细胞术评估每个指示组中的指示CD8 T细胞比例。n=5,所有数据均以均值+/- SD表示。p值来自单因素方差分析。
对 MHCII 高 CD206 低和 MHCII 低 CD206 高巨噬细胞的影响传递到了 CD8 T 细胞和 NK 细胞的变化上(图 4D,补充图 6F)。在 T22 共移植模型中,PMX53 组合治疗显著增加了 CD3 和 CD8 T 细胞,对 NK 细胞数量的影响有限(图 4D)。在未经治疗的 T127 共移植模型中,PMX53 诱导了 CD3+ T 细胞的增加,并在组合治疗中得以维持。奥拉帕尼治疗后 T127 中 NK 细胞水平的降低被 PMX53 逆转(图 4D)。支持 CD8 T 细胞在 PMX53 与奥拉帕尼组合治疗中的作用,抗 CD8 治疗显著减弱了组合治疗的疗效(图 4E)。
为了直接测试来自 T127 的 C5aR1 高表达巨噬细胞是否在共移植模型中参与了对 T22 的耐药性传递,我们从 T127 肿瘤负荷小鼠的血液中分离了 C5aR1 高表达细胞(主要是 TAM_C3)并将其转移到 T22 肿瘤负荷小鼠中(补充图 6G)。显著的是,来自 T127 肿瘤负荷小鼠血液的 C5aR1 高表达细胞足以使 T22 对奥拉帕尼耐药(图 4F,补充图 6H、I)。正如预期的那样,流式细胞术显示 C5aR1 高表达细胞转移后,T22 肿瘤中 MHC-II 低 C5aR1 高巨噬细胞的水平更高。与 8 天奥拉帕尼处理的 T22 肿瘤的 scRNA-seq 数据一致,奥拉帕尼处理后 MHC-II 低 C5aR1 高促肿瘤巨噬细胞的轻微减少仅在未转移模型中观察到,而在 C5aR1 高表达细胞转移模型中未观察到。C5aR1 高表达细胞移植显著减少了 T22 中的 CD3 和 CD8 比例(图 4G)。相反,C5aR1 高表达细胞的转移并未改变 NK 细胞水平。
综上所述,这些数据一致表明 C5aR1 在 T127 和 T22 共移植模型中起到了重要作用。PMX53 和奥拉帕尼治疗小鼠中 MHCII 低 CD206 高巨噬细胞比例的降低以及 CD8 T 细胞的增加可能促成了 PMX53 和奥拉帕尼组合在 T127 和 T22 共移植模型中诱导的肿瘤生长抑制。
经奥拉帕尼处理的 T127 培养的 C5aR1 高表达巨噬细胞在体外改变 T 细胞活性¶
为了研究 T127 培养的 C5aR1 高表达巨噬细胞对体外 T 细胞功能的影响,我们首先从载体或奥拉帕尼处理的小鼠中分离 T127 肿瘤细胞,如图 1 所述。然后将纯化的 T127 肿瘤细胞与从肿瘤无感染小鼠脾细胞中分离的 CD11b+C5aR1 低或 CD11b+C5aR1 高巨噬细胞一起培养,培养时是否加入 PMX53(40 nM)在 transwell 系统中进行 24 小时(详情见方法)。然后将预培养的巨噬细胞(无 PMX53)与从肿瘤无感染小鼠脾细胞中分离的 CD8+ T 细胞共培养。预培养的 C5aR1 低和 C5aR1 高巨噬细胞与载体处理的 T127 细胞共同培养未显著改变 CD8 活性,如颗粒酶 B 或 IFNγ 表达,尽管穿孔素(PRF1)略有增加(图 5A 和补充图 6J)。PMX53 未改变颗粒酶或穿孔素表达,但略微增加了 IFNγ 表达(图 5A)。相反,经奥拉帕尼处理的 T127 肿瘤细胞预培养的 C5aR1 高巨噬细胞减少了颗粒酶 B(GZMB)和穿孔素表达(图 5B)。目前尚不清楚原因的情况下,CD8 T 细胞中 IFNγ 表达增加。C5aR1 高巨噬细胞预培养的 T127 肿瘤细胞与奥拉帕尼共同培养期间 C5aR1 抑制逆转了对颗粒酶 B 和穿孔素表达的影响,而与奥拉帕尼处理的 T127 肿瘤细胞预培养相比,IFNγ 未进一步增加(图 5B)。
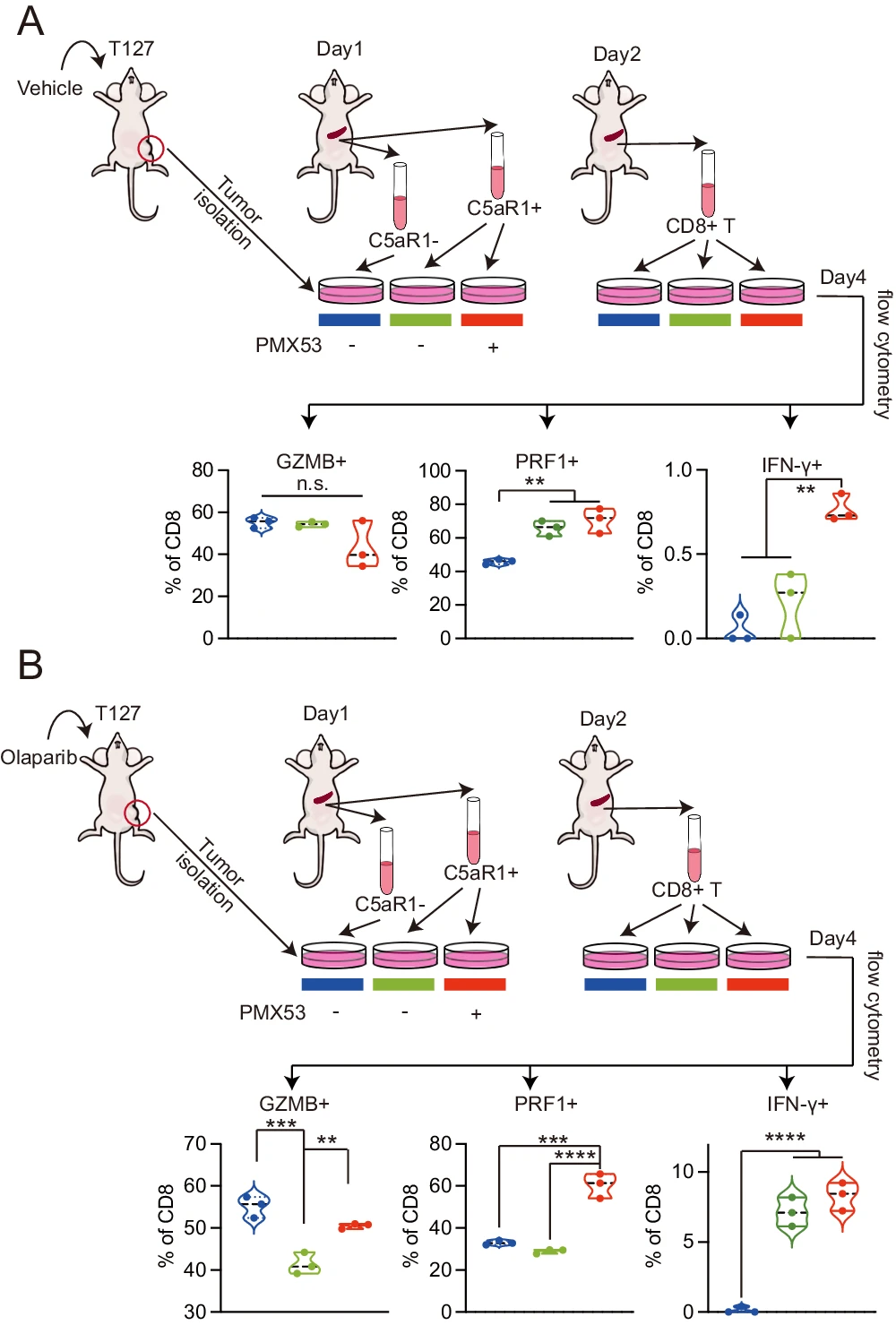
A 工作流程:如图 1 所述,从对照小鼠中纯化的 T127 肿瘤细胞、从无肿瘤小鼠中分离的 CD11b+C5aR1−/+ 巨噬细胞以及从无肿瘤小鼠中分离的 T 细胞的体外共培养实验(详情见方法)。简而言之,在第 1 天,1×10^6 从对照处理的小鼠中分离的 T127 肿瘤细胞与 5×10^5 CD11b+C5aR1−/+ 巨噬细胞(有或没有 PMX53(40 nM))共同培养。在第 2 天,分离 CD11b+C5aR1−/+ 巨噬细胞,然后与 5×10^5 分离的 CD8 T 细胞共同培养。如方法中所述,从 CD11b+ 阳性脾细胞中分离 CD11b+C5aR1−/+ 巨噬细胞。通过流式细胞术评估左侧每个指示组中 GZMB+、PRF1+、IFNγ+ CD8+ T 细胞的比例。n=3。数据以平均值 ±SD 表示。通过单向 ANOVA 进行比较。* p < 0.05,** p < 0.01,*** p < 0.001,**** p < 0.0001。
接下来,我们将我们的发现扩展到 PARP1 选择性抑制剂 AZD5305(补充图 6K)。我们从未经治疗的小鼠中分离 T127 肿瘤细胞。纯化的 T127 肿瘤细胞经过 DMSO 或 AZD5305 处理 96 小时,然后与从肿瘤无感染小鼠脾细胞中分离的 CD11b+C5aR1 高巨噬细胞共同孵育,在 transwell 系统中是否加入 PMX53(40 nM)共孵育 24 小时,如上所述。然后我们将预孵育的巨噬细胞(无 PMX53)与从肿瘤无感染小鼠脾细胞中分离的 CD8+ T 细胞共培养(补充图 6K)。在体外用 AZD5305 处理的 T127 肿瘤细胞,然后在 transwell 系统中与 C5aR1 巨噬细胞共同培养,导致 C5aR1 巨噬细胞抑制 T 细胞激活,这通过 GMZB、PRF1 或 IFNγ 的阳性来评估(补充图 6K)。这种效果被 PMX53 治疗逆转。因此,AZD5305 处理的 T127 肿瘤细胞产生了免疫抑制性细胞表型,这种表型被 PMX53 治疗逆转。
C5aR1 促进 TAM_C3 极化¶
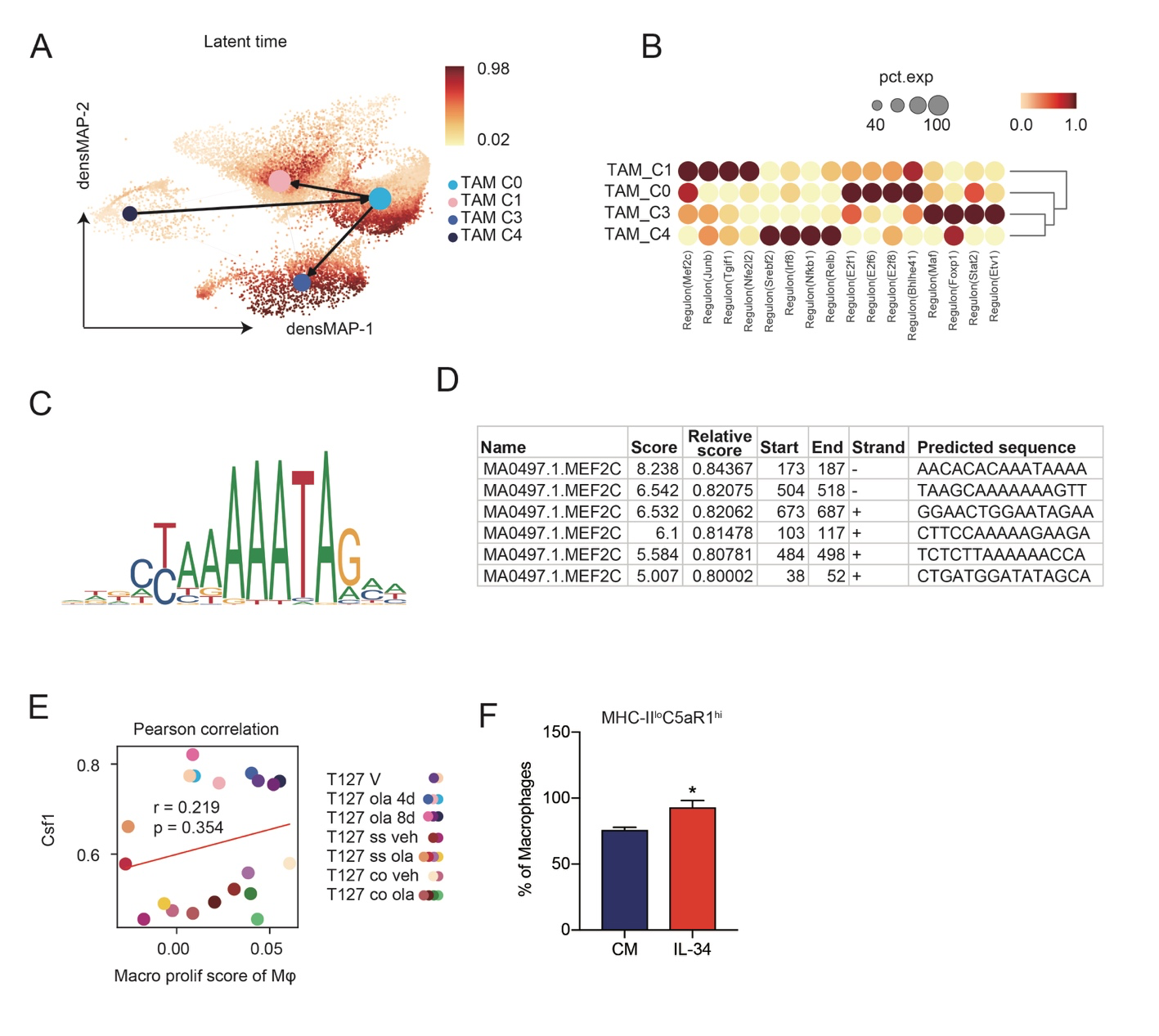
SupFig7 (A) 使用scVelo中的潜在时间在基于分区的图抽象(PAGA)表示中的细胞亚簇被映射在密度保持的统一流形近似和投影图(densMAP)上,按图1和图3中提到的所有肿瘤的巨噬细胞各亚簇的潜在时间得分进行着色。 (B) 点图显示由pySCENIC在(A)中提到的所有肿瘤的汇总巨噬细胞的每个巨噬细胞亚型中富集的指示转录因子的AUC。 (C) Jaspar预测的Mef2c在C5ar1上的结合位点的基序。 (D) Jaspar预测的Mef2c在C5ar1转录调控区域的结合潜力。 (E) (A)中相同肿瘤中的恶性细胞表达的Csf1 mRNA与巨噬细胞增殖评分之间的相关性。V:短期载体处理的肿瘤。veh:载体。ola:奥拉帕利。p值来自Pearson分析。 (F) 在有或没有IL34的完全培养基中处理后的BMDM细胞的巨噬细胞总数中MHCIIloC5aR1hi巨噬细胞的比例。CM:完全培养基。n=3。组间比较采用双侧学生t检验。来源数据和确切的p值作为来源数据文件提供。来源数据作为来源数据文件提供。
为了在转录组重塑的背景下可视化细胞分化的进程,我们使用 scVelo RNA 速度 densMAP 分析开发了一种基于潜在时间的模型,该模型与 TAM_C4 向 TAM_C0 再向 TAM_C1 或 TAM_C3 巨噬细胞的转变一致(补充图 7A)。DensMAP 使用细胞密度来开发轨迹,并提供比 UMAP 更强的细胞状态表示。这与 TAM 表型具有相对可塑性一致。C5ar1 表达与极化潜在时间轴相关(图 6A)。这表明 C5ar1 可能促进 TAM_C3 的极化。
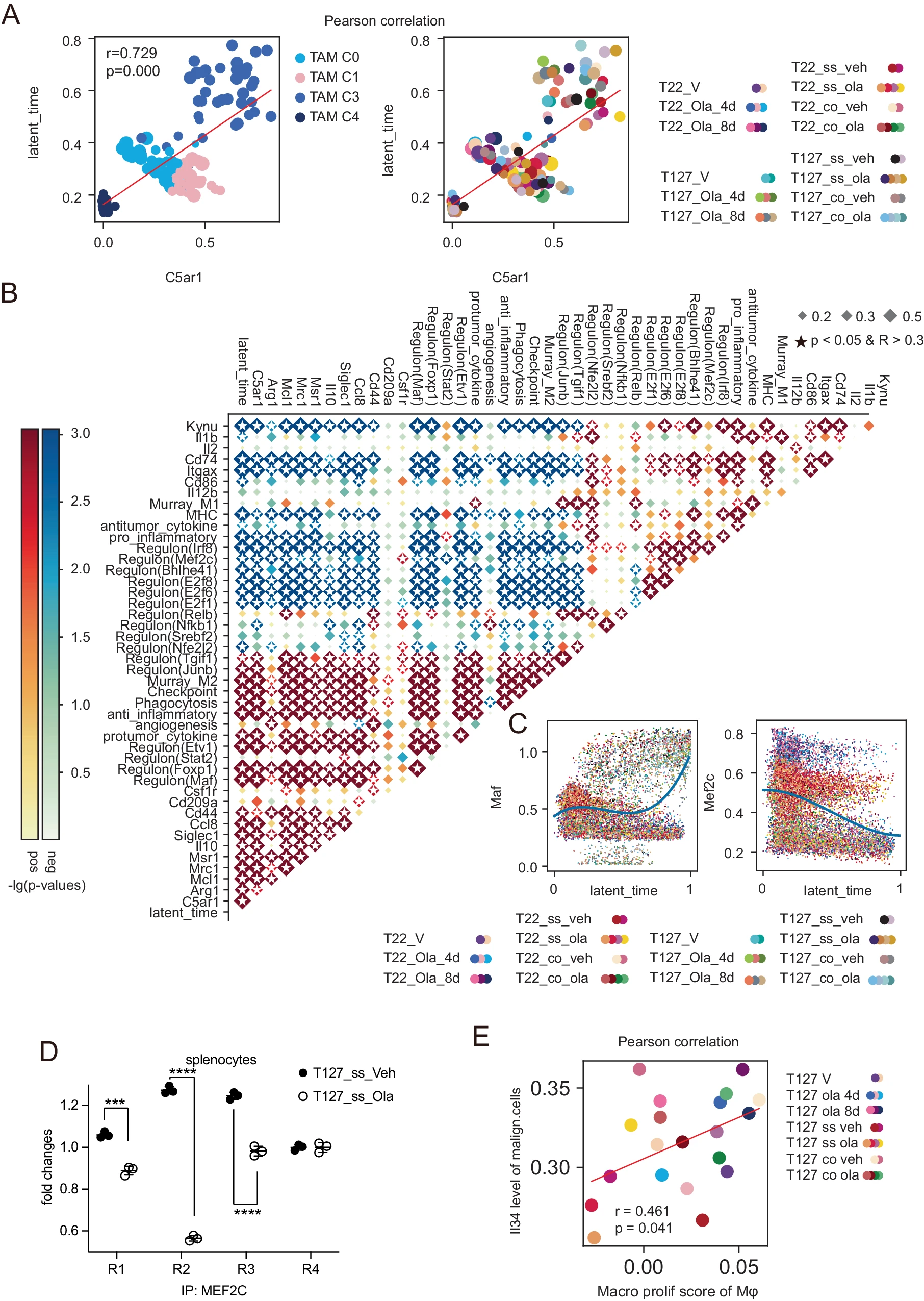
A 各肿瘤模型中巨噬细胞亚群的潜伏时间与 C5ar1 表达的相关性,包括图 1 和图 2 中的所有巨噬细胞亚群。细胞按每个处理组的亚群分组,见右侧框图。点的大小表示每组中的细胞数量。ss:单株移植模型。co:共移植模型。veh:载体。ola:奥拉帕尼。p 值来自皮尔逊分析。
B 热图显示了通过伪散装分析在(A)中合并的巨噬细胞中 SCENIC 富集的巨噬细胞功能标志物、通路和调控子之间的相关性。用于功能通路评分的基因列表列于补充表 2 中。
C Maf mRNA 相对表达的 AUC 与潜伏时间的相关性(左)和 Mef2c mRNA 相对表达的 AUC 与潜伏时间的相关性(右),在 B 中的巨噬细胞群体中。点按下图所示的处理组着色。ss:单株移植模型。co:共移植模型。Veh:载体。Ola:奥拉帕尼。
D 从如图 1 所示的对照或奥拉帕尼(50 mg/kg,每日口服)处理的 T127 单株模型中提取的脾细胞,进行 ChIP 与抗兔 IgG 或抗 MEF2C 的结合。目标的倍数变化通过减去正常兔 IgG 抗体检测到的背景结合后,归一化到β- 肌动蛋白表达水平计算得出。n=3。数据以平均值±SD 表示。组间比较使用双侧学生 t 检验。** P < 0.01。
E 恶性细胞的 Il34 mRNA 表达与来自同一肿瘤的巨噬细胞增殖评分之间的相关性。点按右图所示的样本和亚型着色。ss:单株移植模型。co:共移植模型。V:短期载体处理的肿瘤。veh:载体。ola:奥拉帕尼。p 值来自皮尔逊分析。
随后,我们分析了潜在时间和巨噬细胞亚型功能标志物,包括 C5ar1 之间的相关性。Mrc1、Msr1、Ccl8、Il10、Siglec1 与促肿瘤活性、促肿瘤细胞因子生产、抗炎、吞噬和检查点活动相关,它们与 C5ar1 表达和潜在时间显著正相关,同样,Msr1、Mrc1、Mcl1、Ccl8、Il10 和 Siglec1 促肿瘤标志物与促肿瘤细胞因子生产、抗炎、吞噬和检查点活动显著正相关(图 6B)。C5ar1 和潜在时间与 Kyun、Cd74、MHCII 和与促炎活性相关的基因显著负相关(图 6B)。Il1b 与与抗肿瘤细胞因子生产相关的基因高度相关(图 6B)。这与 C5ar1 抑制与炎症和抗肿瘤活性相关的基因表达一致。为了识别可能促进巨噬细胞极化的转录因子(TF),我们应用单细胞调控网络推断和聚类(SCENIC),并确定了 Mef2c、Junb、Tgif1 和 Nfe2l2 作为促进 TAM_C1 极化的候选转录因子,而 Maf、Foxp1、Stat2 和 Etv1 是促进 TAM_C3 极化的候选转录因子(补充图 7B)。C5ar1 表达与 Maf、Foxp1 和 Etv1 TAM_C3 巨噬细胞调控子候选者的 AUC 相关,但与似乎促进 TAM_C1 巨噬细胞极化的四个调控子的 AUC 负相关。此外,拟议的 TAM_C1 巨噬细胞调控子的 AUC 共同变化,但与 TAM_C3 巨噬细胞调控子负相关(图 6B)。C5ar1 与与促肿瘤细胞因子生产相关的转录组高度相关。支持这一点的是,Maf 的 mRNA 水平在潜在时间轴上逐渐增加,而 Mef2c 则减少(图 6C)。
MEF2C 被认为是促肿瘤抑制因子,显示了 JASPAR 最高的结合潜力。因此,我们探讨了 Mef2c 是否可能促进 C5ar1 的表达。在 C5ar1 启动子区域存在多个 Mef2c 共识结合序列(补充图 7C、D)。在 T127 单一模型的脾细胞中,使用 MEF2C 抗体和 C5ar1 启动子区域引物进行的 ChIP-qPCR 显示,在奥拉帕尼存在下,4 个 C5ar1 启动子区域中的 3 个 MEF2C 结合显著减少(图 6D)。这与 TAM_C3 巨噬细胞中 C5ar1 的增加以及奥拉帕尼处理的 T127 中 TAM_C3 巨噬细胞的增加一致。
IL34 和 CSF1 都被报道可以促进巨噬细胞增殖。由于增殖的 TAM_C0 巨噬细胞似乎是向 TMA_C1 和 TAM_C3 巨噬细胞过渡的一个步骤(补充图 7A),我们随后确定了增殖的 TAM 部分是否与恶性细胞中的 Il34 和 Csf1 mRNA 水平相关。有趣的是,T127 中恶性细胞的 Il34 但不是 Csf1 表达与较高的巨噬细胞增殖评分相关(图 6E 和补充图 7E)。支持 IL34 在 TAM_C0 巨噬细胞向 TAM_C3 巨噬细胞过渡中的作用,IL34 上调了骨髓来源髓细胞中的 C5aR1 表达(补充图 7F)。
综上所述,C5aR1 与 TAM_C3 转录程序的激活相关,这似乎是通过 MEF2C 抑制的丧失而增加的。
C5aR1 与人类 TNBC 肿瘤中的 T 细胞功能障碍相关¶
为了确定 C5aR1 是否可以预测 TNBC 患者的预后,我们评估了免疫激活标志物在 C5aR1 高或低水平的人类 TNBC 肿瘤中的预后关联。令人惊讶的是,CD8A、PD-1、KLRD1、NKG7、CD226、CD96、HLA-DOA、IL-12B、IFNG 和 PRF1 与免疫浸润和 TNBC 预后改善相关,这些标志物预测 C5aR1 低队列中的预后改善,但在 C5aR1 高队列中没有(图 7A)。这表明高水平的 C5aR1 限制了免疫细胞有效抗肿瘤活动的能力。CD209 和 CSF1,介导巨噬细胞吞噬和活化,也与 C5aR1 低队列中的更好预后相关,但在 C5aR1 高队列中则没有(图 7A)。有趣的是,促肿瘤细胞死亡的颗粒酶(GZMs)独立于 C5aR1 水平预测预后,这与 C5aR1 在效应 T 细胞的发展而非其活动中的作用一致(图 7A)。CSF1R 水平独立于 C5aR1 水平预测良好的患者预后,原因尚待确定。这些数据与 C5aR1 限制抗肿瘤免疫活性效能的模型一致。
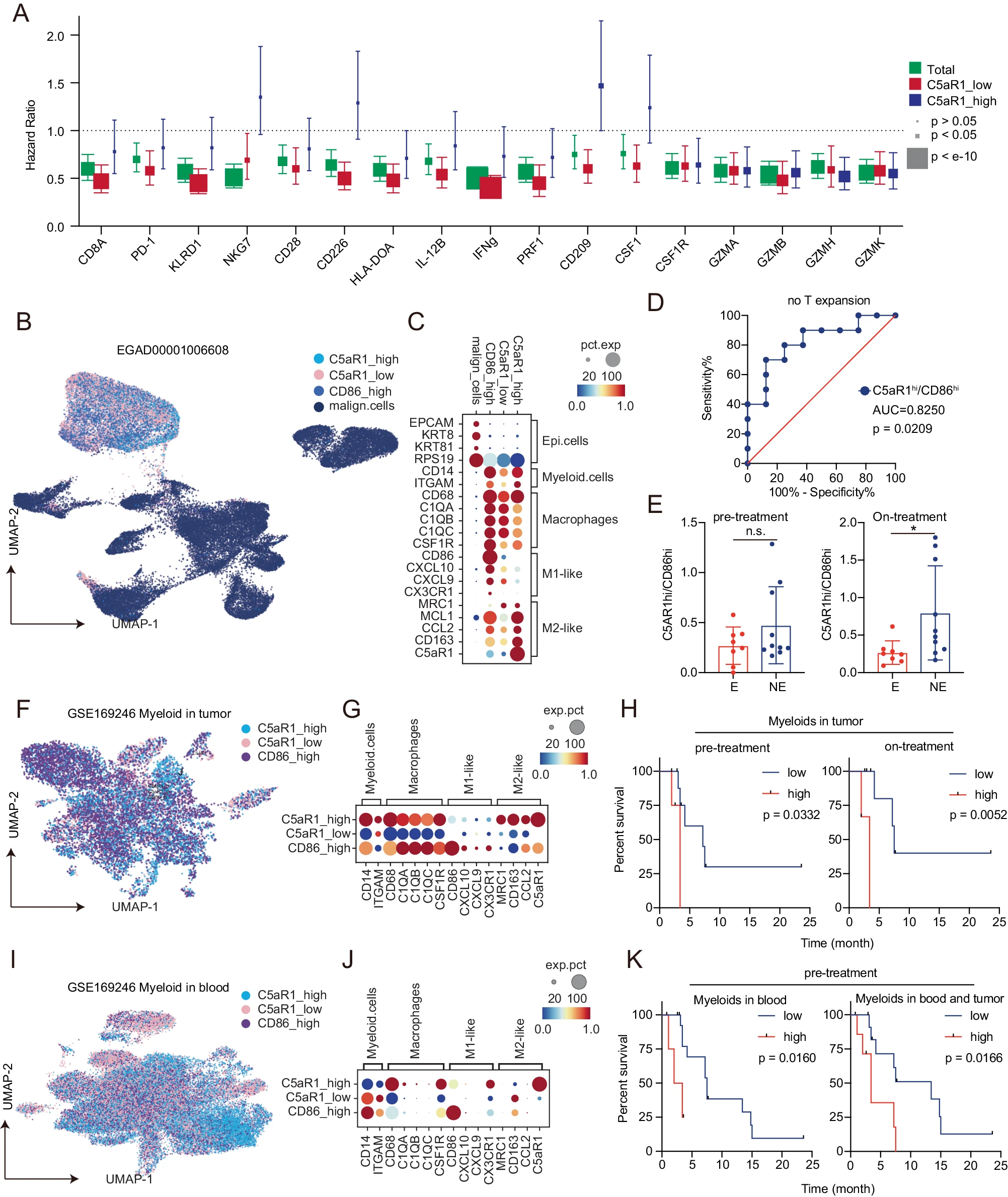
A 指示基因在 TNBC 数据库(953 名患者,2022.07 版本)中的风险比点图(绿色)77,C5aR1 高组(蓝色)或 C5aR1 低组(红色)。C5aR1 组的截断值设置为 C5aR1 的中位表达值。p 值来自对数秩检验。在点图中,中心点显示风险比(HR),线条代表 95% 置信区间(CI)。 B 从 EGAD0000100660846 中人类肿瘤细胞和 TAM 的分层和细胞类型鉴定。恶性细胞注释采用自原始出版物。高=前 25%。n = 18。 C 点图显示细胞类型特异性标记和功能基因的基因表达。 D ROC 曲线展示 C5aR1 高/CD86 高比率预测未能在抗 PD1 治疗后实现 T 细胞扩增的患者的能力。p 值来自受试者工作特征曲线(ROC)。n = 18。 E 展示 T 细胞扩增(E)或未扩增(NE)患者在治疗前和治疗中的 C5aR1 高/CD86 高比率的条形图。数据以平均值±SD 表示。p 值来自双侧学生 t 检验。* p < 0.05。nE = 8,nNE = 10。 F GSE16924644 中人类 TNBC 肿瘤 TAM 的分层和细胞类型鉴定。细胞类型如 B 中所示分类。n = 14。 G 点图显示肿瘤髓系细胞中谱系特异性标记的基因表达。 H GSE16924646 患者在基于 C5aR1 高/CD86 高比率的治疗前(左)和治疗后(右)肿瘤细胞中的无复发生存率 Kaplan-Meier 曲线。高=前 25%。生存信息来自原始出版物 44,86。p 值来自 Mann-Whitney 检验。n = 14。 I GSE169246 中人类 TNBC 血液髓系细胞的分层和细胞类型鉴定。细胞类型如 B 中所示定义。n = 20。 J 点图显示血液髓系细胞中谱系特异性标记的基因表达。 K 基于 GSE169246 中治疗前血液(左,高=前 25%)和血液或肿瘤髓系细胞中的 C5aR1 高/CD86 高比率的患者的无复发生存率 Kaplan-Meier 曲线。生存信息来自原始出版物 44。n = 20。p 值来自双侧 Mann-Whitney 检验。
ICB 针对免疫调节途径的治疗在多种肿瘤类型中重新激活了免疫反应,改善了患者预后。然而,ICB 在 TNBC 临床研究中的活性较为有限。为了确定巨噬细胞上 C5aR1 表达是否可能改变对 ICB 的反应,我们评估了在接受抗 PD-1 治疗的患者中,C5aR1 表达与 T 细胞扩增的相关性。髓系细胞被标注为 C5aR1 高和 CD86 高(基于高表达 CD86 且不表达 C5aR1、CD163、MRC1)和“C5aR1 低”细胞,不表达高水平的 C5aR1 或 CD86。值得注意的是,在治疗样本中 C5aR1 高/CD86 高比率高预测了缺乏 T 细胞扩增对抗 PD-1 的反应(AUC 为 0.8250,p = 0.0209)。正如预期的那样,高 CD163 高/CD86 高比率或 CD163 高/CD86 高比率未显示类似的预测能力。
我们分析了接受化疗或联合阿特珠单抗治疗的患者的另一组 scRNA 数据。在 14 名 TNBC 患者中,高肿瘤 C5aR1 高/CD86 高比率显著与预后较差的无进展生存期(PFS)相关,无论是治疗前(PFS,p = 0.0332)还是治疗后(PFS,p = 0.0052)样本。与我们观察到的内在 PARPi 耐药 T127 肿瘤小鼠血液中 C5aR1 阳性巨噬细胞水平升高一致,20 名患者的血液中 C5aR1 高/CD86 高比率升高也显著与预后较差的 PFS 相关(p = 0.0160),但仅在治疗前样本中。进一步支持这一点的是,无论是在治疗前血液还是肿瘤细胞中高 C5aR1 高/CD86 高比率均显著与预后较差的 PFS 相关(p = 0.0166)。类似的预测能力未在高 CD163 高/CD86 高比率或 CD163 高/CD86 高比率中观察到。
总之,这些数据表明,巨噬细胞上 C5aR1 水平升高与人类癌症患者中的 ICB 耐药性相关。
讨论¶
我们证明了 C5aR1 高表达巨噬细胞介导了对 PARPi 的耐药性,并且高 C5aR1 高 TAM 比例预测了对 PARPi 的耐药性。C5aR1 阳性巨噬细胞介导的 PARPi 耐药性可以转移到远端部位,这表明肿瘤亚克隆产生 C5aR1 高巨噬细胞的能力可能介导在实体肿瘤或潜在转移肿瘤部位的通常 PARPi 敏感的亚克隆的耐药性。
几项研究已确定了促肿瘤巨噬细胞与促炎/促肿瘤巨噬细胞比例与患者预后以及包括 BRCA 突变乳腺癌模型中的 PARPi 在内的多种疗法耐药性之间的相关性。然而,这可能过于简化,因为巨噬细胞具有可塑性,最近在 scRNA Seq 分析中鉴定的巨噬细胞亚群表达与促炎和抗炎/促肿瘤活动相关的基因表达模式的混合。多个临床试验通过抑制 CSF1R 激活和信号传导来靶向巨噬细胞功能。与 CSF1R 研究相比,我们的数据表明,靶向 C5aR1 选择性地减弱了巨噬细胞的促肿瘤功能,同时保留了与促炎功能相关的基因表达模式占主导地位的巨噬细胞群体,从而导致高促炎巨噬细胞/促肿瘤巨噬细胞比例。这与 CD8 T 细胞依赖的 PARPi 耐药肿瘤生长减少相关。C5aR1 以前被认为介导促肿瘤巨噬细胞极化并创造促肿瘤微环境。C5aR1 高表达巨噬细胞被认为增加肿瘤进展并抑制 T 细胞细胞毒性,包括鳞状细胞癌、结肠癌、宫颈癌和乳腺癌。我们的数据支持阻断 C5aR1 将靶向促肿瘤巨噬细胞,同时保留巨噬细胞的促炎功能,这表明靶向 C5aR1 可能使接受 PARPi 和潜在其他疗法的患者受益。支持这一观点的是,高 C5aR1 水平以及人类患者中的 C5aR1 阳性巨噬细胞与免疫检查点阻断的耐药性相关。
最近的一项研究表明,C5aR1+ 中性粒细胞通过分泌 IL1β 和 TNFα 促进糖酵解能力,导致乳腺癌预后不良。这与我们的研究类似,我们确定了 C5aR1 水平与促肿瘤细胞因子基因表达增加之间的相关性。然而,我们的数据集中未检测到 Fut4 表达(编码 CD15)或 Cd177,表明在我们的 scRNA Seq 分析中由于肿瘤分离过程中的脆弱性未能捕获中性粒细胞。
先前尚未阐明调节巨噬细胞中 C5aR1 表达的机制。使用 RNA 速度轨迹分析、转录组分析和 ChIP-PCR,我们指出 MEF2C 在抑制 C5aR1 表达中的作用。MEF2C 以前被认为抑制促肿瘤巨噬细胞功能,同时促进促炎巨噬细胞极化。这与我们的轨迹分析一致,C5aR1 在巨噬细胞表达与促肿瘤功能相关基因的转变中起作用。C5aR1 和 MEF2C 表达与多种其他 TFs 的表达相关,表明它们也可能通过 C5aR1 调控促肿瘤巨噬细胞功能。
我们和其他人已经证明 PARPi 可以激活 STING,导致干扰素产生和功能性适应性免疫反应。有趣的是,STING 激动剂可以将 TAMs 极化为更促炎状态,从而改善对 PARPi 的反应。这与 PARPi 在 T22 中诱导 STING 激活而在 T127 中没有的情况一致。事实上,我们先前已经证明,T127 中存在的 KRAS 突变通过抑制细胞质中双链 DNA(dsDNA)的积累,限制了 PARPi 诱导的 STING 激活,并且 MEKi 的抑制会引起细胞质 dsDNA 积累、STING 激活和有效免疫反应的产生。我们的先前研究涉及 T127 中 PARPi 耐药中的 MDSC/巨噬细胞群体。目前的研究扩展了这些观察,表明 C5aR1 高巨噬细胞群体不仅在 T127 中介导 PARPi 耐药性,还能够将 PARPi 耐药性转移到通常对 PARPi 敏感的 T22 肿瘤。
总之,我们的研究指出 C5aR1 高巨噬细胞在介导 PARPi 以及可能其他治疗剂包括 ICB 的局部和远距离耐药中的作用。我们的研究进一步确认 C5aR1 作为潜在的治疗靶点,提供了一种方法,选择性地清除促肿瘤巨噬细胞功能,同时保留抗肿瘤巨噬细胞功能,从而导致 CD8 T 介导的肿瘤生长抑制。此外,我们的研究表明,通过抑制 C5aR1 作为一种策略来清除促肿瘤巨噬细胞功能,以使肿瘤对 PARPi 以及更广泛范围的治疗剂敏感,值得在临床中进行探索。
单细胞 RNA 测序文库构建¶
通过使用 gentleMACS 试剂盒(Miltenyi Biotec, #130-096-730)分散 MDST 肿瘤,按照生产商说明用 TotelseqA 标签抗体(Biolegend, #394601, #394603, #394605, #394607, #394609, #394611, #394613, #394615)孵育。使用 EasySep Dead Cell Removal(Annexin V)试剂盒(STEMCELL Technologies, #17899)分离活细胞。单细胞悬液根据 10xGenomics scRNAseq 样本制备协议(Chromium Single Cell 3’ v3.1 Reagent Kit, 10xGenomics)处理。简而言之,2×10^6 活细胞的单细胞悬液与 100 μL 抗体标签寡核苷酸(10 μg/μL)在冰上孵育 30 分钟。用 3 mL 含 10% FBS 的 PBS 洗涤 3 次后,将 5×10^5 个带标签的活细胞混合在一起,并使用死细胞去除柱(STEMCELL, 17899)富集活细胞。测序由 Novaseq 6000 进行。每个样本目标为 10000 个细胞。
单细胞 RNA 测序数据分析¶
由 10xGenomics 生成的单细胞 RNA 测序数据通过 Cell Ranger count pipeline(v6.0.0)处理,命令参数为“include introns”,以过滤低质量的读数并将读数对齐到小鼠参考基因组(GRCm38, vM32),分配细胞条形码并生成唯一分子标识符(UMI)。输出的 BAM 文件通过 Velocyto 进一步处理,生成包含剪接 RNA、未剪接 RNA 和模糊的 UMI 的 LOOM 文件。检测到的基因少于 1000 个(UMI > 1000)或来自线粒体基因的 UMI 超过 20% 的细胞被 SCANPY 排除。通过 Scrublet 以预期的双胞胎率 6 和双胞胎评分大于 95% 去除双胞胎。使用 SCANPY 和 scVelo 对具有 4000 个高可变基因(HVG)的过滤数据进行标准化和缩放,以去除不需要的变异来源,例如批次效应和细胞周期效应。通过无监督的 Louvain 聚类对细胞多重寡核苷酸矩阵进行单细胞样本的去重操作,分辨率为 1.2,Louvain 社区仅包含独特的细胞多重寡核苷酸,分配给相关的处理组。通过 SCENPY 中的 Leiden 聚类模块对基因表达矩阵进行无监督聚类,分辨率为 2。通路得分根据补充表 2 中列出的基因通过 SCANPY 的得分模块计算。使用 densVis 包进行保持密度的降维,dens_lambda = 0.755。使用 pySCENIC 在默认参数下进行调控子富集。基于配体 - 受体的细胞 - 细胞通讯通过 CellChat 在超过 10% 细胞表达的基因和基因表达排名前 80% 的细胞之间进行。配体 - 受体对数据库从 CellphoneDB 下载。
细胞亚型分配¶
细胞标注根据图 1E 中列出的细胞标记进行。简而言之,表达 Cd45、Cd3、Cd4 和 Tcf 的细胞被分配为中央记忆 T 细胞。表达 Cd45、Cd3、Cd4 和 Foxp3 的细胞被分配为调节性 T 细胞。表达 Cd45、Cd3、Cd8 和 Ccr7 的细胞被分配为初始 CD8 T 细胞。表达 Cd45、Cd3 和 Mki67 的细胞被分配为增殖性 T 细胞。表达 Cd45、Ncr1 和 Nkg7 的细胞被分配为 NK 细胞。表达 Cd79a 和 Cd19 的细胞被分配为 B 细胞。表达 Clec10a、Siglec 和 Cd300c 的细胞被分配为浆细胞样树突细胞。表达 Dcn 或 Acta2 的细胞被分配为成纤维细胞。表达 Epas1 的细胞被分配为内皮细胞。具有拷贝数变异的细胞(CaSpEr)被分配为恶性细胞。表达 Itgam、Cd14 和 Adgre1 的细胞被分配为单核细胞或巨噬细胞。通过无监督 Leiden 聚类对单核细胞、巨噬细胞和成纤维细胞进行重新聚类,并根据之前报告的不同亚群标记和每个 Leiden 簇的顶级 DEG 进行标注。
标签转移¶
标签转移用于验证巨噬细胞表型。将一个全癌症肿瘤微环境项目(GSE154763)的髓系原始计数与 Seurat 中的参考基础整合工作流整合。简而言之,数据集首先通过 SCTransform 处理,然后通过 FindTransferAnchors 找到转移锚点,随后通过 IntregrateData 进行整合。在整合全癌症髓系数据集后,通过 TransferData 将全癌症的标签信息转移到我们的髓系数据集,另一个由 FindTransferAnchors 识别的全癌症髓系数据集和我们的髓系数据集之间的转移锚点集。预测分数低于 0.35 的预测标签被视为信心水平不足。
通过 scGPT 进行细胞交叉注释¶
从基因表达综合(GEO)数据库获取人类全癌症髓系数据集,使用接入号 GSE154763。通过 scGPT 对小鼠髓系亚簇和人类全癌症肿瘤髓系簇进行交叉验证。将小鼠标准化数据集进行对数转换和分箱处理,然后在 scGPT 中使用默认参数进行模型微调。训练数据集包含 25408 个细胞和 2425 个基因。查询数据集包含 9748 个细胞和 3000 个基因。在数据处理期间选择了 4000 个 HVG。
互动计数估计¶
为了估计包含独立重复的两个不同条件之间的相互作用,我们开发了一个“互动计数”估计公式。条件 A 中的每个肿瘤(a)与条件 B 中的每个肿瘤(b)进行比较,产生 a×b 的比较对。然后,互动计数估计通过对每个比较对中增加的互动求和,并减去那些在每个比较对中减少的互动来计算。如果一个受体有多个配体,互动计数计算为所有配体的互动计数估计之和。
轨迹分析¶
通过 scVelo 使用动态模式进行 RNA 速度分析。计算的 RNA 速度向量嵌入低维空间,使用 scVelo 包中的基于分区的图抽象(PAGA)模块。巨噬细胞发育的潜在时间由 scVelo 分配给每个巨噬细胞亚簇,较低的潜在时间表示初始事件,较高的潜在时间表示终末事件。
使用 Kaplan-Meier Plotter 在线工具进行生存分析¶
三阴性乳腺患者整体 RNA 序列数据库的 Kaplan-Meier 图由 Kaplan-Meier Plotter 在线工具生成,限制为仅“基底”亚型。生存分析通过 Kaplan-Meier Plotter 提供的自动选择最佳截断值的单变量 Cox 回归进行评估。对于 C5aR1 高和低亚群的生存分析,患者首先按 C5aR1 的中位表达进行分层,然后在上述亚群中分别通过单变量 Cox 回归进行生存分析。GSE169246 的生存分析通过 Graphpad Prism 进行单变量 Cox 回归。
TCGA 基底乳腺癌队列的生存分析¶
从 UCSC Xena 下载的 TCGA 全癌症基因表达数据的基底乳腺癌数据。通过 UCell(v2.2)使用每个簇的前 3 个 DEG 计算每个亚簇的细胞签名得分。通过样本内每个细胞亚簇的 Z 分数计算细胞签名得分排名。每个得分排名的截止值由 Survival R 包的 cutpoint 函数设置。使用 Survival R 包(v3.3-1)进行危险比(HRs)和 log-rank 检验。
伪批量相关性分析¶
为了可视化所有 40 个 MDST 肿瘤中巨噬细胞特异性基因和途径共表达模式的相关性,共表达相关性分析通过取每个样本中每个巨噬细胞亚簇的平均基因表达或途径得分进行计算,并计算包含所有 40 个 MDST
肿瘤的数据集中的皮尔逊相关性。相关系数高于 0.3 且 p 值小于 0.05 的相关性被认为是显著的。
公共数据分析¶
从 EGAD00001006608 和 GSE169246 获得接受 ICB 联合或不联合化疗的 TNBC 患者的处理人类数据。细胞标注基于 C5aR1(C5aR1 高)和 CD86(CD86 高)的表达,以及“不表达高水平的 CD86 或 C5aR1”的“其他”。队列的 C5aR1 高/CD86 高比率的上四分位数被用作患者分组的截断值。生存分析通过 Graphpad Prism 使用单变量 Cox 回归评估。
本研究中使用的软件¶
使用的软件包括:R(https://www.r-project.org/;RRID:SCR_001905,v4.1.0),SCANPY(https://scanpy.readthedocs.io/en/stable/,v1.7.1),scVelo(https://scvelo.readthedocs.io/en/stable/,v0.2.3),densVis(https://github.com/hhcho/densvis/tree/master/densmap),Cell Ranger(https://www.10xgenomics.com/support/software/cell-ranger/latest,v6.0.0),GSVA(https://github.com/rcastelo/GSVA,1.40.1),Survival R(https://cran.r-project.org/web/packages/survival/survival.pdf,v3.3-1),samtools(https://github.com/samtools/samtools,v1.11),CellChat(https://github.com/sqjin/CellChat,v1.1.3),CellphoneDB(https://www.cellphonedb.org/),pySCENIC(https://github.com/aertslab/pySCENIC,v0.11.2),Seurat(https://satijalab.org/seurat/,4.3.0.1),UCell(https://github.com/carmonalab/UCell,v2.2),scGPT(https://github.com/bowang-lab/scGPT v0.2.1),ImageJ(v1.52a),Prism(http://www.graphpad.com/faq/viewfaq.cfm?faq=1362,v8.0.2),FlowJo(https://www.flowjo.com/,v10)。所有统计分析均通过 GraphPad Prism 软件进行。非正态分布数据通过 Mann-Whitney 检验评估。配对数据使用 Wilcoxon 匹配对符号秩检验。多组比较通过单向 ANOVA 进行。
报告总结¶
关于研究设计的进一步信息,请参阅链接到本文的《Nature Portfolio Reporting Summary》。
数据可用性¶
本研究中生成的小鼠衍生同种移植测序数据已存储在 GEO 数据库中,访问编号为 GSE215908。本研究中使用的癌细胞系数据可在 GEO 数据库中获得,访问编号为 GSE157220 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157220]。本研究中使用的 TNBC 人类公共数据可在欧洲基因组 - 表型档案(EGA)中获得,访问编号为 EGAD00001006608 [https://ega-archive.org/studies/EGAS00001004809],以及在 GEO 数据库中,访问编号为 GSE169246。来自公共数据库 The Cancer Genome Atlas(TCGA, National Cancer Institute(NCI),Bethesda, MD, USA)的乳腺癌公共 RNA-seq 数据可从 GDC 门户下载(https://www.cbioportal.org/study/summary?id=brca_tcga_pub)。其余数据可在本文、补充信息或源数据文件中获得。本文附带提供了源数据。
代码可用性¶
本研究未生成自定义代码,研究中使用的封装代码可从 https://github.com/xili3367/Left-and-right-co-transplantation(版本:01/21/21)免费下载。