1.2 Raw data processing¶
在本节中,我们讨论了单细胞和单核 RNA 测序(sc/snRNA-seq)数据的“预处理”过程中一些基本问题。尽管这是常用术语,但它似乎有些用词不当,因为这个过程涉及几个步骤,这些步骤对如何处理和表示数据做出重要决策,从而能够或排除后续分析。在这里,我们主要将这个处理阶段称为“原始数据处理”,我们的重点是从巷道解复用的 FASTQ 文件开始,到以计数矩阵结束,这个矩阵代表每个基因在每个量化细胞中产生的不同分子数量的估计值,可能会根据每个分子的推断剪接状态进行分层(参见{numref}raw-proc-fig-overview)。
在原始数据处理阶段的几个关键步骤包括:
- 质量控制:通过质量控制步骤识别和过滤低质量的读数和细胞,以确保数据的准确性和可靠性。
- 读数对齐:将测序读数比对到参考基因组或转录组,以确定每个读数的来源基因或转录本。
- 去重和定量:去除 PCR 重复读数,并量化每个基因在每个细胞中的表达水平。这通常涉及到唯一分子标识符(UMIs)的使用,以减少扩增偏差的影响。
- 剪接状态推断:根据读数的分布和特征推断每个分子的剪接状态,以提供更详细的转录本信息。
这个处理阶段的输出通常是一个计数矩阵,表示在每个细胞中每个基因的表达水平。这些数据为后续的下游分析提供了基础,例如细胞类型的识别、基因表达模式的研究和细胞间关系的探索。
在这个过程中做出的决策对后续分析的准确性和可靠性具有重大影响,因此需要特别关注和谨慎处理。
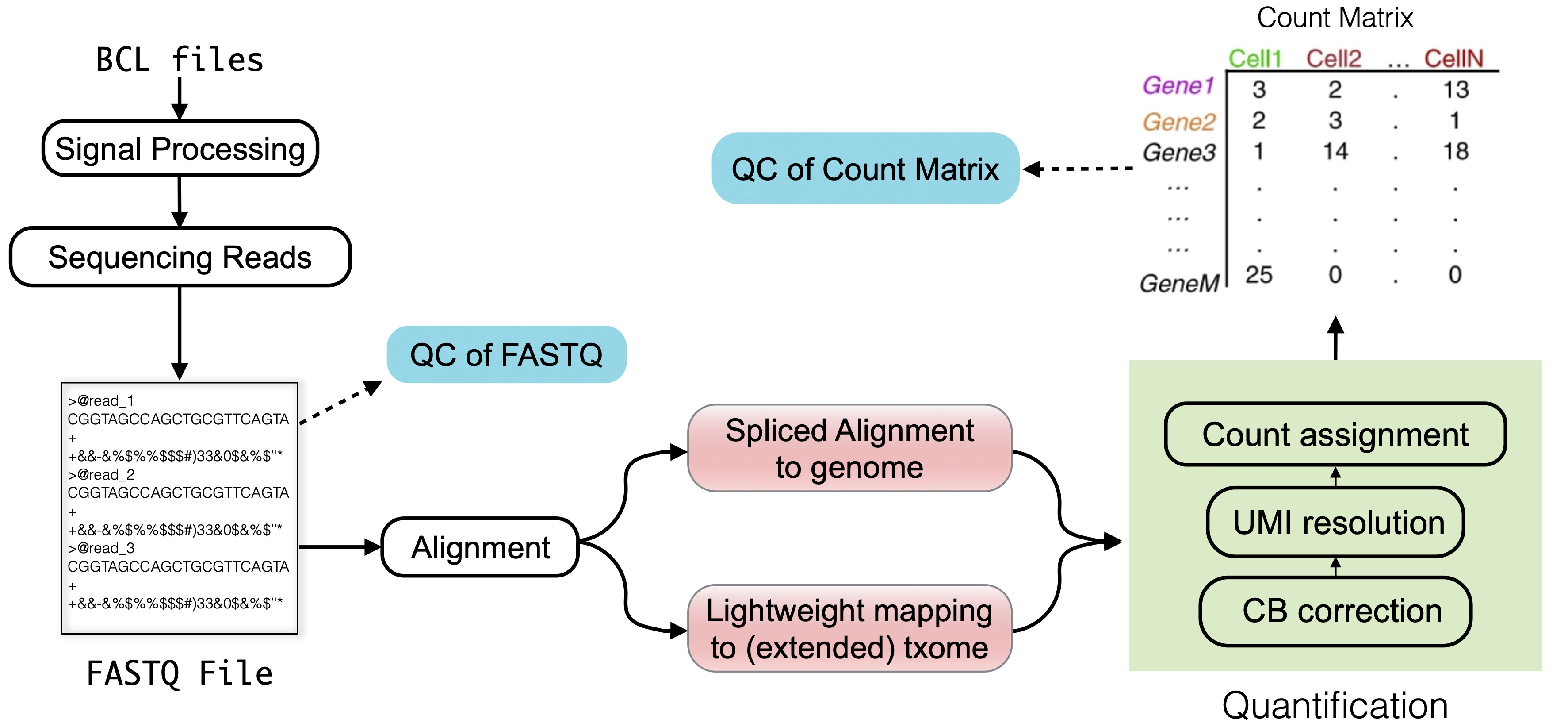
Fig1 本章讨论主题概览:在图表中,“txome”代表转录组。
这个计数矩阵随后作为使用 scRNA-seq 数据进行各种分析方法的输入,这些方法包括规范化、整合和过滤的方法,以及推断细胞类型、发育轨迹和表达动态的方法【Zappia2021】。由于计数矩阵是所有这些分析的起点,因此对其进行稳健和准确的估计是支持和实现准确可靠的后续分析的基础和关键步骤。原始数据处理中的基本误估可能会导致高级分析中的错误推断,并可能阻碍基于原始数据中信号的发现,这些信号可能由于原始数据处理而被遗漏、减弱或扭曲。正如我们将在本节中看到的,尽管我们希望从处理管道中的这一步骤中获得直观的输入和输出,但在原始数据处理中会出现几个重要且困难的挑战,处理这些挑战的改进计算方法仍在积极开发中。特别是,我们将涵盖原始数据处理中的基本步骤,包括读数比对/映射、细胞条形码(CB)识别和校正,以及通过解析唯一分子标识符(UMIs)来估计分子计数。我们还将提到在执行这些处理步骤时出现的选择和挑战。
A note on what precedes raw data processing
决定从哪里开始讨论原始数据处理有些任意。在这里,我们从巷道解复用的FASTQ文件作为原始输入开始讨论,但即便如此,这些数据已经经过处理和转换,不再是最初的测量数据。此外,在生成FASTQ文件之前的一些处理与后续处理过程中可能出现的挑战相关。例如,碱基调用和碱基质量估计过程会影响FASTQ文件中测序片段的错误率,以及仪器对每个调用核苷酸置信度的估计。此外,在生成FASTQ文件之前可能会出现问题,例如索引跳跃【Farouni2020】。尽管这些问题可以通过计算机方法【Farouni2020】和增强的协议(如双重索引)在很大程度上得到缓解。
然而,在本节中,我们将不考虑这些上游效应,而是将例如通过适当工具从BCL文件生成的FASTQ文件作为考虑的原始输入。
原始数据质量控制¶
一旦获得了原始 FASTQ 文件,可以通过运行质量控制(QC)工具,如 FastQC,快速诊断读数本身的质量,以评估读数质量、序列内容等。如果运行成功,FastQC 会为每个输入的 FASTQ 文件生成 QC 报告。总体而言,该报告总结了质量评分、碱基含量和某些相关统计数据,以发现可能源自文库制备或测序的问题。
尽管如今单细胞原始数据处理工具内部会评估一些对单细胞数据重要的质量检查,例如序列的 N 含量和映射读数的比例,但在处理数据之前快速进行质量检查仍然是一个好习惯,因为它评估了其他有用的 QC 指标。对于有兴趣了解来自 RNA-seq 样本的 FASTQ 文件的 FastQC 报告的读者,在以下切换内容中,我们使用 FastQC手册网页 提供的 良好 和 糟糕 的 Illumina 数据示例报告,以及 MSU 的 RTSF、HBC 培训项目 和 QC Fail 网站 的教程和描述,来演示 FastQC 报告中的模块。这些模块的详细描述可以在 FastQC手册网页 上找到。尽管这些教程并非专门为单细胞数据制作,但其中许多结果对于单细胞数据仍然相关,以下描述了一些需要注意的地方。在切换部分,除非特别提到,所有图表均来自 FastQC手册网页 上的示例报告。最后,此类质量控制报告中报告的大多数指标仅对单细胞数据集的“生物”读数有意义(即从基因转录本中抽取的读数)。例如,对于 10x Chromium v2 和 v3 数据集,这将是包含 R2 在其文件名中的读数。这是因为技术读数主要由条形码和 UMI 序列组成,这些序列不是从底层转录组中抽取的,因此我们不期望它们具有典型的生物学上合理的序列或 GC 含量,尽管某些指标如 N 碱基调用的比例仍然相关。
0. 摘要
HTML 报告左侧的摘要面板显示报告中包含的模块名称,以及用于快速评估模块结果的符号。然而,由于 FastQC 对所有类型的测序平台和基础生物材料生成的文件使用统一的阈值,我们有时会看到对于良好数据的警告(橙色感叹号)和失败(红色叉号),以及对可疑数据的通过(绿色勾)。因此,在得出数据质量结论之前,应仔细评估所有模块。

The summary panel of a bad example.
1. 基本统计
基本统计模块包含输入 FASTQ 文件中读数的概览信息和统计数据,例如文件名、序列总数、低质量序列数、序列长度以及所有序列中所有碱基的总体 GC 含量。良好的单细胞数据通常具有很少的低质量序列,并且大多数情况下具有统一的序列长度。GC 含量应与测序物种的基因组或转录组的总体 GC 含量相匹配。
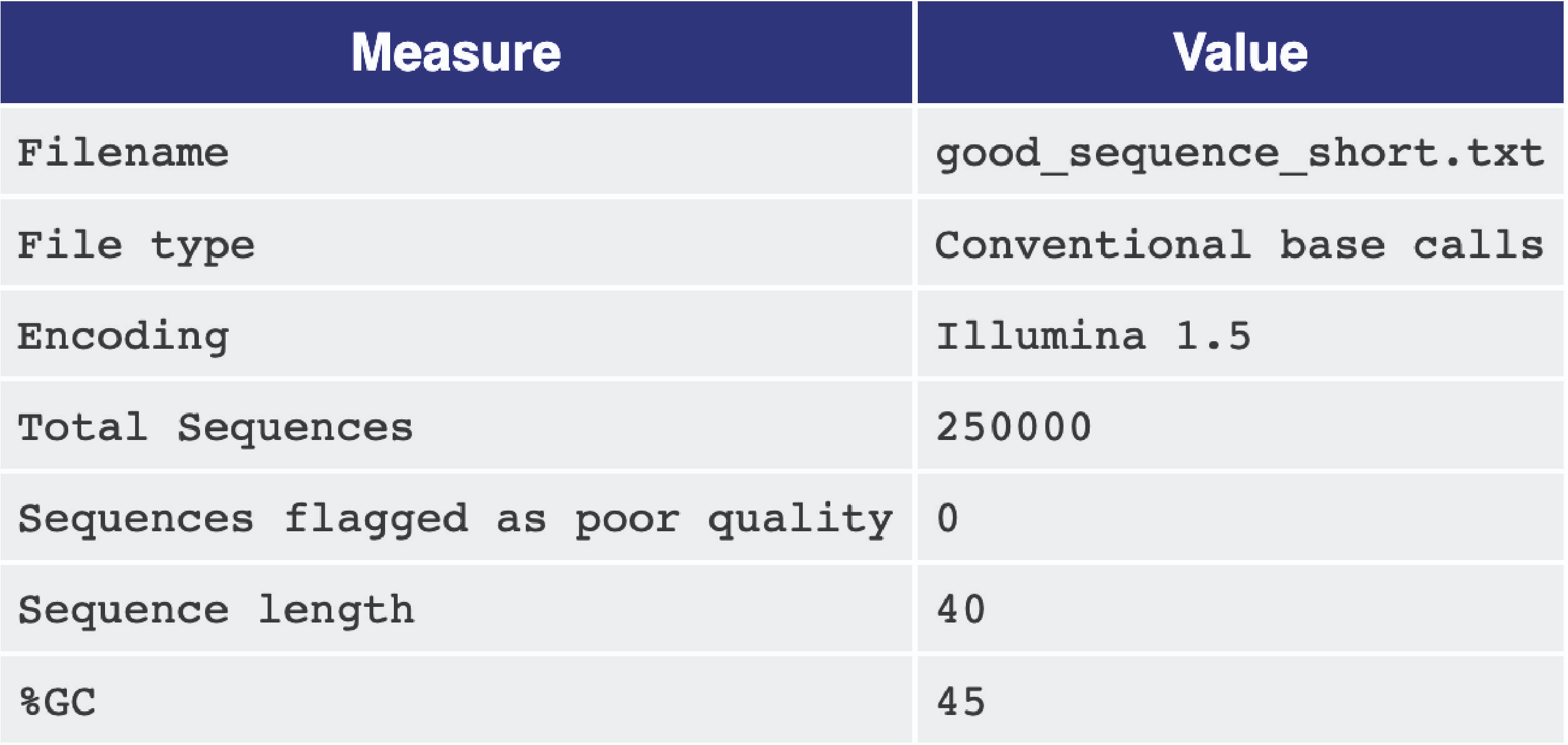
A good basic statistics report example.
2. 每碱基序列质量
每碱基序列质量视图为读数中的每个位置包含一个箱线图,显示文件中所有读数的每个位置上所有碱基的质量分数范围。x 轴表示读数中的位置,y 轴显示质量分数。对于高质量的单细胞数据,视图中的所有黄色箱体(代表位置质量分数的四分位间距)应该落在绿色(高质量调用)区域内。所有代表分布的 10% 和 90% 的须线也应如此。质量分数沿读数主体下降,最后几个位置的某些碱基调用落入橙色(合理质量调用)区域是可以预期的,因为在大多数合成测序方法中,信噪比的下降是常见现象。不过,箱体应该在红色(低质量调用)区域之外。如果观察到低质量调用,可以考虑对读数进行质量修剪。有关测序错误概况的详细解释由 HBC 培训项目 提供【详细说明】(https://hbctraining.github.io/Intro-to-rnaseq-hpc-salmon/lessons/qc_fastqc_assessment.html)。
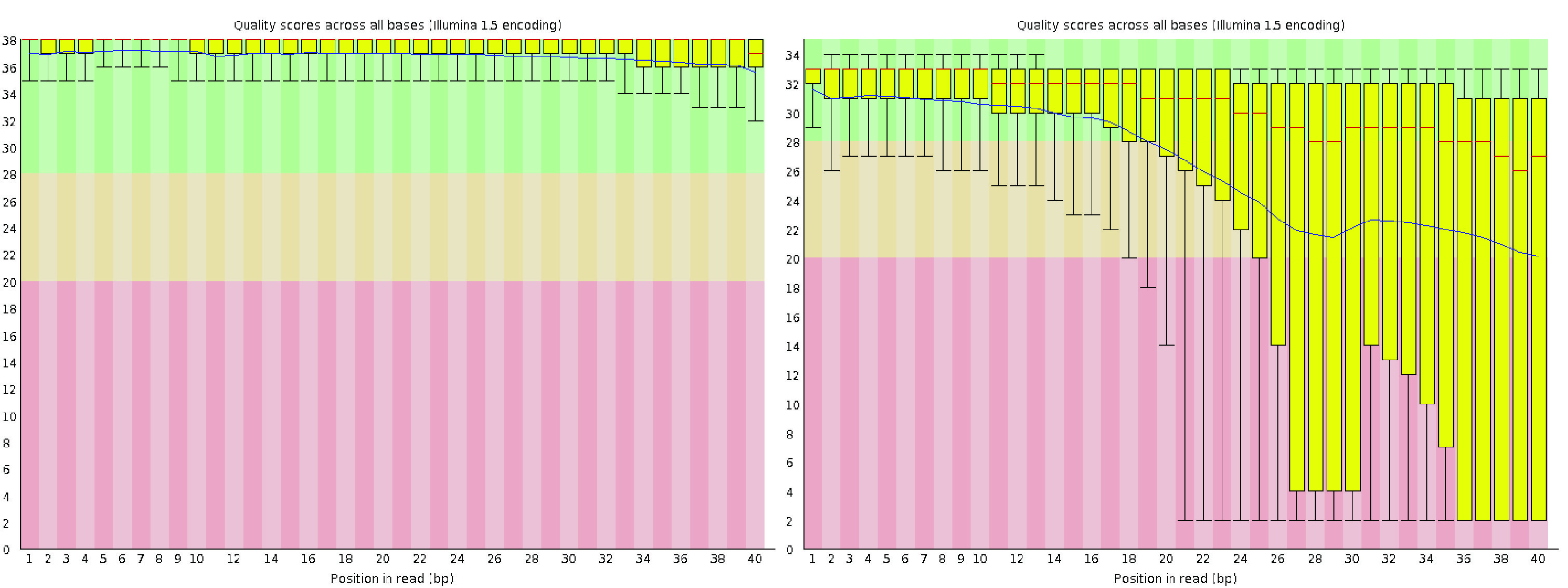
A good (left) and a bad (right) per-read sequence quality graph.
3. 每 patch 序列质量
使用 Illumina 文库时,每瓦片序列质量图显示了每个流动槽瓦片中测序读数相对于平均质量的偏差。颜色越“热”,偏差越大。对于高质量数据,我们应看到整个图中都是蓝色,这意味着流动槽中所有瓦片的质量一致。如果流动槽的某部分质量较差,图中会出现一些热色。在这种情况下,测序步骤可能遇到了暂时性问题,例如气泡通过流动槽或流动槽道内有污点和碎屑。有关进一步诊断,请参阅 QC Fail 和 FastQC 提供的 警告的常见原因。
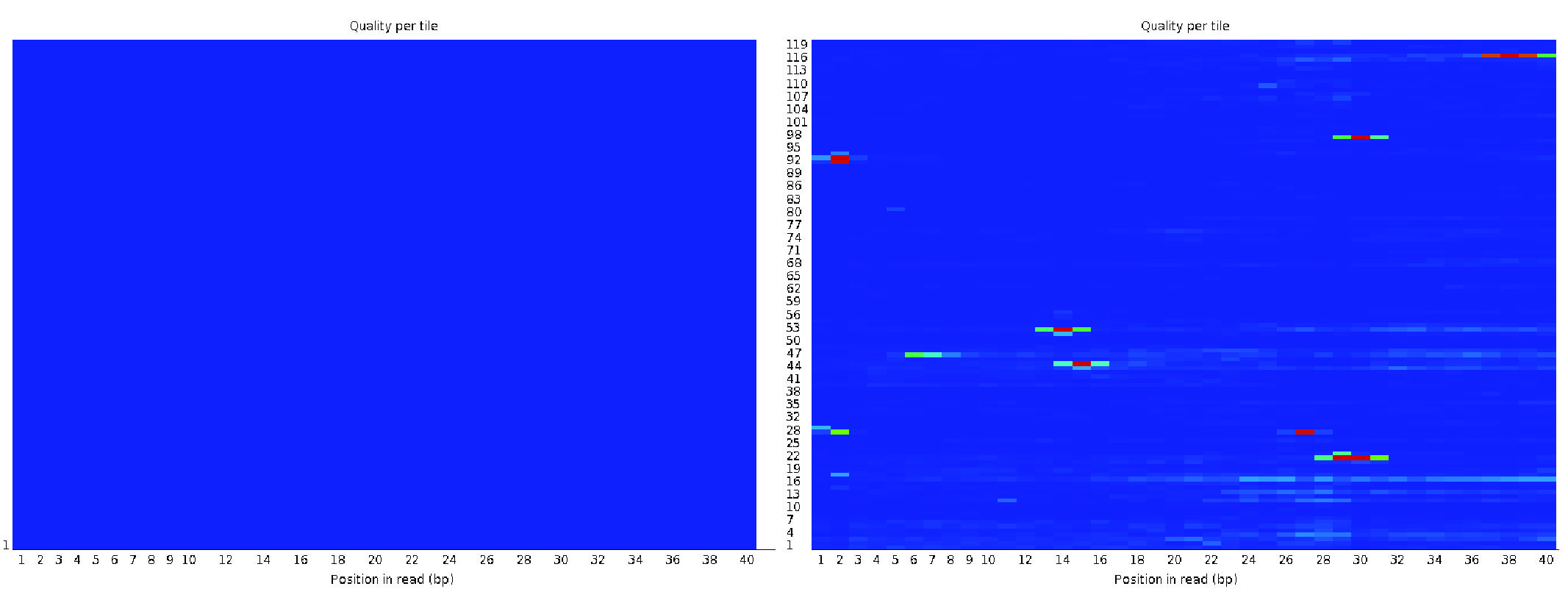
A good (left) and a bad (right) per tile sequence quality view.
4. 每序列质量分数
每序列质量分数图显示文件中每个读数的平均质量分数分布。x 轴显示平均质量分数,y 轴表示每个质量分数的频率。对于高质量数据,此图应只有一个位于尾部的峰值。如果出现其他峰值,可能表示某些读数存在质量问题。
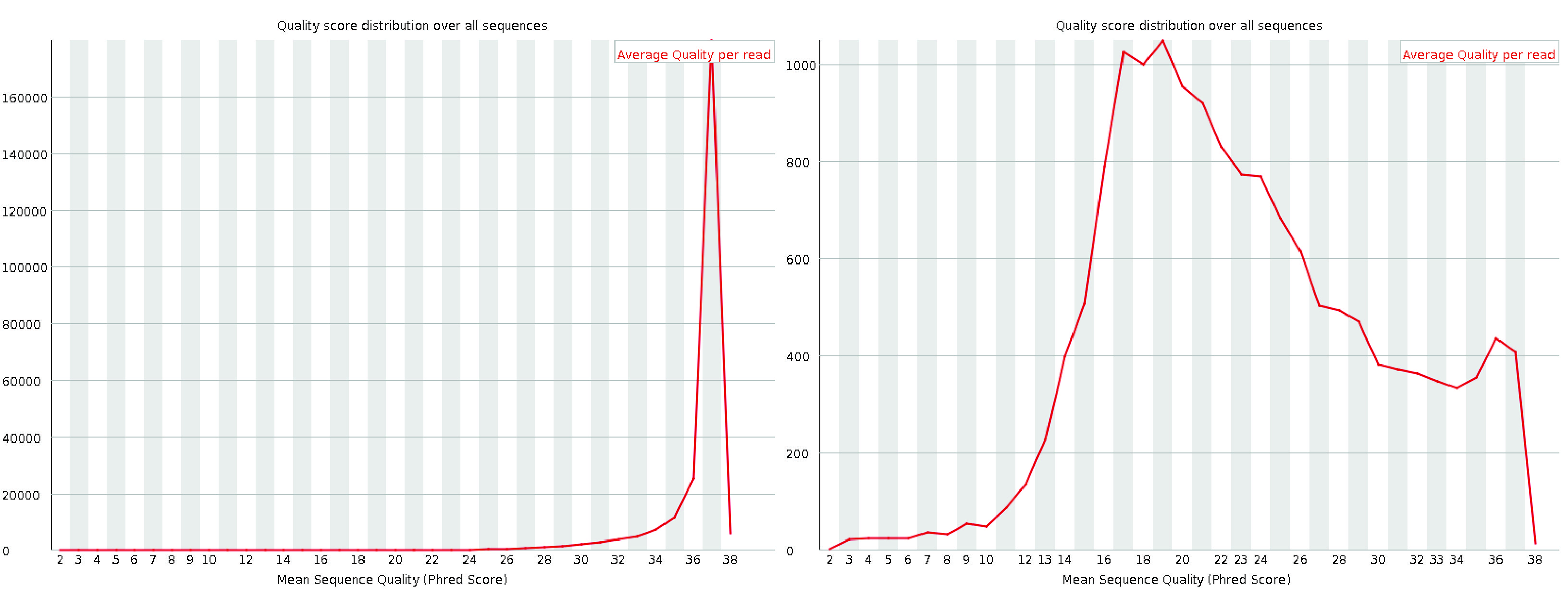
A good (left) and a bad (right) per sequence quality score plot.
5. 每碱基序列内容
每碱基序列内容图报告文件中所有读数的每个碱基位置上四种核苷酸的百分比。对于单细胞数据,看到行开始部分的波动并不罕见,因为读数的前几个碱基代表引物位点的序列,这些序列可能并不完全随机,正如 QC Fail 网站 所解释的那样。这在 RNA-seq 文库中经常发生,尽管 FastQC 会发出警告或失败。
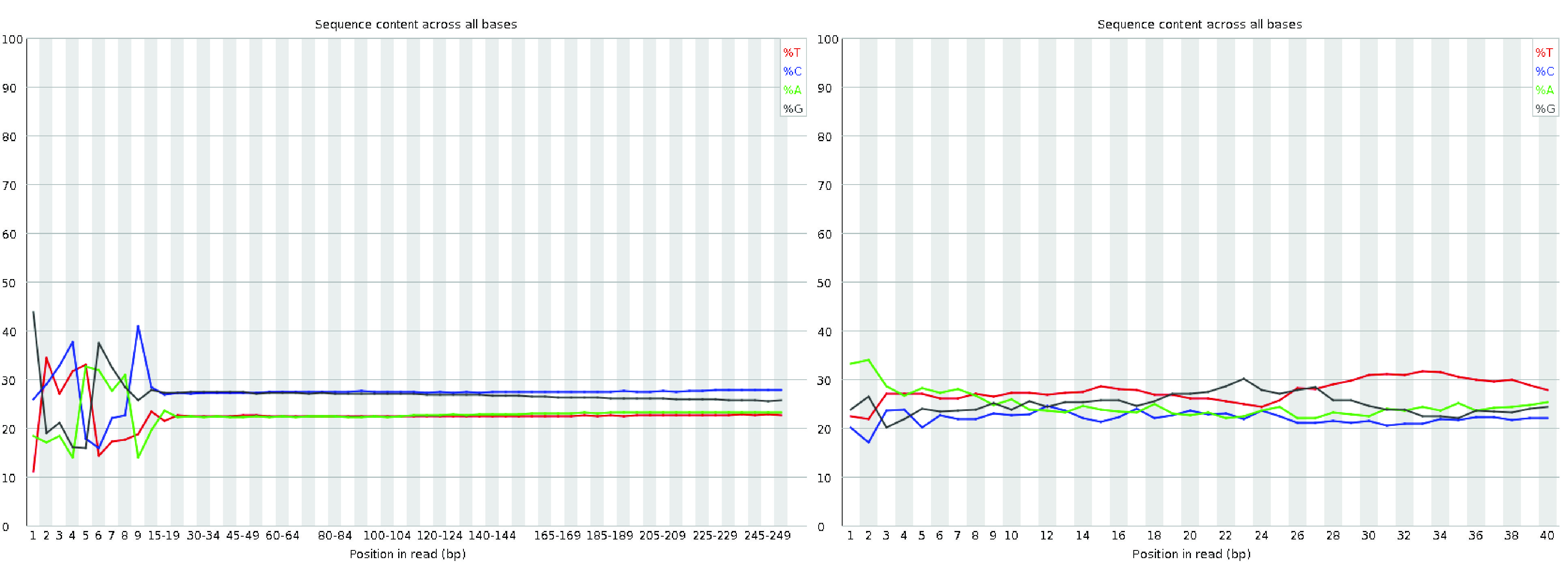
A good (left) and bad (right) per base sequence content plot.
6. 每序列 GC 含量
每序列 GC 含量图显示了所有读数的 GC 含量分布(红色)和理论(预期)分布(蓝色)。观察到的分布的中心峰应对应于底层转录组的总体 GC 含量。有时,我们会看到比理论分布更宽或更窄的分布,因为转录组的 GC 含量可能与基因组不同,理论上应遵循蓝色显示的分布。这种分布范围的差异并不罕见,尽管它可能会触发警告或失败。然而,复杂的分布通常表示文库受到污染。值得注意的是,在转录组学实验中,GC 含量图的解释可能有些复杂,因为预期的 GC 含量分布不仅取决于底层转录组的序列内容,还取决于样本中基因的表达情况,这些情况是未知的。
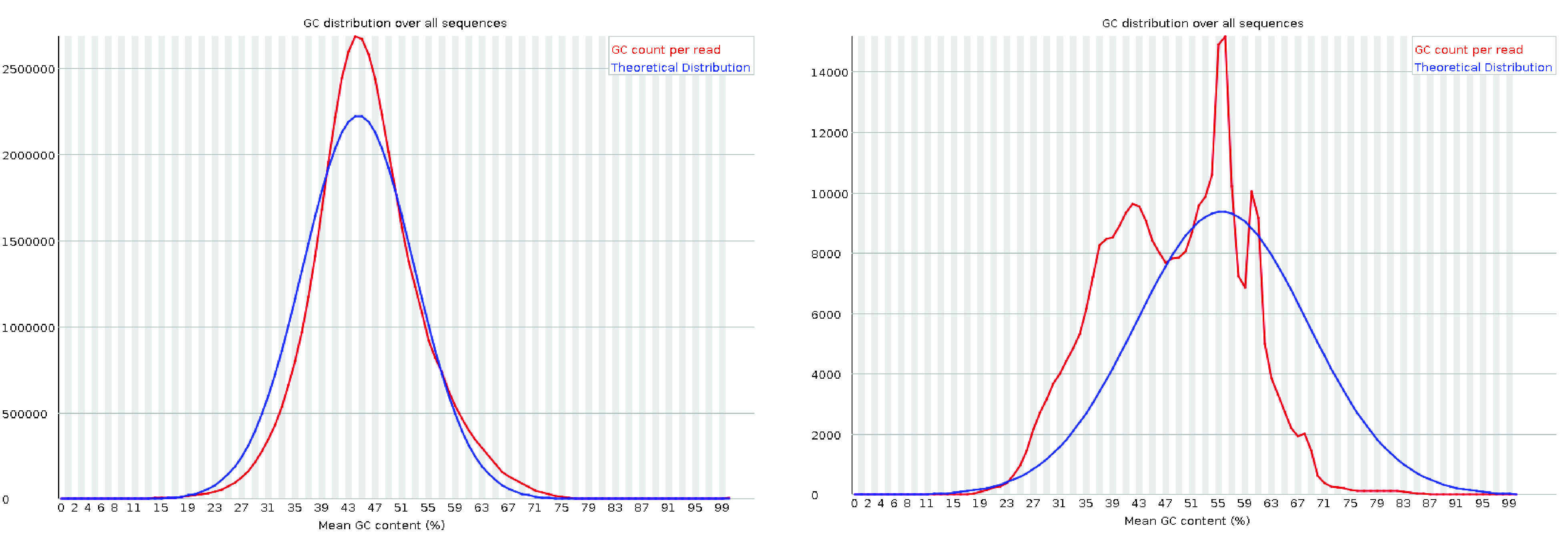
A good (left) and a bad (right) per sequence GC content plot. The plot on the left is from the RTSF at MSU. The plot on the right is taken from the HBC training program.
7. 每碱基 N 含量
每碱基 N 含量图显示每个位置上被调用为 N 的碱基百分比,当测序仪没有足够信心进行碱基调用时,会出现这种情况。在高质量的文库中,我们不应该期望在整个行中出现任何显著的非零 N 含量。
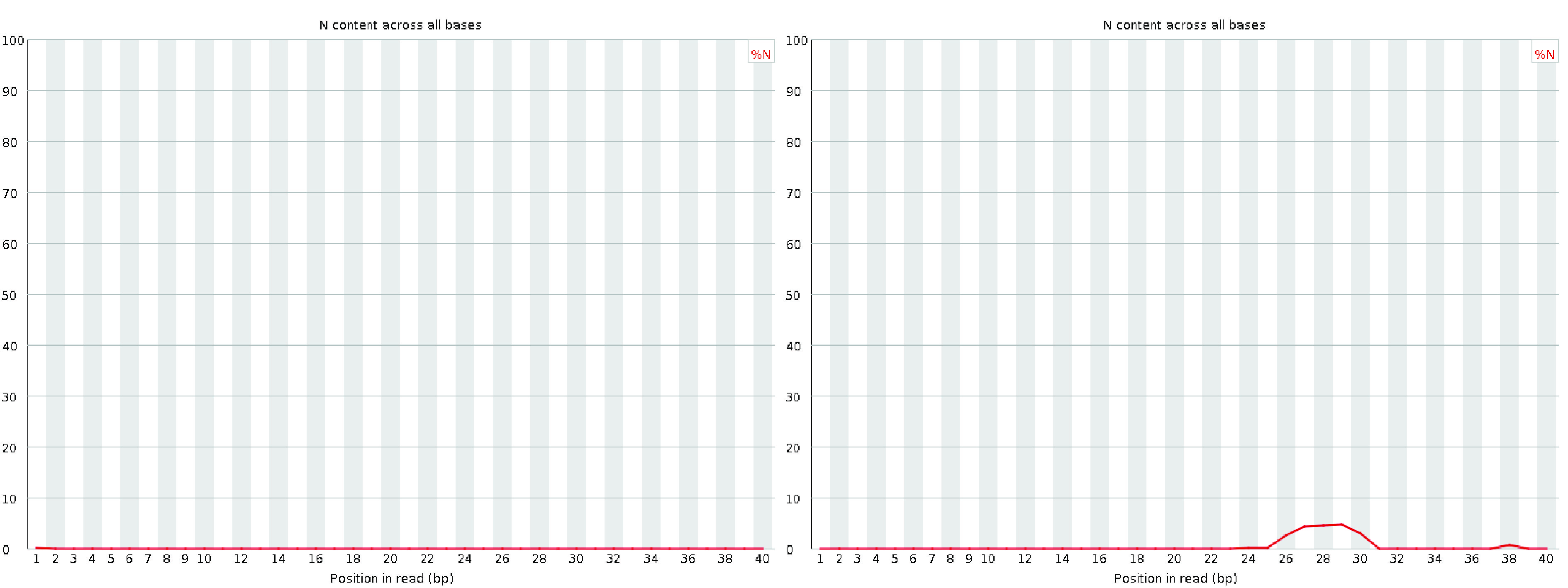
A good (left) and a bad (right) per base N content plot.
8. 序列长度分布
序列长度分布图显示读数长度的分布。在大多数单细胞化学方法中,所有读数的长度都是相同的。如果在质量评估之前进行了修剪,读数长度可能会有一些小的变化。
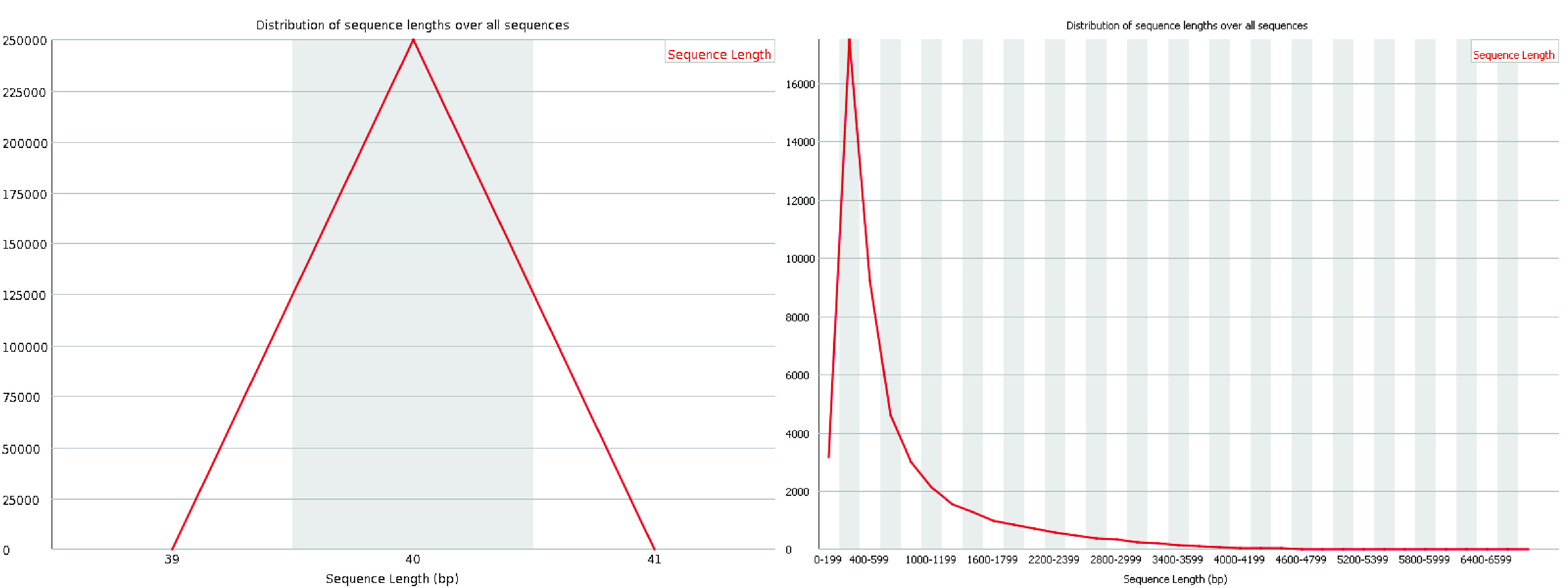
A good (left) and a bad (right) sequence length distribution plot.
9. 序列重复水平
序列重复水平图显示读数序列的重复程度分布(蓝线)以及去重后的分布。由于大多数单细胞平台需要多轮 PCR 扩增,高表达基因通常会产生大量转录本,并且 FastQC 本身不识别 UMI,因此一小部分序列可能会有大量重复。这可能会触发该模块的警告或失败,但不一定代表数据质量问题。不过,大多数序列应具有较低的重复水平。
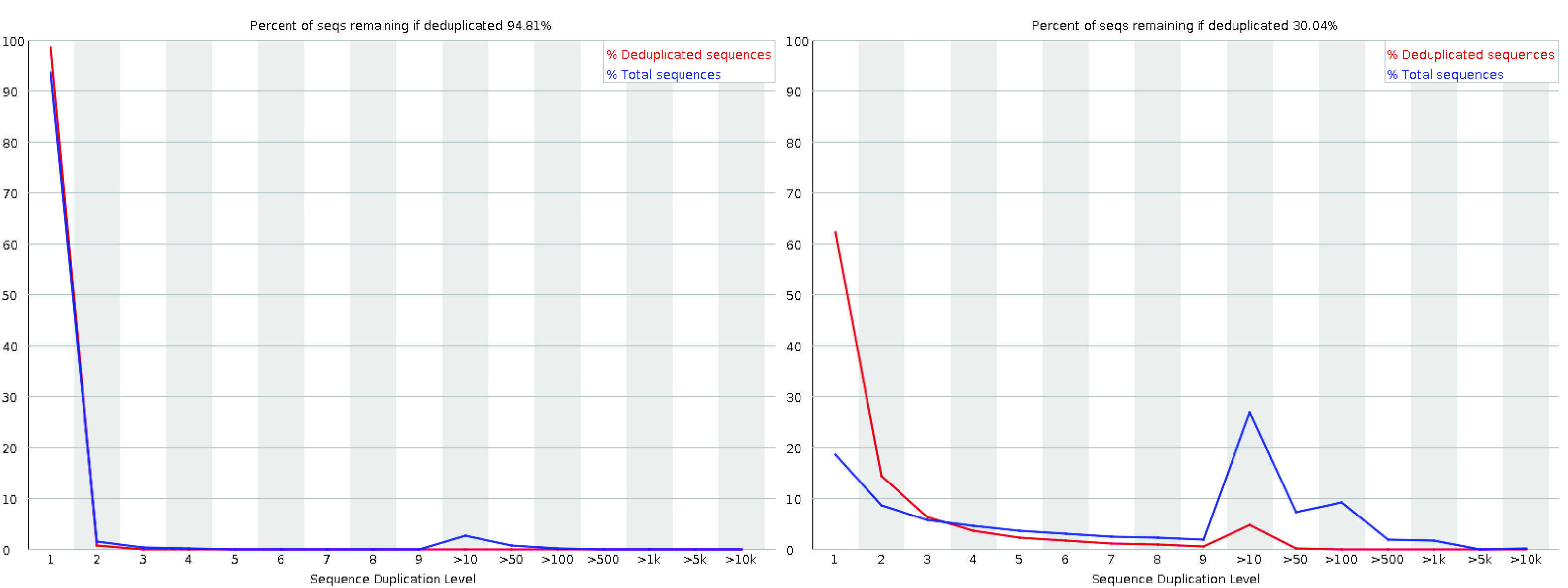
A good (left) and a bad (right) per sequence duplication levels plot.
10. 过度代表序列
过度代表序列模块列出了所有占总序列数超过 0.1% 的读数序列。经过 PCR 扩增后,我们可能会看到一些来自高表达基因的过度代表序列,但大多数序列不应过度代表。请注意,如果过度代表序列的可能来源不是“无匹配”,则可能表明文库被相应类型的来源污染。
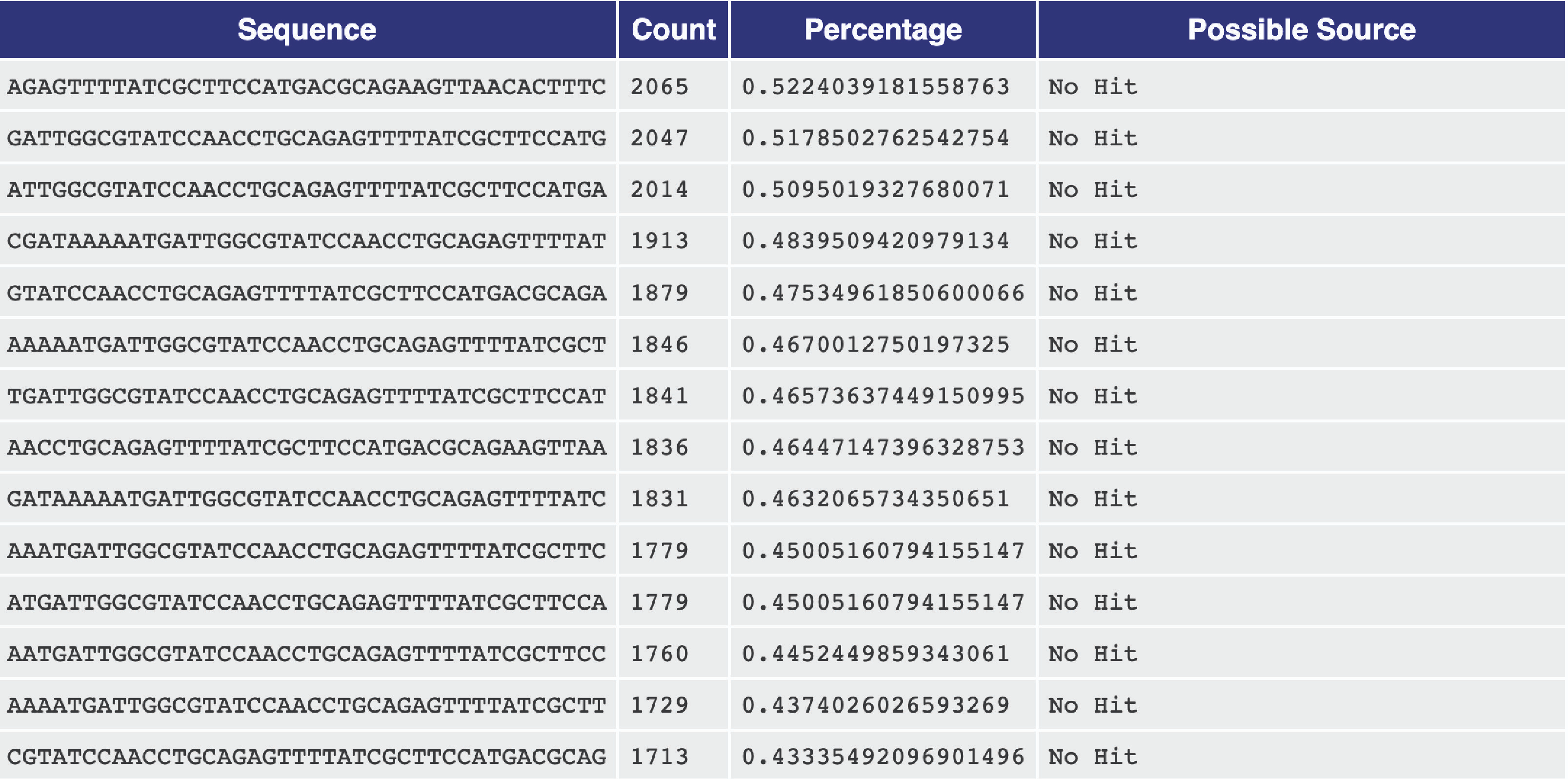
An overrepresented sequence table.
11. 接头序列含量
接头序列含量模块显示在每个碱基位置观察到的各接头序列的累积百分比。理想情况下,我们不应该在数据中看到任何丰富的接头序列。
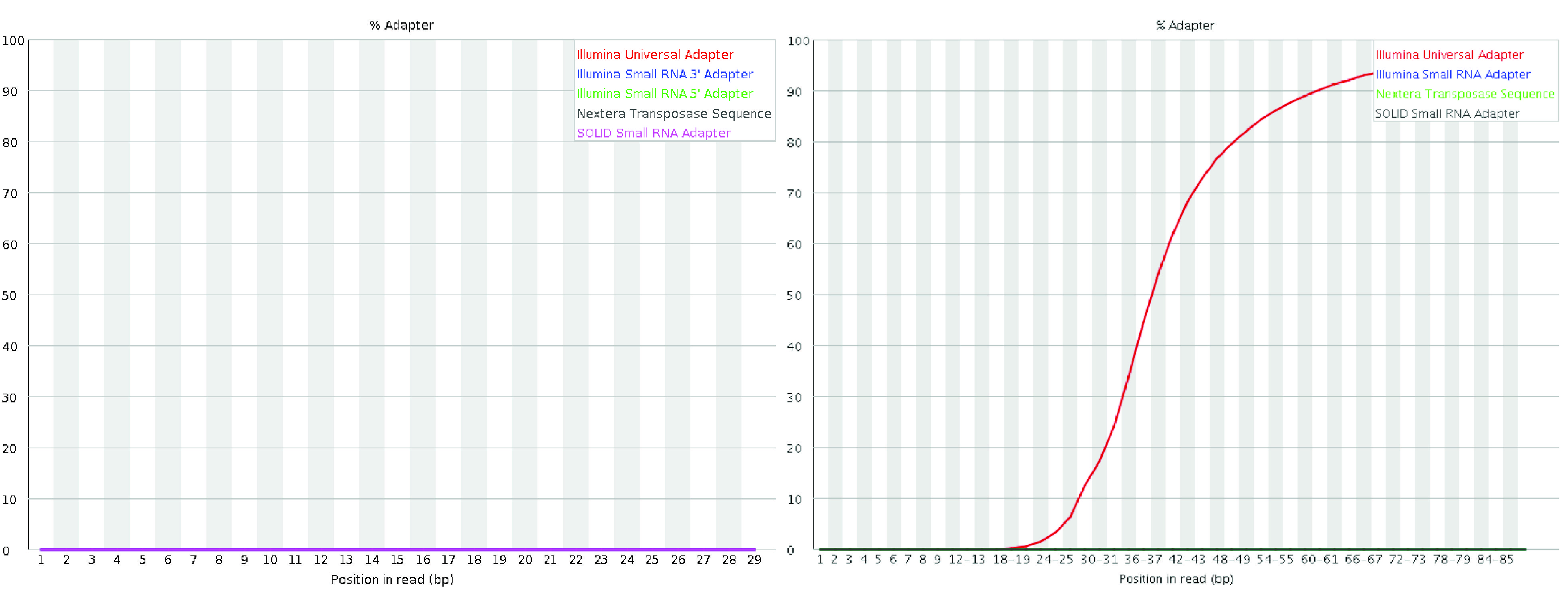
A good (left) and a bad (right) per sequence quality score plot. The plot on the right is from the QC Fail website.
比对和映射¶
比对或映射是单细胞原始数据处理中最基本的一步。它指的是确定每个测序片段可能的起源位置的过程(例如,与读数相似的一组基因组或转录组位置)。根据测序协议,生成的原始序列文件包含细胞级信息,通常称为细胞条形码(CB)、唯一分子标识符(UMI)和从分子生成的原始 cDNA 序列(读数序列)。作为单细胞样本原始数据处理的第一步(参见{numref}raw-proc-fig-overview),准确执行比对或映射对所有下游分析至关重要,因为错误的读数到转录本/基因的映射会导致错误或不准确的矩阵。
虽然将读数序列比对到参考序列的操作远早于 scRNA-seq 的出现,但当前快速增长的 scRNA-seq 样本规模(通常为数亿到数十亿读数)使得这一步骤特别需要计算资源。此外,使用不考虑特定 scRNA-seq 协议(如细胞条形码长度和位置、UMI 等)的现有 RNA-seq 比对器需要使用单独的工具来执行其他步骤,如解复用和 UMI 解析【Smith2017】。这种额外的开销在计算上可能很麻烦。此外,它通常需要大量磁盘空间来存储中间文件,并且处理这些中间文件所需的额外输入和输出操作进一步增加了运行时间要求。
为此,已经开发了几种专门用于比对或映射 scRNA-seq 数据的工具,这些工具能够自动和/或内部处理这些额外的处理要求。工具如 Cell Ranger(10x Genomics 的商业软件)【Zheng2017】、zUMIs【zumis】、alevin【Srivastava2019】、RainDrop【niebler2020raindrop】、kallisto|bustools【Melsted2021】、STARsolo【Kaminow2021】和 alevin-fry【He2022】提供了专门用于比对 scRNA-seq 读数以及解析技术读数内容(CB 和 UMI)的处理方法,以及解复用和 UMI 解析的方法。虽然所有这些工具都为用户提供了相对简化的界面,但这些工具在内部使用了各种不同的方法,有些生成传统的中间文件(例如 BAM 文件),然后处理它们,而其他工具则完全在内存中工作或使用简化的中间表示来减少输入/输出负担。
这些工具的另一个好处是它们依赖于预先存在的比对/映射引擎。因此,预先存在的用户群和教程使这些工具对新用户更友好。
虽然不同比对和映射方法使用的具体算法、数据结构以及时间和空间权衡可以有很大差异,但我们可以大致沿着两个轴对现有方法进行分类:
- 它们执行的比对类型
- 它们比对读数的参考序列类型。
映射类型¶
在这里,我们考虑三种主要的映射算法,这些算法最常应用于 sc/snRNA-seq 数据的映射:剪接比对、连续比对和轻量级映射的变种。
首先,我们在这里区分基于比对的方法和基于轻量级映射的方法(参见{numref}raw-proc-fig-alignment-mapping)。比对方法应用一系列不同的启发式方法来确定读数可能来自的潜在位置,然后在每个假设位置对读数和参考之间的最佳核苷酸级别比对进行评分,通常使用动态规划方法。使用动态规划算法解决比对问题有着悠久而丰富的历史。使用的具体动态规划算法类型取决于所需的比对类型。全局比对适用于查询序列和参考序列的全部进行比对的情况。另一方面,局部比对适用于可能只有查询的子序列期望匹配参考的子序列的情况。通常,最适用于短读数比对的模型既不是完全全局也不是完全局部,而是属于半全局比对,其中期望查询的大部分比对到参考的某个子字符串(这通常称为“拟合”比对)。此外,有时允许比对的软剪切,这样在读数的开头或结尾出现的错配、插入或删除的惩罚被忽略或降低权重。这通常使用扩展比对来完成。尽管这些修改改变了动态规划递归和回溯中使用的具体规则,但它们并没有从根本上改变其整体复杂性。
除了这些一般的比对技术,许多更复杂的修改和启发式方法已经被设计出来,使得在基因组测序读数比对中更加实际和有效。例如,带状比对是许多流行工具中包含的流行启发式方法,如果用户对低于某个阈值的比对分数不感兴趣,它可以避免计算大部分的动态规划表。同样,其他启发式方法如 X-drop 和 Z-drop 在早期修剪不太可能的比对中很流行。最近的算法进展,如波前比对,允许在渐近上(和实际上)更短的时间和更小的空间内确定最佳比对,如果存在高分(低差异)比对。最后,还投入了大量精力来优化数据布局和计算,以利用指令级并行,并将比对动态规划递归表达为高度适合数据并行和向量化的方式(例如,在差异编码中)。大多数广泛使用的比对工具都使用高度优化和向量化的比对实现。
除了比对分数,还可以获得产生此分数的实际比对的回溯。这些信息通常在输出的 SAM 或 BAM 文件中编码为 CIGAR 字符串(即“简洁特有的间隔比对报告”的缩写),这是比对的压缩字母数字表示形式。CIGAR 字符串的一个例子是 3M2D4M,它表示比对有三个匹配或错配,随后是两个长度的删除(参考中存在但读数中不存在的碱基),然后是四个更多的匹配或错配。CIGAR 字符串的其他变体也可以区分匹配或错配。例如,3=2D2=2X 是编码相同比对的有效扩展 CIGAR 字符串,除了它明确表示删除前的三个碱基构成匹配,并且删除后有两个匹配碱基,后跟两个错配碱基。有关 CIGAR 字符串格式的详细描述可以在 SAMtools 手册 或 UMICH 的 SAM wiki 页面 中找到。
通过计算这些分数,比对方法知道每个潜在读数比对的质量,然后它们可以使用这些分数来过滤那些与参考比对良好的读数,而不是那些由于低复杂性或“虚假”匹配而产生的映射。比对方法包括传统的“全比对”方法,如 STAR 和 STARsolo 中实现的,以及在 salmon 和 alevin 中实现的选择性比对方法,这些方法对映射进行评分,但省略了最佳比对回溯的计算。
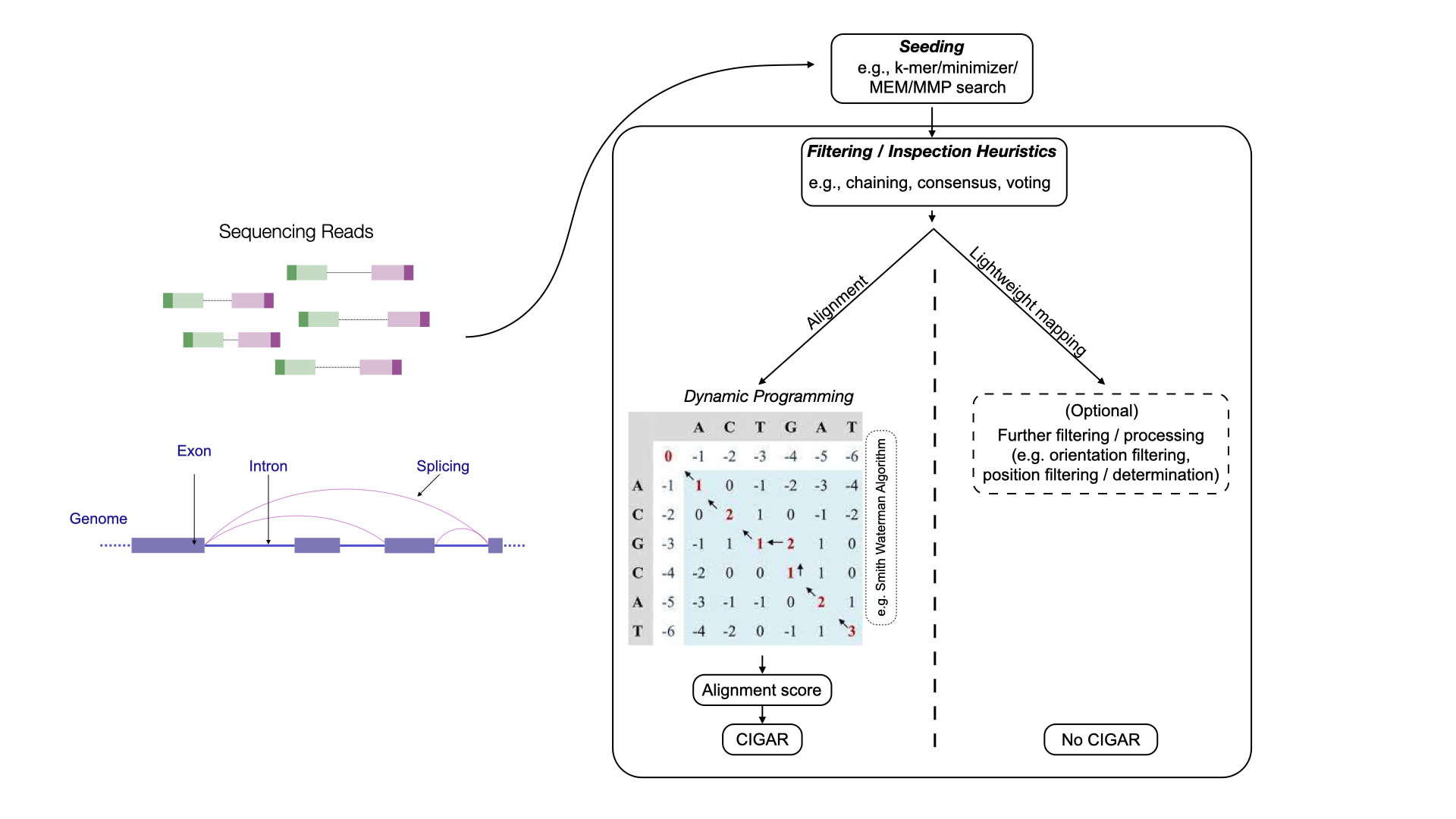
An abstract overview of the alignment-based method and lightweight mapping-based method.
基于比对的方法可以进一步分为剪接比对和连续比对(目前,没有执行剪接映射的轻量级映射方法)。剪接比对方法能够将一个序列读数与参考的多个不同片段对齐,允许在参考的比对区域之间存在较大的间隙。这些比对方法通常在将 RNA-seq 读数比对到基因组时应用,因为比对过程必须能够将跨越一个或多个剪接位点的读数对齐,在读数中连续的序列在参考中可能被内含子和外显子子序列分隔开(可能跨越数千个碱基序列)。剪接比对是一个具有挑战性的问题,特别是在读数只有一小部分区域跨越剪接位点的情况下,因为只有少量的信息序列可用于定位跨越剪接位点的读数片段。另一方面,连续比对则寻找一个与读数良好对齐的参考的连续子串。在这种比对问题中,虽然可以允许小的插入和删除,但通常不允许像剪接比对中观察到的那样的大间隙。
最后,我们可以区分上述基于比对的方法和轻量级映射方法,后者包括伪比对、准映射和带有结构约束的伪比对。这些方法通常通过避免读数和参考序列之间的核苷酸级比对来实现更快的速度。相反,它们基于一套单独的规则和启发式方法来确定报告的读数映射位置,这些规则和启发式方法可能仅查看匹配的 k-mer 集合或其他类型的精确匹配(例如,通过共识规则),并可能查看它们在读数和参考上的方向和相对位置(例如,链式匹配)。这可以显著提高速度和映射吞吐量,但也排除了基于评分的评估,即读数和参考之间的匹配是否构成支持高质量比对的良好匹配。
针对不同参考序列的映射¶
除了在映射算法本身上可以做出不同选择外,还可以对读数映射的参考序列进行不同选择。我们考虑了三种主要的参考序列类别:
- 完整的(可能已注释的)参考基因组
- 注释过的转录组
- 增强的转录组
还值得注意的是,目前并不是所有映射算法和参考序列的组合都是可能的。例如,目前不存在能够对参考基因组执行剪接映射的轻量级映射算法。
映射到完整基因组¶
第一种参考序列类型包括目标生物体的整个基因组,通常在映射过程中会考虑已注释的转录本。像 zUMIs【zumis】、Cell Ranger【Zheng2017】和 STARsolo【Kaminow2021】这样的工具采用这种方法。由于许多读数来自剪接转录本,这种方法还需要使用剪接感知比对算法,其中一个读数的比对可以跨越一个或多个剪接位点。这种方法的主要优点是可以考虑来自基因组任何位置的读数,而不仅仅是来自已注释的转录本的读数。由于这些方法需要构建全基因组索引,因此不仅可以报告比对到已知剪接转录本的读数,还可以报告重叠或比对到内含子的读数,使得使用这种方法的比对成本对于单细胞和单核数据非常相似。最后一个优点是,即使是位于注释转录本、外显子和内含子之外的读数也可以被这种方法考虑,从而可以在事后扩充量化位点的集合(例如,通过在样本特定和数据驱动的方式中将表达的 UTR 扩展添加到转录本注释中),并可能提高基因检测和量化的灵敏度。
另一方面,针对完整基因组执行剪接比对的策略的多功能性也带来了一些权衡。首先,在单细胞领域采用这种策略的最常用比对工具具有较大的内存需求。由于其速度和多功能性,这些工具大多基于 STAR 比对器。然而,对于人类规模的基因组,构建和存储索引可能需要超过 32 GB 的内存。使用稀疏 后缀数组 可以将最终索引大小减少接近一半,但这会降低比对速度,并且仍需要较大的内存进行初始构建。其次,与连续比对相比,剪接比对的难度增加,以及当前剪接比对工具必须显式计算每个读数比对的分数,这意味着这种方法的计算成本高于替代方法。最后,这种方法要求被研究生物体的基因组可用。虽然这对最常研究的参考生物体来说不是问题,并且很少出现问题,但对于只有转录组组装可用的非模式生物体来说,使用这些工具可能会很困难。
映射到剪接转录组¶
为了减少对基因组进行剪接比对的计算开销,广泛采用的替代方法是仅使用注释转录本的序列作为参考。由于大多数单细胞实验是从模式生物(如小鼠或人类)中生成的,这些模式生物具有良好的转录组注释,因此这种基于转录组的量化方法可能提供与基因组基于方法相似的读数覆盖率。与基因组相比,转录组序列的大小要小得多,从而最大限度地减少运行映射管道所需的计算资源。此外,由于相关的剪接模式已经在转录本序列本身中表示出来,这种方法不需要解决复杂的剪接比对问题。相反,可以简单地搜索读数的连续比对或映射。由于只需发现连续映射,基于比对和轻量级映射技术都适用于转录组参考,并且这两种方法都已被采用剪接转录组作为参考映射目标的流行工具所使用。
然而,虽然这种方法可以大大减少比对和映射所需的内存和时间,但它无法捕获来自剪接转录组之外的读数。因此,在处理单核数据时,这种方法是不足够的。即使在单细胞实验中,来自剪接转录组之外的读数也可能构成所有数据的相当一部分,并且越来越多的证据表明,这些读数应该纳入后续分析中。此外,当与轻量级映射方法结合使用时,剪接转录组和真正产生读数的参考序列之间共享的短匹配可能导致虚假的读数映射,从而导致某些基因的表达估计出现虚假甚至生物上不合理的结果【Kaminow2021,Bruning2022Comparative,raw:He2022】。
映射到增强的转录组¶
为了解决测序读数可能来自剪接转录本之外的问题,可以将剪接转录本序列与其他可能产生读数的参考序列(例如,完整长度的未剪接转录本或切除的内含子序列)结合起来增强。将转录组参考序列与内含子等序列增强后,可以生成比全基因组所需索引更小的参考索引,同时保留仅搜索连续读数比对的能力。这意味着相比于对全基因组进行映射,这种方法可以实现更快且占用内存更少的比对,同时仍然能够考虑来自剪接转录组之外的许多读数。最后,通过映射到扩展的参考序列集合,不仅可以比仅映射到剪接转录组恢复更多的读数位置,而且使用轻量级映射方法时,可以大大减少虚假映射【raw:He2022】。这种扩展或增强的转录组通常用于需要量化单核数据或为 RNA 速度分析准备数据的方法中(那些不映射到全基因组的方法)【Soneson2021Preprocessing】。因此,可以为所有不使用全基因组剪接比对的常见方法构建这种增强的参考【Srivastava2019,Melsted2021,raw:He2022】。
【raw:He2022】认为,即使在处理标准单细胞 RNA-seq 数据时,这种方法也是有价值的,并建议构建增强的splici(即剪接 + 内含子)参考进行映射和量化。splici参考通过使用剪接转录组序列以及包含与注释基因的内含子对应的合并区间的序列来构建。然后,每个参考都被标记其注释的剪接状态,并且映射到此参考时会结合剪接状态感知的方法进行 UMI 解析。该方法的主要优点是允许使用轻量级映射方法,同时大大减少虚假映射的发生率。它能够检测剪接和未剪接来源的读数,这可以提高后续分析的灵敏度【technote_10x_intronic_reads,Pool2022】。此外,由于在量化过程中追踪并在输出中单独报告剪接状态,它统一了单细胞、单核和 RNA 速度预处理的处理流程。
细胞条形码校正¶
基于液滴的单细胞分离系统,如 10x Genomics 提供的系统,已成为研究细胞异质性原因和结果的重要工具。在这种分离系统中,每个捕获细胞的 RNA 材料在基于水的液滴包裹中与条形码珠子一起提取。这些珠子使用称为细胞条形码(CBs)的独特寡核苷酸标记各个细胞的 RNA 内容,这些条形码随后与从 RNA 内容反转录的 cDNA 片段一起测序。珠子包含高多样性的 DNA 条形码,能够实现细胞分子内容的并行条形码化和测序读数的计算机解复用到单个细胞容器中。## 细胞条形码校正
基于液滴的单细胞分离系统,如 10x Genomics 提供的系统,已成为研究细胞异质性原因和结果的重要工具。在这种分离系统中,每个捕获细胞的 RNA 材料在基于水的液滴包裹中与条形码珠子一起提取。这些珠子使用称为细胞条形码(CBs)的独特寡核苷酸标记各个细胞的 RNA 内容,这些条形码随后与从 RNA 内容反转录的 cDNA 片段一起测序。珠子包含高多样性的 DNA 条形码,能够实现细胞分子内容的并行条形码化和测序读数的计算机解复用到单个细胞容器中。
```{admonition} A note on alignment orientation
根据所分析样本的化学特性和用户提供的处理选项,并非所有与参考序列对齐的测序片段都会被考虑用于量化和条形码校正。一个常用的过滤标准是比对方向。具体来说,某些化学特性规定的协议要求对齐的读数应该仅从特定方向(即回到)底层转录本。例如,在10x Genomics的3' Chromium化学体系中,我们期望生物读数与底层转录本的正向链对齐,尽管确实存在反义读数【technote_10x_intronic_reads】。因此,用户可以自行决定忽略或过滤与底层序列反向互补的读数映射。如果某种化学体系遵循这种所谓的“链特异性”协议,则应有文档说明。
### 条形码错误类型
单细胞分析的标记、测序和解复用方法通常效果很好。然而,在基于液滴的文库中观察到的细胞条形码(CB)数量可能与最初封装的细胞数量有显著差异,通常相差数倍。导致这种现象的几个主要错误来源包括:
- 双胞/多胞:单个条形码可能与两个或更多细胞相关联,导致细胞计数不足。
- 空液滴:液滴可能没有封装细胞,其中环境 RNA 被条形码标记并可以被测序,这导致细胞计数过多。
- 序列错误:PCR 扩增或测序过程中可能出现错误,导致细胞计数既有不足也有过多。
用于将 RNA-seq 读数解复用到细胞特定存储桶的计算工具使用广泛的诊断指标来过滤伪造或低质量数据。例如,有许多方法可以去除环境 RNA 污染、检测双胞和基于核苷酸序列相似性校正条形码序列错误。
几种常见策略用于识别和校正细胞条形码:
- 针对已知的潜在条形码列表进行校正:某些化学体系(如 10x Chromium)从已知的潜在条形码序列池中提取 CB。因此,任何样本中观察到的条形码集合应该是这个已知列表的子集,通常称为“白名单”、“许可列表”或“通过列表”。在这种情况下,常见策略是假设每个与已知列表中的某个元素完全匹配的条形码是正确的,并对所有其他条形码根据已知条形码列表进行校正(即找到许可列表中与观察到的条形码有小 Hamming 距离或编辑距离的条形码)。这种方法利用已知的许可列表来有效校正可能被损坏的许多条形码。然而,这种方法的一个困难是给定的损坏条形码可能在许可列表中有多个可能的校正(即其校正可能是模棱两可的)。实际上,如果考虑一个从 10x Chromium v3 许可列表中提取并在一个位置突变为列表中不存在的条形码,它有约 81% 的机会与许可列表中的两个或更多条形码有 Hamming 距离 1。通过仅考虑对许可列表中的条形码进行校正(即使是那些在给定样本中精确出现的条形码,或仅那些在给定样本中超过某个名义频率阈值的条形码),可以减少这种冲突的概率。此外,可以使用“校正”位置的碱基质量等信息来在模棱两可的校正情况下打破平局。然而,随着测定细胞数量的增加,潜在细胞条形码集合中的序列多样性不足会增加模棱两可校正的频率,并且标记有模棱两可校正条形码的读数最常被丢弃。
- 基于拐点或肘部的方法:如果潜在条形码集合未知——甚至如果已知,但希望直接从观察数据本身进行校正而不咨询外部列表——可以采用基于观察的样本中“真实”或高质量条形码的列表可能是与最大读数数相关联的那些条形码的方法。为此,可以构建条形码的累积频率图,其中条形码按与其相关联的不同读数或 UMI 的数量降序排列。通常,这个累积频率图将包含一个“拐点”或“肘部”——一个拐点,可以用来表征频繁出现的条形码与不频繁(因此可能是错误的)条形码之间的差异。许多方法存在以试图识别这样的拐点作为正确捕获的细胞和空液滴之间的可能区分点。随后,出现在“拐点”上方的条形码集合可以被视为许可列表,对其余条形码进行校正,如上述第一种方法所列。这种方法具有灵活性,因为它可以应用于具有外部许可列表的化学体系和没有外部许可列表的化学体系。拐点查找算法的进一步参数可以改变,以产生更多或更少限制性的选定条形码集合。然而,这种方法可能有某些缺点,比如过于保守的倾向,有时在没有明显拐点的样本中无法稳健工作。
- 基于用户提供的预期细胞数进行过滤和校正:这些方法在 CB 频率分布可能没有明确的拐点或由于技术伪影而表现出双峰性的情况下,旨在估计高质量或存在的条形码的可靠列表。在这种方法中,用户提供测定细胞数的估计值。然后,条形码按频率降序排列,在预期细胞计数附近的可靠分位索引处获得频率 f,并将所有频率在 f 的一个小常数倍数内的细胞(例如,≥ f/10)视为有效条形码。再次,剩余条形码根据这个有效列表进行校正,试图基于序列相似性唯一校正到这些有效条形码之一。
- 基于固定数量有效细胞的过滤:也许最简单的方法,尽管可能存在问题,是用户直接提供在排序频率图中将条形码视为有效的索引。所有频率大于或等于所选索引处频率的条形码被视为有效,并构成许可列表。然后,剩余的条形码集合使用上述方法中的相同方法与此列表进行校正。如果有足够多的不同条形码与用户要求的细胞数量相同,则始终会选择这么多的细胞。当然,这种方法仅在用户有充分理由相信阈值频率应设置在提供的索引周围时才是合理的。
### 未来的挑战
尽管高通量单细胞分析的细胞条形码方法取得了巨大成功,但随着实验规模的不断扩大,仍然存在一些挑战。例如,从所有观察中选择高质量细胞条形码的鲁棒方法的设计仍然是一个活跃的研究领域,在单细胞和单核实验之间会出现不同的挑战。此外,随着单细胞技术的发展,分析的细胞数量不断增加,CB 序列中的序列多样性不足可能导致序列校正引起的 CB 碰撞。解决这个问题可能需要更智能的条形码设计方法,并不断增加用于细胞条形码的寡核苷酸的长度。
## UMI 解析
在细胞条形码(CB)校正之后,读数要么被丢弃,要么被分配到一个校正后的 CB。随后,我们希望量化每个校正后 CB 中每个基因的丰度。
由于在转录本量化中讨论的扩增偏差,必须根据 UMI 对读数进行去重,以评估采样分子的真实计数。此外,在尝试执行这种估计时,还存在其他一些复杂因素。
UMI 去重步骤旨在识别在实验中捕获并测序的每个细胞中每个原始 PCR 前分子派生的读数和 UMI 集合。这个过程的结果是将分子计数分配给每个细胞中的每个基因,随后在下游分析中用作该基因的原始表达估计值。我们将这种观察 UMI 及其相关映射读数并尝试推断每个基因的观察分子原始数量的过程称为 UMI 解析。
为了简化说明,在以下文本中,映射到参考序列(例如,基因的基因组位点)的读数称为该参考序列的读数,其 UMI 标签称为该参考序列的 UMI;由 UMI 标记的读数集合称为该 UMI 的读数。一个读数只能被一个 UMI 标记,但如果它映射到多个参考序列,则可以属于多个参考序列。此外,由于 scRNA-seq 中每个细胞的分子条形码过程通常是隔离和独立的(除了前面提到的与准确解析细胞条形码相关的问题),在不损失一般性的情况下,将为特定细胞解释 UMI 解析。通常相同的程序将独立应用于所有细胞。
### UMI 解析的必要性
在理想情况下,正确(未修改)的 UMIs 标记读数,每个 UMI 的读数唯一地映射到一个共同的参考基因,并且 UMIs 与 PCR 前分子之间存在一一对应关系。因此,UMI 去重过程在概念上是简单的:一个 UMI 的读数是来自单个 PCR 前分子的 PCR 重复。每个基因捕获和测序的分子数量是观察到的该基因的不同 UMI 的数量。
然而,实践中遇到的问题使得上述简单规则不足以普遍识别 UMI 的基因来源,因此需要开发更复杂的模型。这里,我们主要关注两个挑战。
- 第一个问题是 UMI 中的错误。这些错误发生在读数的测序 UMI 标签在 PCR 或测序过程中引入错误时。常见的 UMI 错误包括 PCR 期间的核苷酸替代和测序期间的读数错误。不解决这些 UMI 错误会导致估计的分子数量膨胀【Smith2017,ziegenhain2022molecular】。
- 第二个问题是多重映射,发生在一个读数或 UMI 属于多个参考序列的情况下,例如多基因读数/UMIs。这种情况发生在一个 UMI 的不同读数映射到不同基因,一个读数映射到多个基因,或者两者都有。这个问题的后果是多基因读数/UMIs 的基因来源是模糊的,这导致对这些基因的 PCR 前分子计数的采样不确定性。简单地丢弃多基因读数/UMIs 会导致数据丢失或对倾向于产生多重映射读数的基因(如序列相似的基因家族)之间的偏差估计【Srivastava2019】。
!!! note "A note on UMI errors"
UMI错误,特别是由于核苷酸替换和错误调用引起的错误,在单细胞实验中普遍存在。研究【Smith2017】表明,测试单细胞实验中观察到的UMI序列之间的平均碱基差异数(编辑距离)低于随机采样的UMI序列,低编辑距离的富集与PCR扩增的程度密切相关。多重映射也存在于单细胞数据中,且根据所考虑的基因,可能发生在非微不足道的频率下。研究【Srivastava2019】表明,丢弃多重映射读数会对预测的分子计数产生负面偏差。
还存在其他我们在这里不重点讨论的挑战,例如“汇聚”和“分歧”UMI 碰撞。我们认为,在同一个细胞中,用于标记来自同一基因的两个不同 PCR 前分子的相同 UMI 是汇聚碰撞。当来自同一个 PCR 前分子的两个或更多不同 UMI 出现时,例如由于从该分子采样了多个引物位点,我们认为这是分歧碰撞。我们预期汇聚 UMI 碰撞是罕见的,因此它们的影响通常很小。此外,有时可以使用转录本级别的映射信息来解决这种碰撞【Srivastava2019】。分歧 UMI 碰撞主要发生在未剪接转录本的内含子中【technote_10x_intronic_reads】,解决它们引发的问题的方法是一个活跃的研究领域【technote_10x_intronic_reads,Gorin2021】。
鉴于 UMI 的使用在高通量 scRNA-seq 协议中几乎无处不在,并且解决这些错误可以改进基因丰度的估计,最近的文献中对 UMI 解析问题给予了很多关注【Islam2013,Bose2015,raw:Macosko2015,Smith2017,Srivastava2019,Kaminow2021,Melsted2021,raw:He2022,calib,umic,zumis】。
### 基于图的 UMI 解析
由于尝试解析 UMI 时遇到的问题,已经开发了许多方法来解决 UMI 解析的问题。尽管存在各种不同的 UMI 解析方法,我们将重点介绍一种基于 UMI 图的框架,这种框架修改自【Smith2017】最初提出的框架。每个图的连通组件代表一个子问题,其中某些 UMI 子集被折叠(即,被解析为同一 PCR 前分子的证据)。许多流行的 UMI 解析方法可以通过简单地修改图的精细化方式和在图上执行折叠或解析过程的方式在此框架中解释。
在单细胞数据的背景下,UMI 图 $G(V,E)$ 是一个有向图,具有节点集 $V$ 和边集 $E$。每个节点 $v_i \in V$ 代表读数的等价类(EC),边集 $E$ 编码 EC 之间的关系。定义在读数上的等价关系 $\sim_r$ 基于它们的 UMI 和映射信息。我们说读数 $r_x$ 和 $r_y$ 是等价的,即当且仅当它们具有相同的 UMI 标签并映射到相同的参考集合时,$r_x \sim_r r_y$。UMI 解析方法可以将“参考”定义为基因组位点【Smith2017】,转录本【Srivastava2019,raw:He2022】或基因【raw:Zheng2017,Kaminow2021】。其他 UMI 解析方法也存在,例如无参考模型【umic】和矩估计法【Melsted2021】,但它们可能不容易在此框架中表示,因此在此不做进一步讨论。
在 UMI 图框架中,UMI 解析方法可以分为三个主要步骤,每个步骤有不同的选项,可以由不同的方法模块化组成。这三个步骤分别是定义节点、定义邻接关系和解析组件。此外,这些步骤有时可能会在设计用于丢弃或启发式分配(通过修改报告的参考映射集合)显示某些类型的映射歧义的读数和 UMI 的过滤步骤之前(和/或之后)进行。
#### 定义节点
如上所述,节点 $v_i \in V$ 是读数的等价类。因此,$V$ 可以基于完整或过滤的映射读数集合及其相关的 _ 未经校正的 _UMI 进行定义。所有满足其参考集合和 UMI 标签的等价关系 $\sim_r$ 的读数都与同一顶点 $v \in V$ 相关联。如果 UMI 是多基因 UMI,则一个 EC 是多基因 EC。一些方法将通过在创建节点之前进行过滤或启发式分配读数来避免创建这样的 EC,而其他方法将保留和处理这些模糊顶点,并尝试通过简约、概率分配或基于相关规则或模型来解析它们的基因来源【Srivastava2019,Kaminow2021,raw:He2022】。
#### 定义邻接关系
在创建 UMI 图的节点集 $V$ 之后,根据 UMI 序列之间的距离(通常是 Hamming 距离或编辑距离)和它们相关的参考集合的内容来定义 $V$ 中节点的邻接关系。
在这里我们定义以下关于节点 $v_i \in V$ 的函数:
- $u(v_i)$ 是 $v_i$ 的 UMI 标签。
- $c(v_i) = |v_i|$ 是 $v_i$ 的基数,即与 $v_i$ 相关联的在 $\sim_r$ 下等价的读数的数量。
- $m(v_i)$ 是 $v_i$ 中编码的映射信息的参考集合。
- $D(v_i, v_j)$ 是 $u(v_i)$ 和 $u(v_j)$ 之间的距离,其中 $v_j \in V$。
根据这些函数定义,任何两个节点 $v_i, v_j \in V$ 当且仅当 $m(v_i) \cap m(v_j) \ne \emptyset$ 且 $D(v_i,v_j) \le \theta$ 时将具有双向边,其中 $\theta$ 是距离阈值,通常设置为 $\theta=1$【Smith2017,Kaminow2021,Srivastava2019】。此外,如果 $c(v_i) \ge 2c(v_j) -1$ 或反之,则可以将双向边替换为从 $v_i$ 到 $v_j$ 的有向边【Smith2017,Srivastava2019】。尽管这些边的定义是最常见的,但其他定义也是可能的,只要它们完全由 $u$,$c$,$m$ 和 $D$ 函数定义。现在,使用 $V$ 和 $E$,UMI 图 $G = (V,E)$ 已定义。
#### 定义图解析方法
给定已定义的 UMI 图,可以应用许多不同的解析方法。解析方法可以简单到找到连通组件、聚类图、贪婪地折叠节点或收缩边缘【Smith2017】,或通过某些规则(例如,单色树【Srivastava2019】)覆盖图来减少图的规模。结果是,简化的 UMI 图中的每个节点,或者在图未动态修改的情况下的覆盖集合的每个元素,表示一个 PCR 前分子。折叠的节点或覆盖集合被视为该分子的 PCR 重复。
不同的定义邻接关系的规则和不同的图解析方法可以寻求保留不同的特性,并可以定义各种不同的总体 UMI 解析方法。请注意,对于旨在概率性解决多重映射引起的歧义的方法,解析的 UMI 图可能包含多基因 EC,它们的基因来源将在后续步骤中解析。
#### 量化
UMI 解析的最后一步是使用解析的 UMI 图量化每个基因的丰度。对于丢弃多基因 EC 的方法,当前处理的细胞中的基因分子计数向量(简称计数向量)是通过计数标有每个基因的 EC 的数量生成的。另一方面,处理多基因 EC 而不是丢弃它们的方法通常通过应用一些统计推断程序来解决歧义。例如,研究【Srivastava2019】引入了一种期望最大化(EM)方法,用于概率分配多基因 UMI,并且相关的 EM 算法也被引入为后续工具的可选步骤【Melsted2021,Kaminow2021,raw:He2022】。在这个模型中,折叠 EC 到基因的分配是潜变量,基因的去重分子计数是主要参数。直观地说,来自基因唯一 EC 的证据将用于帮助概率性分配多基因 EC。EM 算法寻找生成观察到的 EC 的可能性最高的参数。
通常,上述描述的 UMI 解析和量化过程将分别对每个细胞(由校正后的 CB 表示)执行,以创建所有细胞中所有基因的完整计数矩阵。然而,高通量单细胞样本中每个细胞的信息相对贫乏,限制了进行 UMI 解析时可用的证据,这反过来限制了基于模型的解决方案(如上述描述的统计推断程序)的潜在有效性。因此,这里的进一步研究是非常必要的。例如,研究【Srivastava2020-lf】引入了一种允许在转录相似细胞之间共享信息的方法,以进一步改善量化结果。
## 计数矩阵质量控制
生成计数矩阵后,进行质量控制(QC)评估非常重要。通常有几种不同的评估被归入质量控制范畴。通常记录和报告基本的全局指标,以帮助评估测序测量本身的整体质量。这些指标包括映射读数的总比例、每个细胞观察到的不同 UMI 的分布、UMI 去重率的分布、每个细胞检测到的基因的分布等。这些和类似的指标通常由量化工具本身记录【raw:Zheng2017,Kaminow2021,Melsted2021,raw:He2022】,因为它们自然产生并可以在读数映射、细胞条形码校正和 UMI 解析过程中计算出来。同样,还存在一些工具来帮助组织和可视化这些基本指标,例如 [Loupe浏览器](https://support.10xgenomics.com/single-cell-gene-expression/software/visualization/latest/what-is-loupe-cell-browser)、[alevinQC](https://github.com/csoneson/alevinQC) 或 [kb_python报告](https://github.com/pachterlab/kb_python),具体取决于使用的量化管道。除了这些基本的全局指标外,在分析的这个阶段,QC 指标主要旨在帮助确定哪些细胞(CB)已经成功测序,以及哪些显示出需要过滤或校正的伪影。
### 空滴检测
其中一个最初的质量控制步骤是确定哪些细胞条形码对应于“高置信度”的测序细胞。在基于液滴的协议中,某些条形码与环境 RNA(而不是捕获细胞的 RNA)相关是很常见的{cite}`raw:Macosko2015`。当液滴未能捕获到细胞时,这种情况会发生。这些空液滴仍然会产生测序读数,但这些读数的特征与正确捕获细胞的条形码相关的读数显著不同。存在许多方法来评估条形码是否可能对应于空液滴。一种简单的方法是检查条形码的累积频率图,其中条形码按与之相关的不同 UMI 的数量降序排列。这个图通常包含一个“膝盖”,可以被识别为正确捕获细胞与空液滴之间的可能分界点{cite}`Smith2017,raw:He2022`。
虽然这种“膝盖”方法直观并且通常可以估计一个合理的阈值,但它有几个缺点。例如,并不是所有的累积直方图都显示出明显的膝盖,并且设计能够稳健地自动检测这种膝盖的算法是非常困难的。最后,仅仅通过条形码关联的总 UMI 数量来确定条形码是否与空或受损细胞相关可能并不是最佳信号。
这导致了几种专门设计用于检测空液滴或受损液滴或一般被认为是“低质量”的细胞的工具的开发{cite}`Lun2019,Heiser2021,Hippen2021,Muskovic2021,Alvarez2020,raw:Young2020`。这些工具结合了细胞质量的各种不同度量,包括不同 UMI 的频率、检测到的基因数量和线粒体 RNA 的比例,通常通过对这些特征应用统计模型来分类高质量细胞与假定的空液滴或受损细胞。这意味着细胞通常可以被评分,并且可以根据细胞不是空的或受损的估计后验概率选择最终的过滤。虽然这些模型通常在单细胞 RNA-seq 数据中表现良好,但在单核 RNA-seq 数据中可能需要应用几个额外的过滤器或启发式方法以获得稳健的过滤{cite}`Kaminow2021,raw:He2022`,如在 `DropletUtils` 中的 [`emptyDropsCellRanger`](https://github.com/MarioniLab/DropletUtils/blob/master/R/emptyDropsCellRanger.R) 函数中所暴露的那样{cite}`Lun2019`。
### 双重体检测
除了确定哪些细胞条形码对应于空液滴或受损细胞外,还可能希望识别那些对应于双重体或多重体的细胞条形码。当一个给定的液滴捕获了两个(双重体)或更多(多重体)细胞时,这可能导致这些细胞条形码在表示的读数和 UMI 数量以及显示的基因表达谱方面的分布出现偏差。也已经开发了许多工具来预测细胞条形码的双重体状态{cite}`DePasquale2019,McGinnis2019,Wolock2019,Bais2019,Bernstein2020`。一旦被检测到,被确定为可能是双重体和多重体的细胞可以在后续分析中被移除或以其他方式调整。
## 计数数据表示
在完成初步的原始数据处理和质量控制并继续后续分析时,重要的是要承认并记住,细胞与基因的计数矩阵充其量只是原始样本中测序分子的近似值。在原始数据处理管道的几个阶段中,应用了启发式方法并进行了简化以生成这个计数矩阵。例如,读数映射是不完美的,细胞条形码校正也是如此。UMI 的精确解析尤其具有挑战性,并且与多重映射读数相关的 UMI 问题通常被忽略,例如在假定单分子对应单 UMI 关系时,多个引物位点特别是在未剪接分子中可能会违背这一假设。
鉴于这些挑战和为解决这些问题而采用的简化方法,关于如何最好地表示预处理数据以供下游工具使用仍然是一个活跃的研究领域。例如,几个量化工具{cite}`Srivastava2019,Melsted2021,Kaminow2021,raw:He2022` 实现了最初由{cite:t}`Srivastava2019` 在此背景下引入的 _ 可选 _ EM 算法,该算法概率地分配与多个基因映射的读数相关联的 UMI。然而,这可能导致某些下游工具可能意外的非整数计数矩阵。或者,可以选择将 UMI 解析到 _ 基因组 _ 而不是单个基因,从而在预处理输出中保留多重映射信息(值得注意的是,类似的表示方法在转录本量水平上已经作为批量 RNA-seq 转录本量化工具的简洁内部表示方法使用了十多年{cite}`Turro2011,Nicolae2011,Patro2014,Bray2016,Patro2017,Ju2017`,并且这样的转录本量表示方法甚至已经被提议用于全长单细胞 RNA-seq 数据的聚类和降维{cite}`Ntranos2016` 和单细胞 RNA-seq 数据的差异表达分析{cite}`Ntranos2019`)。在这种情况下,结果计数矩阵的维度将是 $C \times G$,其中 $G$ 是量化注释中的基因数量,它将具有维度 $C \times E$,其中 $E$ 是给定样本中所有细胞中的不同 _ 基因组 _(通常称为等价类标签)的数量。通过将此信息传递到输出计数矩阵,可以避免应用启发式或概率性 UMI 解析方法,这些方法可能导致下游分析中使用的计数数据丢失或产生偏差。当然,要利用此信息,下游分析方法必须以这种格式期望信息。此外,这些下游方法通常必须有一种方法最终将这些计数解析到基因水平,因为基因组的丰度缺乏基因丰度的直观生物学解释。然而,这种表示方法在向下游分析工具传递更完整和准确的信息方面提供的好处是巨大的,正在开发利用这种表示方法的工具(例如 [DifferentialRegulation](https://github.com/SimoneTiberi/DifferentialRegulation));这是一个仍在积极研究的领域。
## 简要讨论
为了结束本章,我们传达一些观察和建议,这些观察和建议来自最近关于一些常见预处理工具的基准测试和评审研究{cite}`You_2021,Bruning_2022`。当然,重要的是要注意,单细胞和单核 RNA-seq 原始数据处理方法和工具的开发以及这些方法的持续评估是一项正在进行的社区努力。因此,当进行自己的分析时,实验几种不同的工具通常是有用且合理的。
在最粗略的层面上,大多数常见的工具可以稳健且准确地处理数据。有建议表明,对于许多常见的下游分析,如聚类,执行这些分析的方法通常比分析过程中的其他步骤对预处理工具的选择影响更小{cite}`You_2021`。然而,也观察到,将轻量级映射限制在剪接转录本上可以增加虚假映射和基因表达的概率{cite}`Bruning_2022`。
最终,特定工具的选择主要取决于手头的任务以及可用计算资源的限制。如果执行标准的单细胞分析,轻量级映射方法是一个不错的选择,因为它们比现有的基于比对的工具更快(通常快很多)且内存需求更少。如果执行单核 RNA-seq 分析,特别是 `alevin-fry` 是一个有吸引力的选项,因为它仍然节省内存,并且即使在扩展转录组参考以包含未剪接参考序列时,其索引仍然相对较小。另一方面,如果重要的是恢复映射到(扩展)转录组之外的读数,或者如果读数的基因组映射位点对于使用相关的下游工具或分析(例如,与工具如 `sierra` 的差异转录本使用分析)是必要的,则建议使用基于比对的方法。在基于比对的管道中,根据{cite:t}`Bruning_2022`,`STARsolo` 应该优于 `Cell Ranger`,因为前者比后者快得多,所需内存也更少,同时它也能产生几乎相同的结果。
## 一个现实世界的例子
既然我们已经介绍了各种原始数据处理方法的概念,我们现在转向演示如何使用特定工具(在本例中为 `alevin-fry`)来处理一个小的示例数据集。首先,我们需要从单细胞实验中获取的 [FASTQ格式](https://en.wikipedia.org/wiki/FASTQ_format) 的测序读数和用于映射这些读数的参考(例如转录组)。通常,参考包括测序物种的基因组序列和相应的基因注释,分别以 [FASTA](https://en.wikipedia.org/wiki/FASTA_format) 和 [GTF](https://useast.ensembl.org/info/website/upload/gff.html) 格式提供。
在本例中,我们将使用人类基因组的 _ 染色体 5_ 及其相关基因注释作为参考,这是 10x Genomics 参考构建的 [GRCh38 (GENCODE v32/Ensembl 98)参考](https://support.10xgenomics.com/single-cell-gene-expression/software/release-notes/build#GRCh38_2020A) 的一部分。相应地,我们从 10x Genomics 的 [人类脑肿瘤数据集](https://www.10xgenomics.com/resources/datasets/200-sorted-cells-from-human-glioblastoma-multiforme-3-lt-v-3-1-3-1-low-6-0-0) 中提取与生成的参考匹配的读数子集。
[`Alevin-fry`](https://alevin-fry.readthedocs.io/en/latest/){cite}`raw:He2022` 是一个快速、准确且节省内存的单细胞和单核数据处理工具。[Simpleaf](https://github.com/COMBINE-lab/simpleaf) 是一个用 [rust](https://www.rust-lang.org/) 编写的程序,提供了使用 `alevin-fry` 管道处理一些最常见协议和数据类型的统一和简化界面。也存在一个基于 nextflow 的 [工作流](https://github.com/COMBINE-lab/quantaf) 工具,用于处理大量的单细胞数据。在这里,我们将首先展示如何使用两个 `simpleaf` 命令处理单细胞原始数据。然后,我们描述了这些 `simpleaf` 命令对应的完整 `salmon alevin` 和 `alevin-fry` 命令集,以概述本节描述的步骤发生的位置,并传达可能的不同处理选项。这些命令将从命令行运行,并使用 [`conda`](https://docs.conda.io/en/latest/) 安装运行此示例所需的所有软件。
### 准备工作
在开始之前,我们需要在终端中创建一个 conda 环境并安装所需的软件包。`Simpleaf` 依赖于 [`alevin-fry`](https://alevin-fry.readthedocs.io/en/latest/)、[`salmon`](https://salmon.readthedocs.io/en/latest/) 和 [`pyroe`](https://github.com/COMBINE-lab/pyroe)。它们都可以在 `bioconda` 上找到,并且在安装 `simpleaf` 时会自动安装。
```bash
conda create -n af -y -c bioconda simpleaf
conda activate af
Note on using an Apple silicon-based device
Conda does not currently build most packages natively for Apple silicon. Therefore, if you are using a non-Intel-based Apple computer (e.g., with an M1(Pro/Max/Ultra) or M2 chip), you should make sure to specify that your environment uses the Rosetta2 translation layer. To do this, you can replace the above commands with the following (instructions adopted from here):
CONDA_SUBDIR=osx-64 conda create -n af -y -c bioconda simpleaf # create a new environment
conda activate af
conda env config vars set CONDA_SUBDIR=osx-64 # subsequent commands use intel packages
````
Next, we create a working directory, `af_xmpl_run`, and download and uncompress the example dataset from a remote host.
```bash
# Create a working dir and go to the working directory
## The && operator helps execute two commands using a single line of code.
mkdir af_xmpl_run && cd af_xmpl_run
# Fetch the example dataset and CB permit list and decompress them
## The pipe operator (|) passes the output of the wget command to the tar command.
## The dash operator (-) after `tar xzf` captures the output of the first command.
## - example dataset
wget -qO- https://umd.box.com/shared/static/lx2xownlrhz3us8496tyu9c4dgade814.gz | tar xzf - --strip-components=1 -C .
## The fetched folder containing the fastq files is called toy_read_fastq.
fastq_dir="toy_read_fastq"
## The fetched folder containing the human ref files is called toy_human_ref.
ref_dir="toy_human_ref"
# Fetch CB permit list
## the right chevron (>) redirects the STDOUT to a file.
wget -qO- https://raw.githubusercontent.com/10XGenomics/cellranger/master/lib/python/cellranger/barcodes/3M-february-2018.txt.gz | gunzip - > 3M-february-2018.txt
准备好参考文件(基因组 FASTA 文件和基因注释 GTF 文件)和读数记录(FASTQ 文件)后,我们现在可以应用上述的原始数据处理流程来生成基因计数矩阵。
简化的原始数据处理流程¶
Simpleaf 旨在简化单细胞和单核原始数据处理的 alevin-fry 接口。它将整个处理流程封装为两个步骤:
simpleaf index对提供的参考文件进行索引或生成 splici 参考(spliced transcripts + introns)并进行索引。simpleaf quant将测序读数映射到已索引的参考文件,并对映射记录进行定量分析以生成基因计数矩阵。
关于使用 simpleaf 进行映射的更多高级用法和选项可以在 这里 找到。
运行 simpleaf index 时,如果提供了基因组 FASTA 文件 (-f) 和基因注释 GTF 文件 (-g),它将生成一个 splici 参考并进行索引;如果仅提供了转录组 FASTA 文件 (--refseq),它将直接对其进行索引。目前,我们推荐使用 splici 索引。.
# simpleaf needs the environment variable ALEVIN_FRY_HOME to store configuration and data.
# For example, the paths to the underlying programs it uses and the CB permit list
mkdir alevin_fry_home && export ALEVIN_FRY_HOME='alevin_fry_home'
# the simpleaf set-paths command finds the path to the required tools and writes a configuration JSON file in the ALEVIN_FRY_HOME folder.
simpleaf set-paths
# simpleaf index
# Usage: simpleaf index -o out_dir [-f genome_fasta -g gene_annotation_GTF|--refseq transcriptome_fasta] -r read_length -t number_of_threads
## The -r read_lengh is the number of sequencing cycles performed by the sequencer to generate biological reads (read2 in Illumina).
## Publicly available datasets usually have the read length in the description. Sometimes they are called the number of cycles.
simpleaf index.\ \$
-o simpleaf_index.\ \$
-f toy_human_ref/fasta/genome.fa.\ \$
-g toy_human_ref/genes/genes.gtf.\ \$
-r 90.\ \$
-t 8
在输出目录 simpleaf_index 中,ref 文件夹包含 splici 参考文件;index 文件夹包含基于 splici 参考文件构建的 salmon 索引。
下一步,simpleaf quant 将使用索引目录和映射记录的 FASTQ 文件来生成基因计数矩阵。此命令封装了本节讨论的所有主要步骤,包括映射、细胞条形码校正和 UMI 解析。
# Collecting sequencing read files
## The reads1 and reads2 variables are defined by finding the filenames with the pattern "_R1_" and "_R2_" from the toy_read_fastq directory.
reads1_pat="_R1_"
reads2_pat="_R2_"
## The read files must be sorted and separated by a comma.
### The find command finds the files in the fastq_dir with the name pattern
### The sort command sorts the file names
### The awk command and the paste command together convert the file names into a comma-separated string.
reads1="$(find -L ${fastq_dir} -name "*$reads1_pat*" -type f | sort | awk -v OFS=, '{$1=$1;print}' | paste -sd,)"
reads2="$(find -L ${fastq_dir} -name "*$reads2_pat*" -type f | sort | awk -v OFS=, '{$1=$1;print}' | paste -sd,)"
# simpleaf quant
## Usage: simpleaf quant -c chemistry -t threads -1 reads1 -2 reads2 -i index -u [unspliced permit list] -r resolution -m t2g_3col -o output_dir
simpleaf quant.\ \$
-c 10xv3 -t 8.\ \$
-1 $reads1 -2 $reads2.\ \$
-i simpleaf_index/index.\ \$
-u -r cr-like.\ \$
-m simpleaf_index/index/t2g_3col.tsv.\ \$
-o simpleaf_quant
运行这些命令后,生成的定量信息可以在 simpleaf_quant/af_quant/alevin 文件夹中找到。在此目录中有三个文件:quants_mat.mtx、quants_mat_cols.txt 和 quants_mat_rows.txt,它们分别对应计数矩阵、该矩阵每列的基因名称以及该矩阵每行的校正和过滤后的细胞条形码。以下是这些文件的尾行内容。需要注意的是,alevin-fry 运行在 USA 模式(unspliced, spliced, and ambiguous mode)下,因此对每个基因的剪接和未剪接状态都进行了定量分析——生成的 quants_mat_cols.txt 文件的行数将等于注释基因数量的 3 倍,这些行分别对应每个基因的剪接(S)、未剪接(U)和剪接模糊(A)变体使用的名称。
# Each line in `quants_mat.mtx` represents
# a non-zero entry in the format row column entry
$ tail -3 simpleaf_quant/af_quant/alevin/quants_mat.mtx
138 58 1
139 9 1
139 37 1
# Each line in `quants_mat_cols.txt` is a splice status
# of a gene in the format (gene name)-(splice status)
$ tail -3 simpleaf_quant/af_quant/alevin/quants_mat_cols.txt
ENSG00000120705-A
ENSG00000198961-A
ENSG00000245526-A
# Each line in `quants_mat_rows.txt` is a corrected
# (and, potentially, filtered) cell barcode
$ tail -3 simpleaf_quant/af_quant/alevin/quants_mat_rows.txt
TTCGATTTCTGAATCG
TGCTCGTGTTCGAAGG
ACTGTGAAGAAATTGC
We can load the count matrix into Python as an AnnData object using the load_fry function from pyroe. A similar function, loadFry, has been implemented in the fishpond R package.
import pyroe
quant_dir = 'simpleaf_quant/af_quant'
adata_sa = pyroe.load_fry(quant_dir)
默认行为将 Anndata 对象的 X 层加载为每个基因的剪接和模糊计数的总和。然而,最近的研究 {cite}Pool2022 和 更新的实践 表明,即使在单细胞 RNA 测序数据中,包含内含子计数也可能增加灵敏度并有利于下游分析。虽然如何利用这些信息仍在研究中,但由于 alevin-fry 自动量化每个样本中的剪接、未剪接和模糊读数,可以简单地通过以下方式获取包含每个基因总计数的计数矩阵:
import pyroe
quant_dir = 'simpleaf_quant/af_quant'
adata_usa = pyroe.load_fry(quant_dir, output_format={'X' : ['U','S','A']})
完整的 alevin-fry 流程¶
通过 Simpleaf,只需几个命令即可以“标准”方式处理单细胞原始数据。接下来,我们将展示如何通过显式调用 pyroe、salmon 和 alevin-fry 命令生成相同的定量结果。除了教学价值外,了解每个步骤的确切命令在需要重新运行管道的某个部分或指定当前 simpleaf 未公开的一些参数时将非常有帮助。
请注意,{ref}raw-proc:example-prep 部分的命令应提前执行。在安装 simpleaf 时,所有在以下命令中调用的工具 pyroe、salmon 和 alevin-fry 已经安装。
构建索引¶
首先,我们处理基因组 FASTA 文件和基因注释 GTF 文件以获得 splici 索引。以下代码块中的命令类似于上面讨论的 simpleaf index 命令。这包括两个步骤:
- 通过调用
pyroe make-splici使用基因组和基因注释文件构建 splici 参考(spliced transcripts + introns) - 通过调用
salmon index对 splici 参考进行索引
# make splici reference
## Usage: pyroe make-splici genome_file gtf_file read_length out_dir
## The read_lengh is the number of sequencing cycles performed by the sequencer. Ask your technician if you are not sure about it.
## Publicly available datasets usually have the read length in the description.
pyroe make-splici.\ \$
${ref_dir}/fasta/genome.fa.\ \$
${ref_dir}/genes/genes.gtf.\ \$
90.\ \$
splici_rl90_ref
# Index the reference
## Usage: salmon index -t extend_txome.fa -i idx_out_dir -p num_threads
## The $() expression runs the command inside and puts the output in place.
## Please ensure that only one file ends with ".fa" in the `splici_ref` folder.
salmon index.\ \$
-t $(ls splici_rl90_ref/*\.fa).\ \$
-i salmon_index.\ \$
-p 8
The splici index can be found in the salmon_index directory.
(raw-proc:example-quant)=
Mapping and quantification¶
Next, we will map the sequencing reads recorded against the splici index by calling salmon alevin. This will produce an output folder called salmon_alevin that contains all the information we need to process the mapped reads using alevin-fry.
# Collect FASTQ files
## The filenames are sorted and separated by space.
reads1="$(find -L $fastq_dir -name "*$reads1_pat*" -type f | sort | awk '{$1=$1;print}' | paste -sd' ')"
reads2="$(find -L $fastq_dir -name "*$reads2_pat*" -type f | sort | awk '{$1=$1;print}' | paste -sd' ')"
# Mapping
## Usage: salmon alevin -i index_dir -l library_type -1 reads1_files -2 reads2_files -p num_threads -o output_dir
## The variable reads1 and reads2 defined above are passed in using ${}.
salmon alevin.\ \$
-i salmon_index.\ \$
-l ISR.\ \$
-1 ${reads1}.\ \$
-2 ${reads2}.\ \$
-p 8.\ \$
-o salmon_alevin.\ \$
--chromiumV3.\ \$
--sketch
Then, we execute the cell barcode correction and UMI resolution step using alevin-fry. This procedure involves three alevin-fry commands:
- The
generate-permit-listcommand is used for cell barcode correction. - The
collatecommand filters out invalid mapping records, corrects cell barcodes and collates mapping records originating from the same corrected cell barcode. - The
quantcommand performs UMI resolution and quantification.
# Cell barcode correction
## Usage: alevin-fry generate-permit-list -u CB_permit_list -d expected_orientation -o gpl_out_dir
## Here, the reads that map to the reverse complement strand of transcripts are filtered out by specifying `-d fw`.
alevin-fry generate-permit-list.\ \$
-u 3M-february-2018.txt.\ \$
-d fw.\ \$
-i salmon_alevin.\ \$
-o alevin_fry_gpl
# Filter mapping information
## Usage: alevin-fry collate -i gpl_out_dir -r alevin_map_dir -t num_threads
alevin-fry collate.\ \$
-i alevin_fry_gpl.\ \$
-r salmon_alevin.\ \$
-t 8
# UMI resolution + quantification
## Usage: alevin-fry quant -r resolution -m txp_to_gene_mapping -i gpl_out_dir -o quant_out_dir -t num_threads
## The file ends with `3col.tsv` in the splici_ref folder will be passed to the -m argument.
## Please ensure that there is only one such file in the `splici_ref` folder.
alevin-fry quant -r cr-like.\ \$
-m $(ls splici_rl90_ref/*3col.tsv).\ \$
-i alevin_fry_gpl.\ \$
-o alevin_fry_quant.\ \$
-t 8
运行这些命令后,生成的定量信息可以在 alevin_fry_quant/alevin 文件夹中找到。有关映射、CB 校正和 UMI 解析步骤的其他相关信息可以在 salmon_alevin、alevin_fry_gpl 和 alevin_fry_quant 文件夹中找到。
在这里给出的例子中,我们演示了使用 simpleaf 和 alevin-fry 来处理一个 10x Chromium 3' v3 数据集。Alevin-fry 和 simpleaf 还提供了许多其他选项来处理不同的单细胞协议,包括但不限于 Dropseq{cite}raw:Macosko2015、sci-RNA-seq3{cite}raw:Cao2019 以及其他 10x Chromium 平台。有关不同处理阶段的可用选项的更全面列表和描述可以在 alevin-fry 和 simpleaf 文档中找到。alevin-fry 还提供了一个基于 nextflow 的工作流,称为 quantaf,可以方便地从简单定义的样品表中处理多个样品。
当然,本节中引用和描述的许多其他原始数据处理工具也有类似的资源,包括 zUMIszumis、alevinSrivastava2019、kallisto|bustoolsMelsted2021、STARsoloKaminow2021 和 CellRanger。nf-core 的 scrnaseq 管道也提供了一个基于 nextflow 的管道,用于处理使用各种不同化学方法生成的单细胞 RNA-seq 数据,并整合了本节中描述的几种工具。
有用的链接¶
Alevin-fry教程 提供了处理不同类型数据的教程。
Pyroe 和 R 中的 roe 提供了处理 alevin-fry 定量信息的辅助函数。它们还提供了访问 quantaf 中预处理数据集的接口。
Quantaf 是一个基于 nextflow 的 alevin-fry 流程工作流,可以方便地根据输入表格处理大量单细胞和单核数据。公开可用的单细胞数据集的预处理定量信息可以在其 网页 上找到。
Simpleaf 是 alevin-fry 工作流程的包装器,允许使用上例中展示的仅两个命令来执行从创建 splici 参考到定量分析的整个流程。
来自 Galaxy 项目 的处理 scRNA-seq 原始数据的教程可以在 这里 和 这里 找到。
解释和评估 FastQC 报告的教程可以从 MSU,HBC 培训计划,Galaxy 培训 和 QC Fail 网站 获取。