1.3 Analysis frameworks and tools
1.4.1. 单细胞分析框架和联盟¶
在获得计数矩阵后,如前所述,探索性数据分析阶段开始。由于数据的规模和复杂性,需要专门的工具。虽然在单细胞分析的早期,人们使用自定义脚本分析数据,但现在已有专门的框架来应对这一目的。最受欢迎的三个选项是基于 R 的 Bioconductor[Huber et al., 2015] 和 Seurat[Hao et al., 2021] 生态系统,以及基于 Python 的 scverse[scverse, 2022] 生态系统。这些框架不仅在使用的编程语言上不同,在底层数据结构和可用的专业分析工具上也有所区别。
Bioconductor 是一个项目,致力于开发、支持和分享专注于严格和可重复的多种生物检测数据分析的免费开源软件,包括单细胞分析。统一的开发者和用户体验以及丰富的文档和用户友好的小插曲是 Bioconductor 的最大优势。Seurat 是一个专门为单细胞数据分析设计的 R 包,提供了包括多模式和空间数据在内的所有分析步骤工具。Seurat 以其写得很好的小插曲和庞大的用户群而闻名。然而,这两个 R 选项在处理极大规模数据集(超过五十万个细胞)时可能会遇到可扩展性问题,这促使了基于 Python 的社区开发 scverse 生态系统。scverse 是一个致力于生命科学基础工具的组织和生态系统,最初专注于单细胞分析。可扩展性、可扩展性以及与现有 Python 数据和机器学习工具的强大互操作性是 scverse 生态系统的一些优势。
所有这三个生态系统都参与了许多努力,以实现涉及框架的互操作性。这将在“互操作性”章节中讨论。本书始终专注于针对相应问题的最佳工具,因此将针对具体问题使用上述生态系统的混合方法。然而,所有分析的基础将是 scverse 生态系统,原因有二:
- 虽然我们将在本书中经常切换生态系统甚至编程语言,但一致使用数据结构和工具有助于读者专注于概念而非实现细节。
- 一本关于 Bioconductor 生态系统的优秀书籍 已经存在。我们鼓励只想学习 Bioconductor 单细胞分析的用户阅读它。
在接下来的部分中,将更详细地介绍 scverse 生态系统,并重点解释最重要的数据结构。这一介绍无法涵盖数据结构和框架的所有方面。介绍所有可用分析功能也超出了范围。因此,我们在必要时会参考相关框架的教程和文档。
1.4.2. 使用 AnnData 存储单模态数据¶
如前所述,在对基因组数据进行比对和基因注释后,通常将其汇总为特征矩阵。这个矩阵的形状为 number_observations x number_variables,对于 scRNA-seq 来说,观测值是细胞条形码,变量是注释基因。在分析过程中,这个矩阵的观测值和变量会被注释上计算得出的测量值(例如质量控制指标或潜在空间嵌入)以及先验知识(例如来源供体或替代基因标识符)。在 scverse 生态系统中,使用 AnnData[Virshup et al., 2021] 将数据矩阵与这些注释关联起来。为了实现快速和内存高效的转换,AnnData 还支持稀疏矩阵和部分读取。
虽然 AnnData 与 R 生态系统中的数据结构(例如 Bioconductor 的 SummarizedExperiment 或 Seurat 的对象)大体相似,但 R 包使用的是转置的特征矩阵。
在其核心,AnnData 对象在 X 中存储一个稀疏或密集矩阵(在 scRNA-Seq 的情况下为计数矩阵)。这个矩阵的维度为 obs_names x var_names,其中 obs(=观测值)对应于细胞的条形码,var(=变量)对应于基因标识符。这个矩阵 X 被 Pandas 数据框 obs 和 var 包围,分别保存细胞和基因的注释。此外,AnnData 还保存了具有相应维度的观测值(obsm)或变量(varm)的完整计算矩阵。将细胞与细胞或基因与基因关联的图状结构通常保存在 obsp 和 varp 中。任何不适合其他插槽的非结构化数据都作为非结构化数据保存在 uns 中。还可以在 layers 中存储更多的 X 值。例如,未标准化的原始计数数据可以存储在 counts 层中,而标准化的数据存储在未命名的默认层中。
AnnData 主要设计用于单模态(例如仅 scRNA-Seq)数据。然而,本章后面会介绍的 AnnData 扩展,如 MuData,允许高效存储和访问多模态数据。
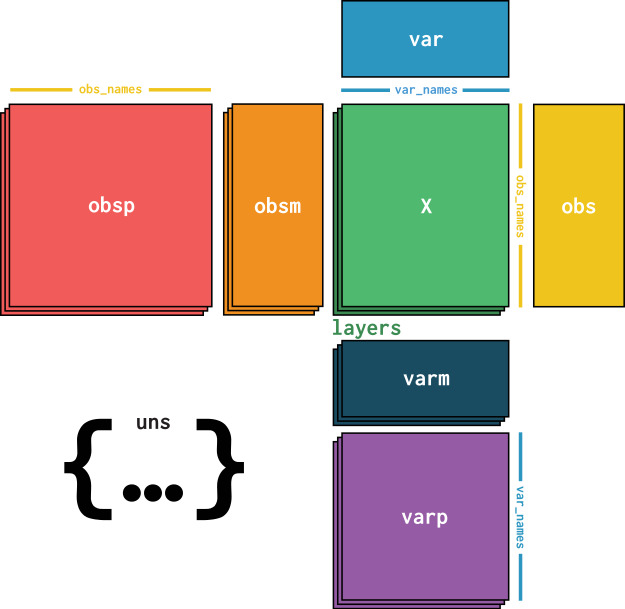
Fig. 4.1 AnnData overview. Image obtained from [Virshup et al., 2021].#
1.4.2.1. 安装¶
AnnData 可以通过 PyPI 和 Conda 安装,使用以下任意一种方法即可完成安装:
pip install anndata
conda install -c conda-forge anndata
1.4.2.2. 初始化 AnnData 对象¶
本节内容参考了 AnnData 的“入门”教程:https://anndata.readthedocs.io/en/latest/tutorials/notebooks/getting-started.html
让我们创建一个包含稀疏计数信息的简单 AnnData 对象,这个对象可以表示基因表达计数。首先,我们导入所需的包。
import numpy as np
import pandas as pd
import anndata as ad
from scipy.sparse import csr_matrix
作为下一步,我们用随机泊松分布的数据初始化一个 AnnData 对象。一个不成文的规定是将分析的主要 AnnData 对象命名为 adata。
counts = csr_matrix(np.random.poisson(1, size=(100, 2000)), dtype=np.float32)
adata = ad.AnnData(counts)
adata
AnnData object with n_obs × n_vars = 100 × 2000
获得的 AnnData 对象有 100 个观测值和 2000 个变量。这相当于 100 个细胞和 2000 个基因。我们传入的初始数据可以作为一个稀疏矩阵,通过 adata.X 访问。
<100x2000 sparse matrix of type '<class 'numpy.float32'>'
with 126413 stored elements in Compressed Sparse Row format>
现在,我们分别使用 .obs_names 和 .var_names 为 obs 和 var 轴提供索引。
adata.obs_names = [f"Cell_{i:d}" for i in range(adata.n_obs)]
adata.var_names = [f"Gene_{i:d}" for i in range(adata.n_vars)]
print(adata.obs_names[:10])
Index(['Cell_0', 'Cell_1', 'Cell_2', 'Cell_3', 'Cell_4', 'Cell_5', 'Cell_6',
'Cell_7', 'Cell_8', 'Cell_9'],
dtype='object')
1.4.2.3. 添加对齐的元数据¶
1.4.2.3.1. 观测值或变量级别¶
我们的 AnnData 对象的核心部分已经就位。下一步,我们在观测值和变量级别添加元数据。请记住,我们在 AnnData 对象的 .obs 和 .var 插槽中分别存储细胞和基因的注释。
ct = np.random.choice(["B", "T", "Monocyte"], size=(adata.n_obs,))
adata.obs["cell_type"] = pd.Categorical(ct) # Categoricals are preferred for efficiency
adata.obs
| cell_type | |
|---|---|
| Cell_0 | B |
| Cell_1 | T |
| Cell_2 | B |
| Cell_3 | T |
| Cell_4 | Monocyte |
| ... | ... |
| Cell_95 | T |
| Cell_96 | T |
| Cell_97 | B |
| Cell_98 | T |
| Cell_99 | Monocyte |
100 rows × 1 columns
如果我们现在再次检查 AnnData 对象的表示,会注意到它也更新了 obs 中的 cell_type 信息。
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
1.4.2.3.2. 使用元数据进行子集化¶
我们还可以使用随机生成的细胞类型对 AnnData 对象进行子集化。AnnData 对象的切片和掩码操作与 Pandas DataFrame 或 R 矩阵的数据访问类似。更多详细信息见下文。
bdata = adata[adata.obs.cell_type == "B"]
bdata
View of AnnData object with n_obs × n_vars = 26 × 2000
obs: 'cell_type'
1.4.2.4. 观测值/变量级别矩阵¶
我们可能还会有多维度的元数据,比如数据的 UMAP 嵌入。对于这种类型的元数据,AnnData 有 .obsm/.varm 属性。我们使用键来标识插入的不同矩阵。.obsm/.varm 的限制是,.obsm 矩阵的长度必须等于观测值的数量,即 .n_obs,而 .varm 矩阵的长度必须等于变量的数量,即 .n_vars。它们各自可以有不同的维度。
让我们从一个随机生成的矩阵开始,这个矩阵可以解释为我们希望存储的数据的 UMAP 嵌入,以及一些随机生成的基因级别元数据。
adata.obsm["X_umap"] = np.random.normal(0, 1, size=(adata.n_obs, 2))
adata.varm["gene_stuff"] = np.random.normal(0, 1, size=(adata.n_vars, 5))
adata.obsm
AxisArrays with keys: X_umap
再次,AnnData 对象的表示已更新。
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
obsm: 'X_umap'
varm: 'gene_stuff'
关于 .obsm/.varm 的几点说明:
- “类似数组”的元数据可以来自 Pandas DataFrame、scipy 稀疏矩阵或 numpy 密集数组。
- 使用 scanpy 时,它们的值(列)不易绘制,相反,来自
.obs的项可以轻松绘制在例如 UMAP 图上。
1.4.2.5. 非结构化元数据¶
如上所述,AnnData 有 .uns,允许存储任何非结构化元数据。这可以是任何内容,例如在数据分析中有用的某些常规信息的列表或字典。尽量只将无法有效存储在其他插槽中的数据存储在此插槽中。
adata.uns["random"] = [1, 2, 3]
adata.uns
OverloadedDict, wrapping:
OrderedDict([('random', [1, 2, 3])])
With overloaded keys:
['neighbors'].
1.4.2.6. 层¶
最后,我们可能会有原始核心数据的不同形式,比如一个是标准化的,一个是未标准化的。这些可以存储在 AnnData 的不同层中。例如,让我们对原始数据进行对数转换并将其存储在一个层中。.
adata.layers["log_transformed"] = np.log1p(adata.X)
adata
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
uns: 'random'
obsm: 'X_umap'
varm: 'gene_stuff'
layers: 'log_transformed'
我们的原始矩阵 X 未被修改,仍然可以访问。我们可以通过比较原始 X 和新的层来验证这一点。
(adata.X != adata.layers["log_transformed"]).nnz == 0
1.4.2.7. 转换为 DataFrame¶
可以从某一层中获取一个 Pandas DataFrame。
adata.to_df(layer="log_transformed")
| Gene_0 | Gene_1 | Gene_2 | Gene_3 | Gene_4 | Gene_5 | Gene_6 | Gene_7 | Gene_8 | Gene_9 | ... | Gene_1990 | Gene_1991 | Gene_1992 | Gene_1993 | Gene_1994 | Gene_1995 | Gene_1996 | Gene_1997 | Gene_1998 | Gene_1999 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell_0 | 0.693147 | 0.000000 | 1.386294 | 1.386294 | 0.693147 | 0.000000 | 0.693147 | 1.098612 | 0.000000 | 1.098612 | ... | 0.693147 | 0.693147 | 0.000000 | 0.693147 | 1.098612 | 0.000000 | 0.000000 | 1.098612 | 0.693147 | 0.000000 |
| Cell_1 | 1.098612 | 1.098612 | 0.000000 | 0.000000 | 0.000000 | 0.693147 | 0.000000 | 0.693147 | 1.386294 | 0.000000 | ... | 1.098612 | 1.386294 | 1.098612 | 0.000000 | 0.693147 | 0.693147 | 1.386294 | 0.000000 | 0.000000 | 0.693147 |
| Cell_2 | 0.693147 | 0.693147 | 1.098612 | 0.693147 | 1.386294 | 1.386294 | 0.693147 | 0.693147 | 0.000000 | 1.098612 | ... | 0.000000 | 0.693147 | 0.693147 | 0.000000 | 1.386294 | 0.693147 | 0.000000 | 0.000000 | 0.693147 | 0.693147 |
| Cell_3 | 0.000000 | 0.000000 | 0.000000 | 0.693147 | 0.693147 | 0.000000 | 1.386294 | 1.098612 | 0.693147 | 0.693147 | ... | 0.693147 | 0.693147 | 0.000000 | 0.693147 | 0.693147 | 0.693147 | 0.693147 | 0.000000 | 1.386294 | 0.693147 |
| Cell_4 | 0.000000 | 0.693147 | 0.000000 | 1.609438 | 1.098612 | 0.693147 | 0.000000 | 0.000000 | 0.693147 | 0.000000 | ... | 1.098612 | 1.098612 | 0.000000 | 1.386294 | 1.791759 | 1.098612 | 1.609438 | 0.693147 | 0.000000 | 0.693147 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| Cell_95 | 0.693147 | 1.098612 | 0.693147 | 1.386294 | 1.098612 | 0.693147 | 0.000000 | 0.000000 | 0.000000 | 0.693147 | ... | 0.000000 | 0.693147 | 0.000000 | 0.000000 | 0.000000 | 0.693147 | 0.000000 | 0.693147 | 1.386294 | 0.693147 |
| Cell_96 | 0.693147 | 0.000000 | 0.000000 | 1.098612 | 0.000000 | 0.693147 | 0.693147 | 1.098612 | 0.000000 | 0.000000 | ... | 1.386294 | 0.693147 | 1.098612 | 0.693147 | 0.000000 | 0.693147 | 1.098612 | 1.386294 | 0.693147 | 0.000000 |
| Cell_97 | 0.693147 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 1.098612 | 0.000000 | 0.000000 | 0.693147 | ... | 0.000000 | 0.693147 | 1.386294 | 0.000000 | 0.000000 | 0.000000 | 0.693147 | 0.693147 | 1.609438 | 0.693147 |
| Cell_98 | 0.693147 | 0.693147 | 0.000000 | 1.098612 | 0.000000 | 0.693147 | 0.693147 | 1.098612 | 0.000000 | 0.000000 | ... | 0.000000 | 0.000000 | 0.693147 | 0.693147 | 0.000000 | 0.693147 | 1.386294 | 0.693147 | 1.098612 | 1.386294 |
| Cell_99 | 1.098612 | 1.791759 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 1.098612 | 1.609438 | 0.693147 | 0.693147 | ... | 0.693147 | 0.000000 | 0.000000 | 0.693147 | 0.693147 | 0.693147 | 0.693147 | 0.000000 | 0.000000 | 1.386294 |
100 rows × 2000 columns
1.4.2.8. 读取和写入 AnnData 对象¶
AnnData 对象可以保存到磁盘上的分层数组存储,如 HDF5 或 Zarr,以便在磁盘和内存中实现类似的结构。AnnData 有自己基于 HDF5 的持久文件格式:h5ad。如果具有少量类别的字符串列尚未转换为分类变量,AnnData 将自动将它们转换为分类变量。现在我们将把 AnnData 对象保存为 h5ad 格式。
adata.write("my_results.h5ad", compression="gzip")
X Group
layers Group
obs Group
obsm Group
uns Group
var Group
varm Group
… and read it back in.
adata_new = ad.read_h5ad("my_results.h5ad")
adata_new
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
uns: 'random'
obsm: 'X_umap'
varm: 'gene_stuff'
layers: 'log_transformed'
1.4.2.9. 高效数据访问¶
1.4.2.9.1. 视图和副本¶
我们来看另一个元数据使用案例。假设观测值来自多个年度研究中的仪器,这些研究使用来自不同地点的不同受试者的样本来表征 10 个读数。我们通常会以某种格式获取这些信息,然后将其存储在 DataFrame 中。
obs_meta = pd.DataFrame(
{
"time_yr": np.random.choice([0, 2, 4, 8], adata.n_obs),
"subject_id": np.random.choice(
["subject 1", "subject 2", "subject 4", "subject 8"], adata.n_obs
),
"instrument_type": np.random.choice(["type a", "type b"], adata.n_obs),
"site": np.random.choice(["site x", "site y"], adata.n_obs),
},
index=adata.obs.index, # these are the same IDs of observations as above!
)
这就是我们如何将读数数据与元数据结合起来。当然,以下调用中第一个参数 X 也可以只是一个 DataFrame。这将导致一个跟踪所有内容的单一数据容器。
adata = ad.AnnData(adata.X, obs=obs_meta, var=adata.var)
adata
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'time_yr', 'subject_id', 'instrument_type', 'site'
对联合数据矩阵进行子集化可以帮助我们关注变量或观测值的子集,或定义机器学习模型的训练 - 测试拆分。
注意 类似于 numpy 数组,AnnData 对象可以包含实际数据或引用另一个 AnnData 对象。在后一种情况下,它们被称为“视图”。子集化 AnnData 对象总是返回视图,这有两个优点:
- 不会分配新的内存
- 可以修改底层的 AnnData 对象
您可以通过对视图调用 .copy() 来获取实际的 AnnData 对象。通常,这是不必要的,因为对视图元素的任何修改(在视图的属性上调用 .[])内部都会调用 .copy() 并使视图成为包含实际数据的 AnnData 对象。请参见下面的示例。
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'time_yr', 'subject_id', 'instrument_type', 'site'
注意 对 AnnData 的索引将假定传递给 [] 的整数参数的行为类似于 pandas 中的 .iloc,而字符串参数的行为类似于 .loc。AnnData 总是假定字符串索引。
adata_view = adata[:5, ["Gene_1", "Gene_3"]]
adata_view
View of AnnData object with n_obs × n_vars = 5 × 2
obs: 'time_yr', 'subject_id', 'instrument_type', 'site'
这是一个视图!这可以通过再次检查 AnnData 对象来验证。
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'time_yr', 'subject_id', 'instrument_type', 'site'
AnnData 对象的维度没有改变,它仍然包含相同的数据。如果我们想要一个在内存中保存数据的 AnnData 对象,我们需要调用 .copy()。
adata_subset = adata[:5, ["Gene_1", "Gene_3"]].copy()
对于视图,我们也可以设置某一列的前 3 个元素。
print(adata[:3, "Gene_1"].X.toarray().tolist())
adata[:3, "Gene_1"].X = [0, 0, 0]
print(adata[:3, "Gene_1"].X.toarray().tolist())
[[0.0], [2.0], [1.0]]
[[0.0], [0.0], [0.0]]
如果您尝试访问 AnnData 视图的某些部分,其内容将自动复制,并生成一个存储数据的对象。
adata_subset = adata[:3, ["Gene_1", "Gene_2"]]
adata_subset
View of AnnData object with n_obs × n_vars = 3 × 2
obs: 'time_yr', 'subject_id', 'instrument_type', 'site'
adata_subset.obs["foo"] = range(3)
Trying to set attribute `.obs` of view, copying.
现在 adata_subset 存储了实际数据,不再只是 adata 的引用。
AnnData object with n_obs × n_vars = 3 × 2
obs: 'time_yr', 'subject_id', 'instrument_type', 'site', 'foo'
显然,您可以使用 pandas 的所有功能来使用序列或布尔索引进行切片。
adata[adata.obs.time_yr.isin([2, 4])].obs.head()
| time_yr | subject_id | instrument_type | site | |
|---|---|---|---|---|
| Cell_0 | 4 | subject 8 | type b | site x |
| Cell_2 | 2 | subject 2 | type b | site y |
| Cell_3 | 4 | subject 8 | type a | site y |
| Cell_5 | 2 | subject 2 | type a | site y |
| Cell_10 | 2 | subject 8 | type b | site x |
1.4.2.9.2. 大数据的部分读取¶
如果单个 h5ad 文件非常大,可以使用支持模式(backed mode)或当前实验性的 read_elem API 将其部分读取到内存中。
adata = ad.read("my_results.h5ad", backed="r")
如果这样做,您需要记住 AnnData 对象与用于读取的文件保持打开连接。
PosixPath('my_results.h5ad')
由于我们在只读模式下使用它,因此不会损坏任何内容。要继续本教程,我们仍然需要明确关闭它。
1.4.3. 使用 scanpy 进行单模态数据分析¶
现在我们已经了解了单模态单细胞分析的基本数据结构,接下来的问题是我们如何实际分析存储的数据。在 scverse 生态系统中,存在用于特定组学数据分析的多种工具。例如,scanpy[Wolf et al., 2018] 提供了面向一般 RNA-Seq 分析的工具,squidpy[Palla et al., 2022] 专注于空间转录组学,scirpy[Sturm et al., 2020] 提供了用于 T 细胞受体(TCR)和 B 细胞受体(BCR)数据分析的工具。尽管存在多种用于各种数据模态的 scverse 扩展,但它们通常在某种程度上使用 scanpy 的预处理和可视化功能。
更具体地说,scanpy 是一个基于 AnnData 构建的 Python 包,用于促进单细胞基因表达数据的分析。通过 scanpy,可以访问多种方法进行预处理、嵌入、可视化、聚类、差异基因表达测试、拟时间和轨迹推断以及基因调控网络模拟。基于 Python 数据科学和机器学习库的高效实现使 scanpy 能够扩展到数百万个细胞。
一般来说,最佳实践的单细胞数据分析是一个互动过程。许多决策和分析步骤依赖于前一步的结果以及实验合作伙伴的输入。scflowKhozoie et al., 2021] 等管道完全自动化了一些下游分析步骤,这类管道直到最近才开始出现。这些管道必须做出假设和简化,可能不会导致最可靠的分析。因此,scanpy 设计用于与例如 Jupyter NotebooksJupyter, 2022] 等进行交互分析。
Fig. 4.2 scanpy overview. Image obtained from [Wolf et al., 2018].#
1.4.3.1. 安装¶
scanpy 可以通过 PyPI 和 Conda 安装,使用以下任意一种方法即可完成安装:
- 使用 PyPI 安装。
- 使用 Conda 安装。
pip install scanpy
conda install -c conda-forge scanpy
1.4.3.2. Scanpy API 设计¶
scanpy 框架的设计方式是将属于同一步骤的功能分组在相应的模块中。例如,所有预处理功能都在 scanpy.preprocessing 模块中,所有不是预处理的数据矩阵转换功能都在 scanpy.tools 中,所有可视化功能都在 scanpy.plot 中。这些模块通常在导入 scanpy 后访问,如 import scanpy as sc,相应的缩写为 sc.pp 代表预处理,sc.tl 代表工具,sc.pl 代表绘图。所有读取或写入数据的模块都是直接访问的。此外,一个用于各种数据集的模块可以作为 sc.datasets 访问。所有函数及其相应的参数和示例图都在 scanpy API 文档中记录 [scverse scanpy, 2022].
请注意,本教程仅涵盖了 scanpy 功能和选项的一小部分。强烈建议读者查阅 scanpy 的文档 以获取更多详细信息。
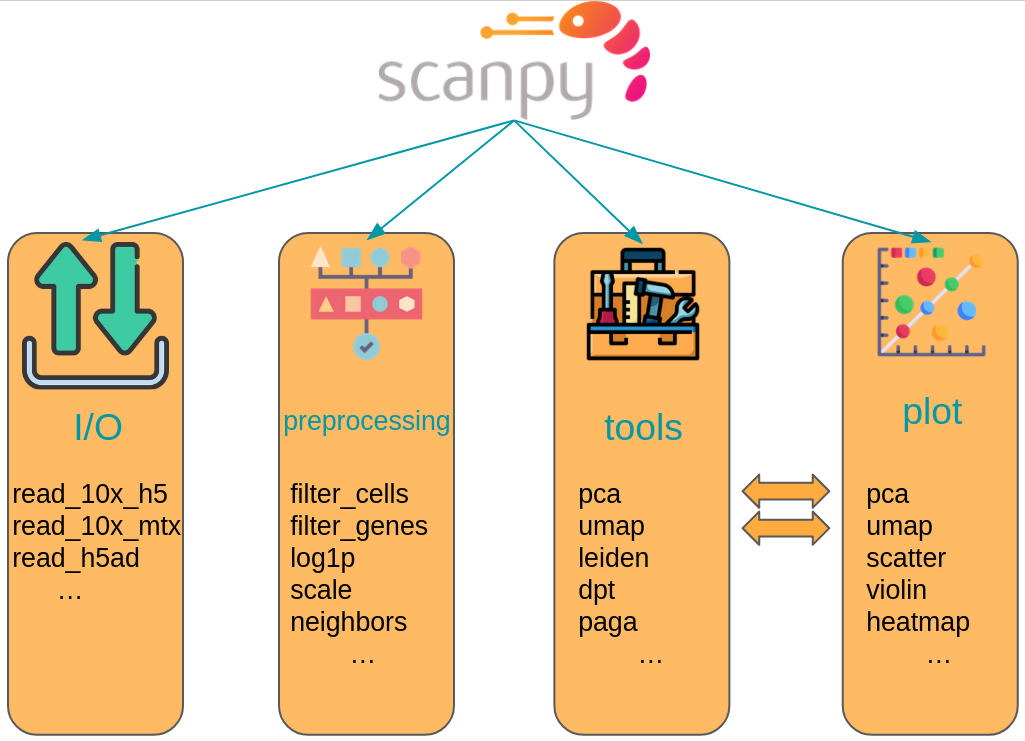
Fig. 4.3 scanpy API overview. The API is divided into datasets, preprocessing (pp), tools (tl) and corresponding plotting (pl) functions.#
1.4.3.3. Scanpy 示例 #¶
在接下来的内容中,我们将简要演示使用 scanpy 进行分析的工作流程。我们不会进行完整的分析,因为具体的分析步骤在相应的章节中介绍。
所选数据集是来自健康供体的 2700 个外周血单个核细胞的数据集,这些细胞在 Illumina NextSeq 500 上进行测序。
第一步,我们导入 scanpy 并为接下来的快速 scanpy 演示定义默认设置。我们使用 scanpy 的设置对象来为所有 scanpy 的绘图设置 Matplotlib 绘图默认值,最后打印 scanpy 的头部信息。这个头部信息包含当前环境中所有相关 Python 包的版本,包括 Scanpy 和 AnnData。这个输出在报告错误给 scverse 团队时特别有用,也有助于结果的可重复性。
import scanpy as sc
sc.settings.set_figure_params(dpi=80, facecolor="white")
sc.logging.print_header()
scanpy==1.9.1 anndata==0.7.8 umap==0.5.2 numpy==1.20.3 scipy==1.7.2 pandas==1.4.3 scikit-learn==1.0.1 statsmodels==0.13.1 python-igraph==0.9.10 pynndescent==0.5.5
接下来,我们使用 scanpy 获取 3k PBMC 数据集作为 AnnData 对象。该数据集会自动下载并保存到路径中,这个路径可以通过 scanpy 的设置对象指定。默认情况下,该路径是文件夹 data。
adata = sc.datasets.pbmc3k()
adata
100%|██████████| 5.58M/5.58M [00:01<00:00, 3.25MB/s]
AnnData object with n_obs × n_vars = 2700 × 32738
var: 'gene_ids'
返回的 AnnData 对象包含 2700 个细胞和 32738 个基因。var 插槽中进一步包含基因 ID。
| gene_ids | |
|---|---|
| index | |
| MIR1302-10 | ENSG00000243485 |
| FAM138A | ENSG00000237613 |
| OR4F5 | ENSG00000186092 |
| RP11-34P13.7 | ENSG00000238009 |
| RP11-34P13.8 | ENSG00000239945 |
| ... | ... |
| AC145205.1 | ENSG00000215635 |
| BAGE5 | ENSG00000268590 |
| CU459201.1 | ENSG00000251180 |
| AC002321.2 | ENSG00000215616 |
| AC002321.1 | ENSG00000215611 |
32738 rows × 1 columns
如上所述,所有 scanpy 的分析功能都可以通过 sc.[pp, tl, pl] 访问。为了首先概览我们的数据,我们使用 scanpy 显示在每个细胞中产生最高计数分数的基因,跨所有细胞。我们简单地调用 sc.pl.highest_expr_genes 函数,传递 AnnData 对象(在几乎所有情况下都是任何 scanpy 函数的第一个参数),并指定我们希望显示表达量最高的 20 个基因。
sc.pl.highest_expr_genes(adata, n_top=20)
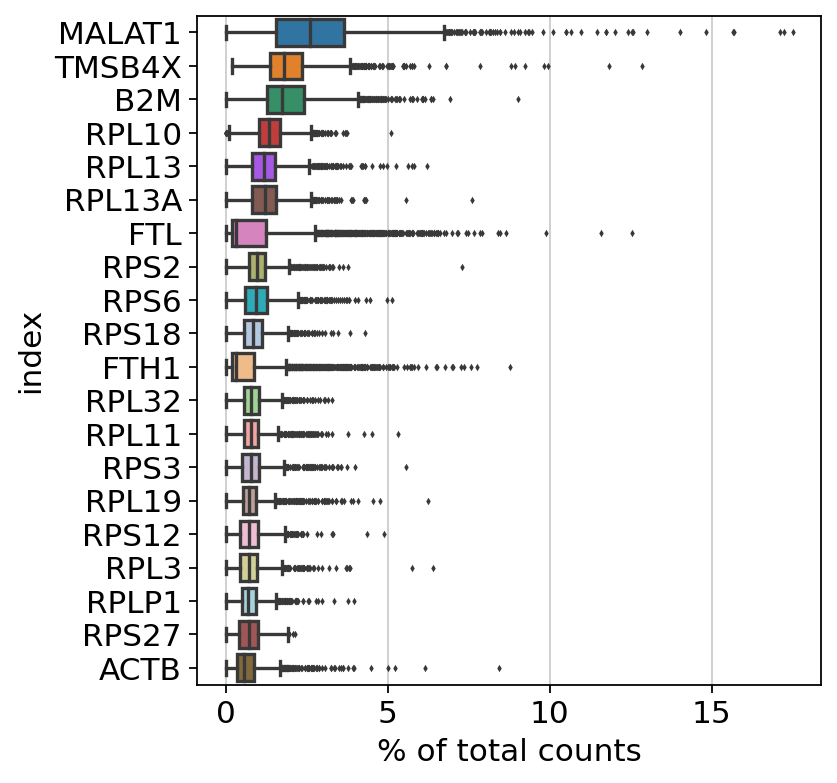
显然,MALAT1 是表达量最高的基因,这在聚合酶捕获的 scRNA-Seq 数据中经常检测到,与实验方案无关。研究表明,这个基因与细胞健康呈负相关,尤其是死亡/濒死细胞的 MALAT1 表达量更高。
现在,我们使用 scanpy 的预处理模块过滤出检测到的基因少于 200 的细胞,以及在少于 3 个细胞中发现的基因,以达到粗略的质量阈值。
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
在单细胞 RNA-Seq 分析中,一个常见的步骤是使用例如主成分分析(PCA)进行降维,以揭示主要的变异轴。这也有助于数据去噪。scanpy 提供了 PCA 作为 preprocessing 或 tools 函数,这两者是等价的。在这里,我们使用 tools 中的版本,没有特别的原因。
sc.tl.pca(adata, svd_solver="arpack")
相应的绘图函数允许我们将基因传递给 color 参数。相应的值会自动从 AnnData 对象中提取。
sc.pl.pca(adata, color="CST3")
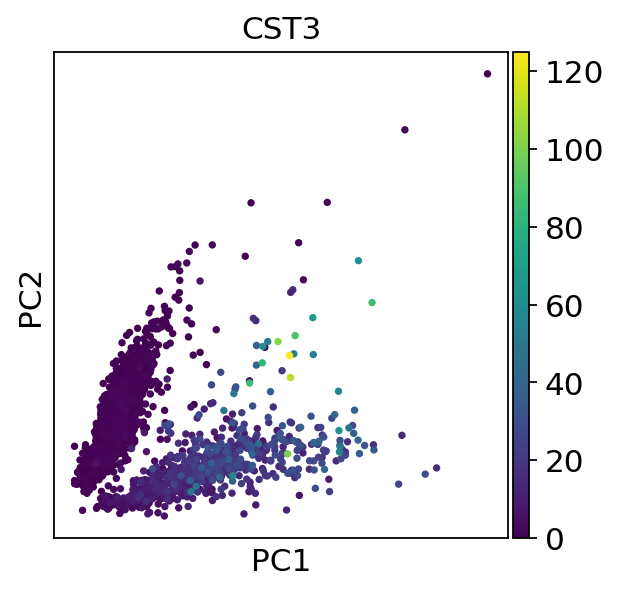
任何高级嵌入和下游计算的一个基本步骤是使用数据矩阵的 PCA 表示计算邻域图。这一步会自动用于需要它的其他工具,例如 UMAP 的计算。
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
现在,我们使用计算出的邻域图通过 UMAP 对细胞进行嵌入,UMAP 是 scanpy 实现的众多高级降维算法之一。
sc.pl.umap(adata, color=["CST3", "NKG7", "PPBP"])
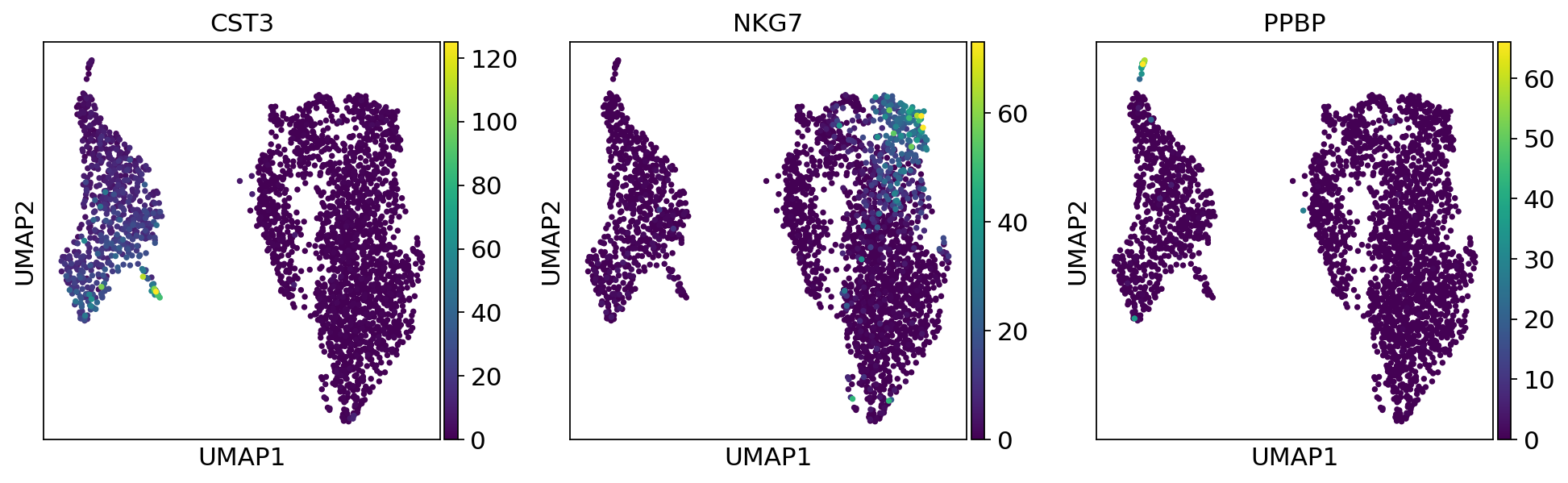
scanpy 的文档还提供了 教程,我们推荐给所有需要复习 scanpy 或新接触 scanpy 的读者。视频教程可在 scverse YouTube 频道 上观看。
1.4.4. 进阶:使用 MuData 存储多模态数据¶
AnnData 主要设计用于存储和处理单模态数据。然而,多模态检测如 CITE-Seq 通过同时测量 RNA 和表面蛋白生成多模态数据。这种数据需要更高级的存储方式,这就是 MuData 的作用所在。MuData 构建在 AnnData 之上,用于存储和处理多模态数据。muon[Bredikhin et al., 2022],一个“scanpy 等价物”和 scverse 的核心包,可以用于分析多模态组学数据。以下部分基于 MuData 快速入门教程 [scverse mudata, 2022] 和多模态数据对象教程 [scverse mudata, 2022].
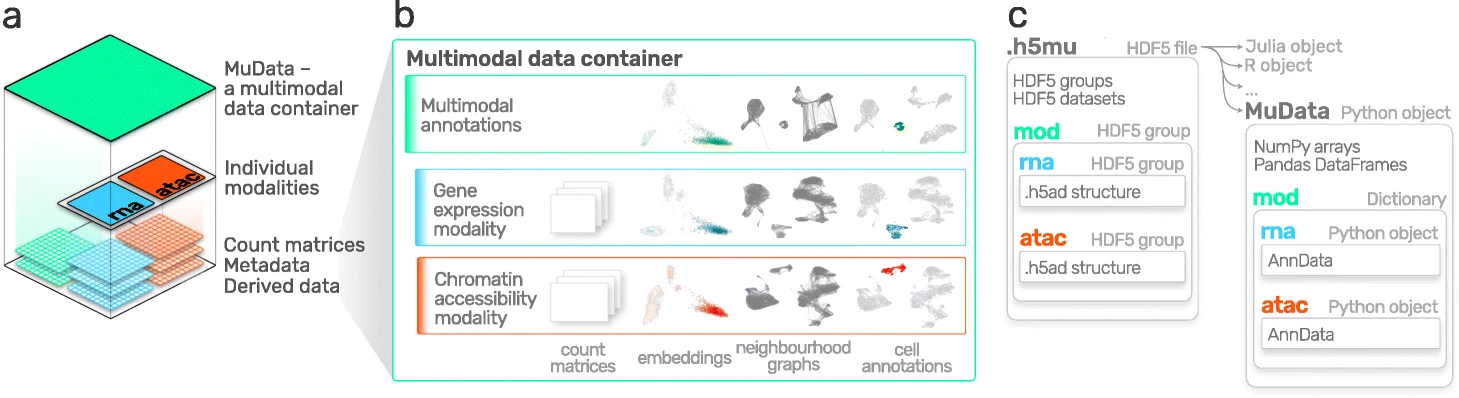
Fig. 4.4 MuData 概览。图片来源于 [Bredikhin et al., 2022].#
1.4.4.1. 安装¶
MuData 可以通过 PyPI 和 Conda 安装,使用以下任意一种方法即可完成安装:
- 使用 PyPI 安装。
- 使用 Conda 安装。
pip install mudata
conda install -c conda-forge mudata
MuData 的主要思想是,MuData 对象包含对单模态数据的单个 AnnData 对象的引用,但 MuData 对象本身也存储多模态注释。因此,可以直接访问 AnnData 对象以执行单模态数据转换,并将结果存储在相应的 AnnData 注释中,也可以聚合模态进行联合计算,并将结果存储在全局 MuData 对象中。从技术上讲,这通过 MuData 对象的 .mod(=模态)属性中的包含 AnnData 对象的字典来实现,每个模态对应一个 AnnData 对象。就像 AnnData 对象本身一样,它们还包含诸如 .obs(观测值注释,如样本或细胞)和 .obsm(多维注释,如嵌入)等属性。
1.4.4.2. 初始化 MuData 对象¶
我们将从 mudata 包中导入 MuData 开始。
要创建一个示例 MuData 对象,我们需要模拟数据。
n, d, k = 1000, 100, 10
z = np.random.normal(loc=np.arange(k), scale=np.arange(k) * 2, size=(n, k))
w = np.random.normal(size=(d, k))
y = np.dot(z, w.T)
y.shape
要创建一个 MuData 对象,我们首先需要单模态的 AnnData 对象。因此,我们创建两个 AnnData 对象,它们具有相同观测值的数据,但 _ 变量不同 _。
adata = ad.AnnData(y)
adata.obs_names = [f"obs_{i+1}" for i in range(n)]
adata.var_names = [f"var_{j+1}" for j in range(d)]
adata
AnnData object with n_obs × n_vars = 1000 × 100
d2 = 50
w2 = np.random.normal(size=(d2, k))
y2 = np.dot(z, w2.T)
adata2 = ad.AnnData(y2)
adata2.obs_names = [f"obs_{i+1}" for i in range(n)]
adata2.var_names = [f"var2_{j+1}" for j in range(d2)]
adata2
AnnData object with n_obs × n_vars = 1000 × 50
这两个 AnnData 对象(两个“模态”)然后可以被包装成一个 MuData 对象。在这里,我们将模态一命名为 A,模态二命名为 B。
mdata = md.MuData({"A": adata, "B": adata2})
mdata
MuData object with n_obs × n_vars = 1000 × 150
2 modalities
A: 1000 x 100
B: 1000 x 50
观测值和变量在 MuData 对象中是全局的,这意味着不同模态中具有相同名称(.obs_names)的观测值被视为相同的观测值。这也意味着变量名称(.var_names)应该是唯一的。这在上面的对象描述中得到了反映:mdata 有 1000 个观测值和 150 = 100 + 50 个变量。
1.4.4.3. MuData 属性¶
MuData 对象包含如 AnnData 对象的注释(如 .obs 或 .var),但通过 .mod 扩展了这种行为,.mod 用于访问单个模态。
模态存储在一个集合中,可通过 MuData 对象的 .mod 属性访问,模态名称为键,AnnData 对象为值。
可以通过 .mod 属性或通过 MuData 对象本身作为简写来访问单个模态。
print(mdata.mod["A"])
print(mdata["A"])
AnnData object with n_obs × n_vars = 1000 × 100
AnnData object with n_obs × n_vars = 1000 × 100
样本(细胞)注释可以通过 .obs 属性访问,默认情况下包括来自各个模态的 .obs 数据框的列的副本。同样,.var 包含变量(特征)的注释。从各个模态复制的观测值列包含模态名称作为前缀,例如 rna:n_genes。变量列也是如此。然而,如果多个模态的 .var 中有相同名称的列,例如 n_cells,这些列将在模态之间合并,不添加前缀。当这些插槽在模态的 AnnData 对象中发生变化时,例如添加新列或过滤出样本(细胞),必须使用 .update() 方法获取更改(见下文)。
样本(细胞)的多维注释可以在 .obsm 属性中访问。例如,这可以是基于所有模态共同学习的 UMAP 坐标。
MuData 对象的形状由两个数字表示,分别计算为各个模态形状的总和——一个用于观测值的数量,一个用于变量的数量。
print(mdata.shape)
print(mdata.n_obs)
print(mdata.n_vars)
默认情况下,变量始终被计为属于单个模态,而具有相同名称的观测值被计为相同的观测值,这些观测值在多个模态中测量了变量。
[adata.shape for adata in mdata.mod.values()]
[(1000, 100), (1000, 50)]
如果模态的形状发生变化,需要运行 MuData.update() 来将相应的更新带入 MuData 对象。
adata2.var_names = ["var_ad2_" + e.split("_")[1] for e in adata2.var_names]
print(f"Outdated variables names: ...,", ", ".join(mdata.var_names[-3:]))
mdata.update()
print(f"Updated variables names: ...,", ", ".join(mdata.var_names[-3:]))
Outdated variables names: ..., var2_48, var2_49, var2_50
Updated variables names: ..., var_ad2_48, var_ad2_49, var_ad2_50
重要的是,单个模态作为对原始对象的引用存储。因此,如果原始 AnnData 发生变化,MuData 对象中也会反映出这些变化。
# Add some unstructured data to the original object
adata.uns["misc"] = {"adata": True}
# Access modality A via the .mod attribute
mdata.mod["A"].uns["misc"]
1.4.4.4. 变量映射¶
在构建 MuData 对象时,会创建观测值与单个模态之间的全局二进制映射,以及变量与模态之间的映射。由于在 mdata 中各个模态之间的所有观测值都是相同的,因此观测值映射中的所有值都设置为 True。
np.sum(mdata.obsm["A"]) == np.sum(mdata.obsm["B"]) == n
然而,对于变量而言,这些是长度为 150 的向量。模态 A 有 100 个 True 值,接着是 50 个 False 值。
array([ True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False])
1.4.4.5. MuData 视图¶
类似于 AnnData 对象的行为,对 MuData 对象进行切片会返回原始数据的视图。
view = mdata[:100, :1000]
print(view.is_view)
print(view["A"].is_view)
对 MuData 对象进行子集化操作具有特殊性,因为它会跨模态进行切片。也就是说,对于一组 obs_names 和/或 var_names 的切片操作将对每个模态执行,而不仅仅是全局多模态注释。这种行为使得工作流在内存使用上更加高效,这在处理大数据集时尤为重要。然而,如果需要修改对象,应创建它的副本,这样它将不再是视图,也不再依赖于原始对象。
mdata_sub = view.copy()
mdata_sub.is_view
1.4.4.6. 共同的观测值¶
虽然一个 MuData 对象包含了两个模态的相同观测值,但在现实世界中并非总是如此,有些数据可能会缺失。设计上,MuData 考虑了这些情况,因为没有保证观测值是相同的,甚至在一个 MuData 实例中是相交的。值得注意的是,其他工具可能会提供方便的函数来处理某些常见的缺失数据场景,例如 muon 中实现的 intersect_obs()。
1.4.4.7. MuData 对象的读取和写入¶
与 AnnData 对象类似,MuData 对象被设计为可以序列化为基于 HDF5 的 .h5mu 文件。所有模态都存储在 .h5mu 文件的 /mod HDF5 组下的各自名称下。每个单独的模态,例如 /mod/A,以与存储在 .h5ad 文件中的方式相同的方式存储。MuData 对象可以按如下方式读取和写入:
mdata.write("my_mudata.h5mu")
mdata_r = md.read("my_mudata.h5mu", backed=True)
mdata_r
MuData object with n_obs × n_vars = 1000 × 150 backed at 'my_mudata.h5mu'
2 modalities
A: 1000 x 100
uns: 'misc'
B: 1000 x 50
各个模态也在 .h5mu 文件中进行备份。
如果原始对象已被备份,则必须在 .copy() 调用中提供文件名,结果对象将在新位置进行备份。
mdata_sub = mdata_r.copy("mdata_sub.h5mu")
print(mdata_sub.is_view)
print(mdata_sub.isbacked)
1.4.4.8. 多模态方法¶
当 MuData 对象准备好后,就可以使用多模态方法来理解数据。最简单和最直接的方法是连接来自多个模态的矩阵,以执行例如降维。
x = np.hstack([mdata.mod["A"].X, mdata.mod["B"].X])
x.shape
我们可以编写一个简单的函数,在这样的连接矩阵上运行主成分分析(PCA)。MuData 对象提供了一个存储多模态嵌入的地方:MuData 的 .obsm。这类似于在单个模态上生成的嵌入的存储方式,只是这次它是存储在 MuData 对象内部,而不是在 AnnData 的 .obsm 中。
例如,为了计算模态联合值的主成分分析(PCA),我们水平堆叠存储在各个模态中的值,然后对堆叠的矩阵执行 PCA。这是可能的,因为各个模态的观测值数量是匹配的(请记住,每个模态的特征数量不必匹配)。
def simple_pca(mdata):
from sklearn import decomposition
x = np.hstack([m.X for m in mdata.mod.values()])
pca = decomposition.PCA(n_components=2)
components = pca.fit_transform(x)
# By default, methods operate in-place and embeddings are stored in the .obsm slot
mdata.obsm["X_pca"] = components
simple_pca(mdata)
print(mdata)
MuData object with n_obs × n_vars = 1000 × 150
obsm: 'X_pca'
2 modalities
A: 1000 x 100
uns: 'misc'
B: 1000 x 50
我们计算的主成分现在存储在 MuData 对象本身中,可以进行进一步的多模态转换。
然而,实际上,具有不同模态通常意味着它们之间的特征来自不同的生成过程,且不可比。这时,特殊的多模态集成方法就派上用场了。对于组学技术,这些方法通常称为多组学集成方法。在以下部分中,我们将介绍 muon,它不仅提供了 RNA-Seq 以外的单模态数据预处理工具,还提供了多组学集成方法。
1.4.5. 进阶:使用 muon 进行多模态数据分析¶
尽管 scanpy 提供了通用的工具,例如 PCA、UMAP 和各种可视化,但它主要设计用于 RNA-Seq 数据分析。muon 填补了这一空白,提供了其他组学数据(如染色质可及性(ATAC)或蛋白质(CITE)数据)的预处理函数。如上所述,muon 还提供了运行多组学算法的工具,这些算法从联合模态中推断知识。例如,用户可以在单个模态上运行 PCA,但 muon 还提供了多组学因子分析算法,这些算法以多种模态作为输入。
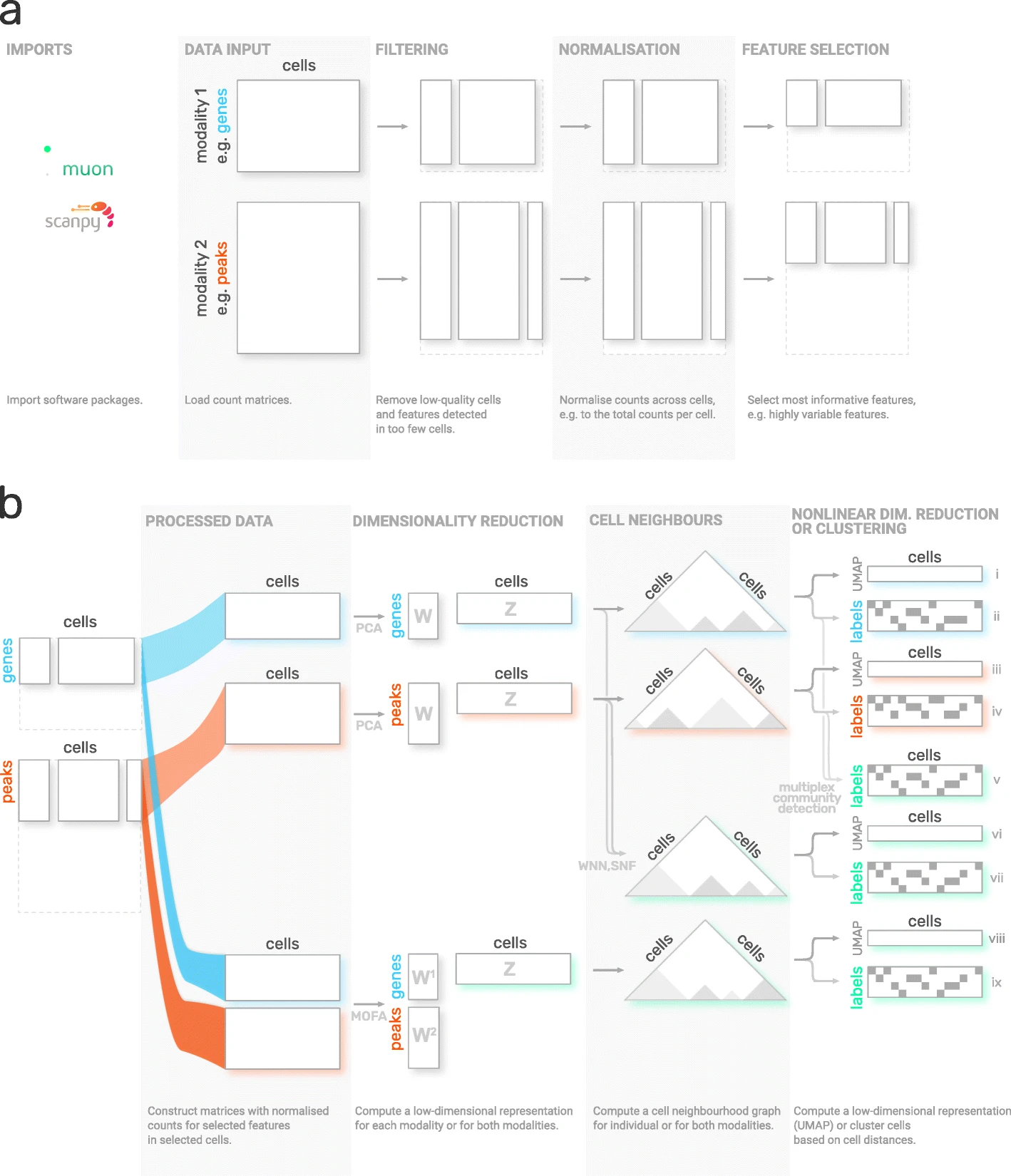
Fig. 4.5 muon overview. Image obtained from [Bredikhin et al., 2022].#
1.4.5.1. 安装¶
muon 可以通过 PyPI 安装:
1.4.5.2. API 概览¶
为了介绍 muon,我们将检查一个多模态数据集的 ATAC 数据。与 scanpy 章节类似,本章仅作为 muon 的快速演示和概览,而不是完整地分析数据集,更不用说提供最佳实践的多组学分析。请阅读相应的章节,了解如何正确地进行此类分析。
muon 以两种方式分隔其模块。首先,类似于 scanpy,通用和多模态函数被分组到预处理(muon.pp)、工具(muon.tl)和绘图(muon.pl)中。其次,单模态工具可以从 muon 的相应部分获得,这些部分再次分为预处理、工具和绘图。例如,所有 ATAC 预处理功能都被分组到 muon.atac.pp 中。这也适用于 CITE-Seq 预处理功能(muon.prot.pp)。
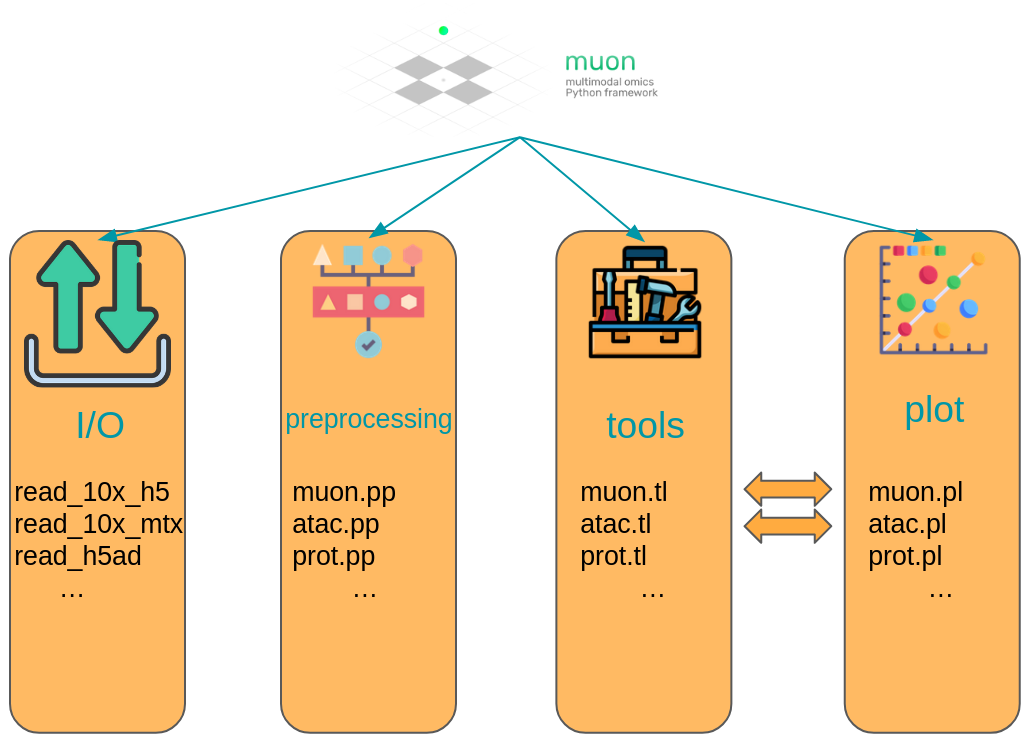
Fig. 4.6 muon API overview. Modality specific functions are provided in correspondingly named modules. Functions for the joint analysis of modalities are available via the muon module directly.#
1.4.5.3. muon API 演示¶
我们演示用的数据集是一个公开可用的 10x Genomics Multiome 数据集,针对人类 PBMCs。让我们导入 muon 并使用 mudatasets 获取该数据集。
import mudatasets as mds
import muon as mu
mdata, _ = mds.load("pbmc10k_multiome", with_info=True, full=True)
mdata.var_names_make_unique()
■ File filtered_feature_bc_matrix.h5 from pbmc10k_multiome has been found at /home/zeth/mudatasets/pbmc10k_multiome/filtered_feature_bc_matrix.h5
■ Checksum is validated (md5) for filtered_feature_bc_matrix.h5
■ File atac_fragments.tsv.gz from pbmc10k_multiome has been found at /home/zeth/mudatasets/pbmc10k_multiome/atac_fragments.tsv.gz
■ Checksum is validated (md5) for atac_fragments.tsv.gz
■ File atac_fragments.tsv.gz.tbi from pbmc10k_multiome has been found at /home/zeth/mudatasets/pbmc10k_multiome/atac_fragments.tsv.gz.tbi
■ Checksum is validated (md5) for atac_fragments.tsv.gz.tbi
■ File atac_peaks.bed from pbmc10k_multiome has been found at /home/zeth/mudatasets/pbmc10k_multiome/atac_peaks.bed
■ Checksum is validated (md5) for atac_peaks.bed
■ File atac_peak_annotation.tsv from pbmc10k_multiome has been found at /home/zeth/mudatasets/pbmc10k_multiome/atac_peak_annotation.tsv
■ Checksum is validated (md5) for atac_peak_annotation.tsv
■ Loading filtered_feature_bc_matrix.h5...
/home/zeth/miniconda3/envs/bp2/lib/python3.9/site-packages/mudatasets/core.py:203: UserWarning: Dataset is in the 10X .h5 format and can't be loaded as backed.
warn("Dataset is in the 10X .h5 format and can't be loaded as backed.")
Variable names are not unique. To make them unique, call `.var_names_make_unique`.
Added `interval` annotation for features from /home/zeth/mudatasets/pbmc10k_multiome/filtered_feature_bc_matrix.h5
Variable names are not unique. To make them unique, call `.var_names_make_unique`.
Added peak annotation from /home/zeth/mudatasets/pbmc10k_multiome/atac_peak_annotation.tsv to .uns['atac']['peak_annotation']
Added gene names to peak annotation in .uns['atac']['peak_annotation']
Located fragments file: /home/zeth/mudatasets/pbmc10k_multiome/atac_fragments.tsv.gz
作为第一步,我们将数据集子集化为 ATAC 模态。
虽然我们现在不处理 RNA-Seq 数据,但可以使用一些 scanpy 的预处理功能,这些功能也可以用于 ATAC 数据。这是因为两种模态具有相似的分布和质量问题。在这里需要记住的是,在具有 ATAC-seq 数据的 AnnData 对象中,基因意味着一个峰。之后,可以使用 muon 的 ATAC 模块进行特定于 ATAC 的预处理。
让我们从一些质量控制开始,通过过滤掉具有太少峰的细胞和在太少细胞中检测到的峰来进行质量控制。现在,我们将过滤掉未通过质量控制的细胞。
sc.pp.calculate_qc_metrics(atac, percent_top=None, log1p=False, inplace=True)
sc.pl.violin(atac, ["total_counts", "n_genes_by_counts"], jitter=0.4, multi_panel=True)
/home/zeth/miniconda3/envs/bp2/lib/python3.9/site-packages/anndata/_core/anndata.py:1228: FutureWarning: The `inplace` parameter in pandas.Categorical.reorder_categories is deprecated and will be removed in a future version. Reordering categories will always return a new Categorical object.
c.reorder_categories(natsorted(c.categories), inplace=True)
... storing 'feature_types' as categorical
/home/zeth/miniconda3/envs/bp2/lib/python3.9/site-packages/anndata/_core/anndata.py:1228: FutureWarning: The `inplace` parameter in pandas.Categorical.reorder_categories is deprecated and will be removed in a future version. Reordering categories will always return a new Categorical object.
c.reorder_categories(natsorted(c.categories), inplace=True)
... storing 'genome' as categorical
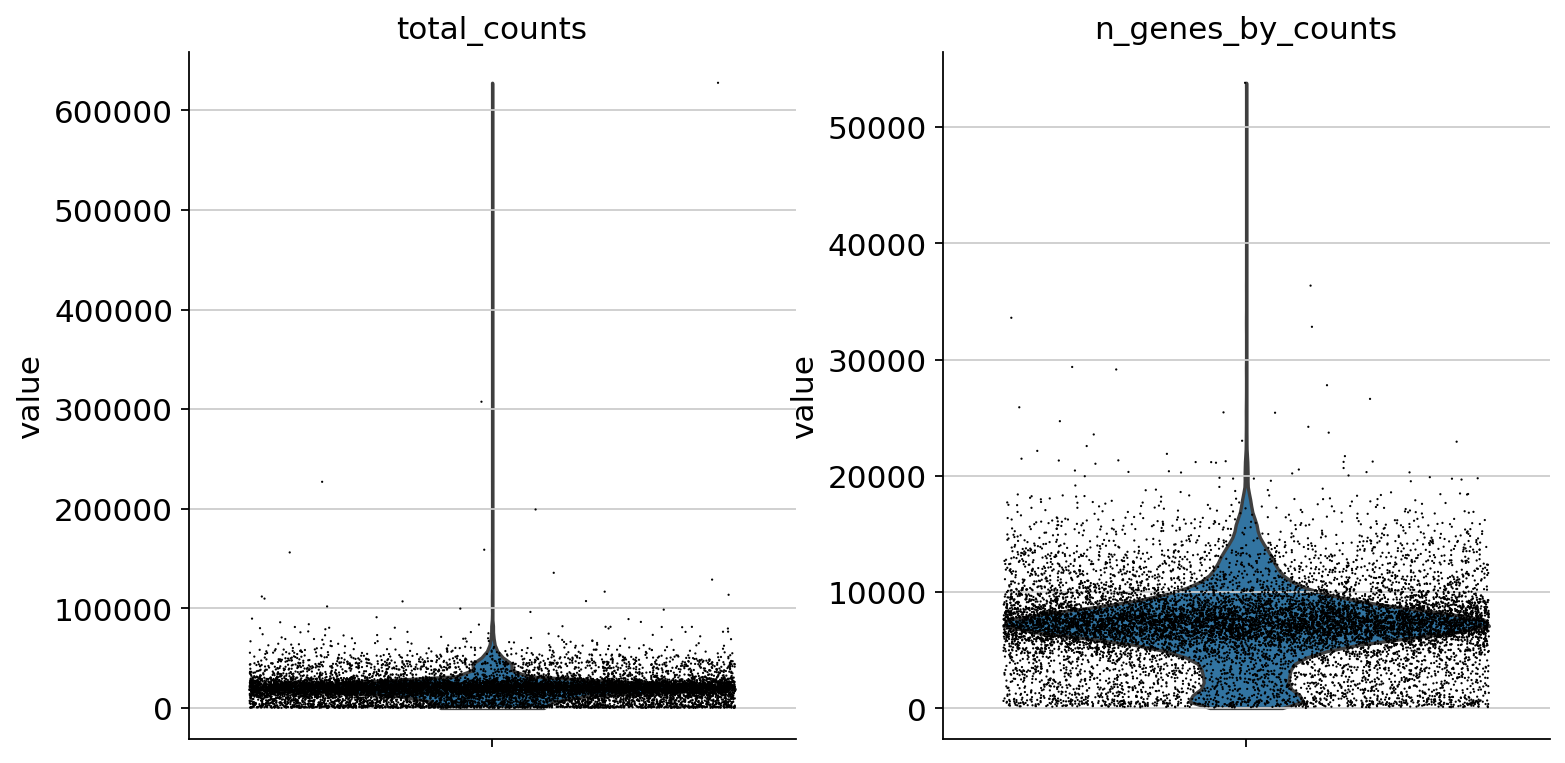
过滤掉未检测到表达的峰。
mu.pp.filter_var(atac, "n_cells_by_counts", lambda x: x >= 10)
# This is analogous to
# sc.pp.filter_genes(rna, min_cells=10)
# but does in-place filtering and avoids copying the object
我们也会过滤掉不合格的细胞。
mu.pp.filter_obs(atac, "n_genes_by_counts", lambda x: (x >= 2000) & (x <= 15000))
# This is analogous to
# sc.pp.filter_cells(atac, max_genes=15000)
# sc.pp.filter_cells(atac, min_genes=2000)
# but does in-place filtering avoiding copying the object
mu.pp.filter_obs(atac, "total_counts", lambda x: (x >= 4000) & (x <= 40000))
sc.pl.violin(atac, ["n_genes_by_counts", "total_counts"], jitter=0.4, multi_panel=True)
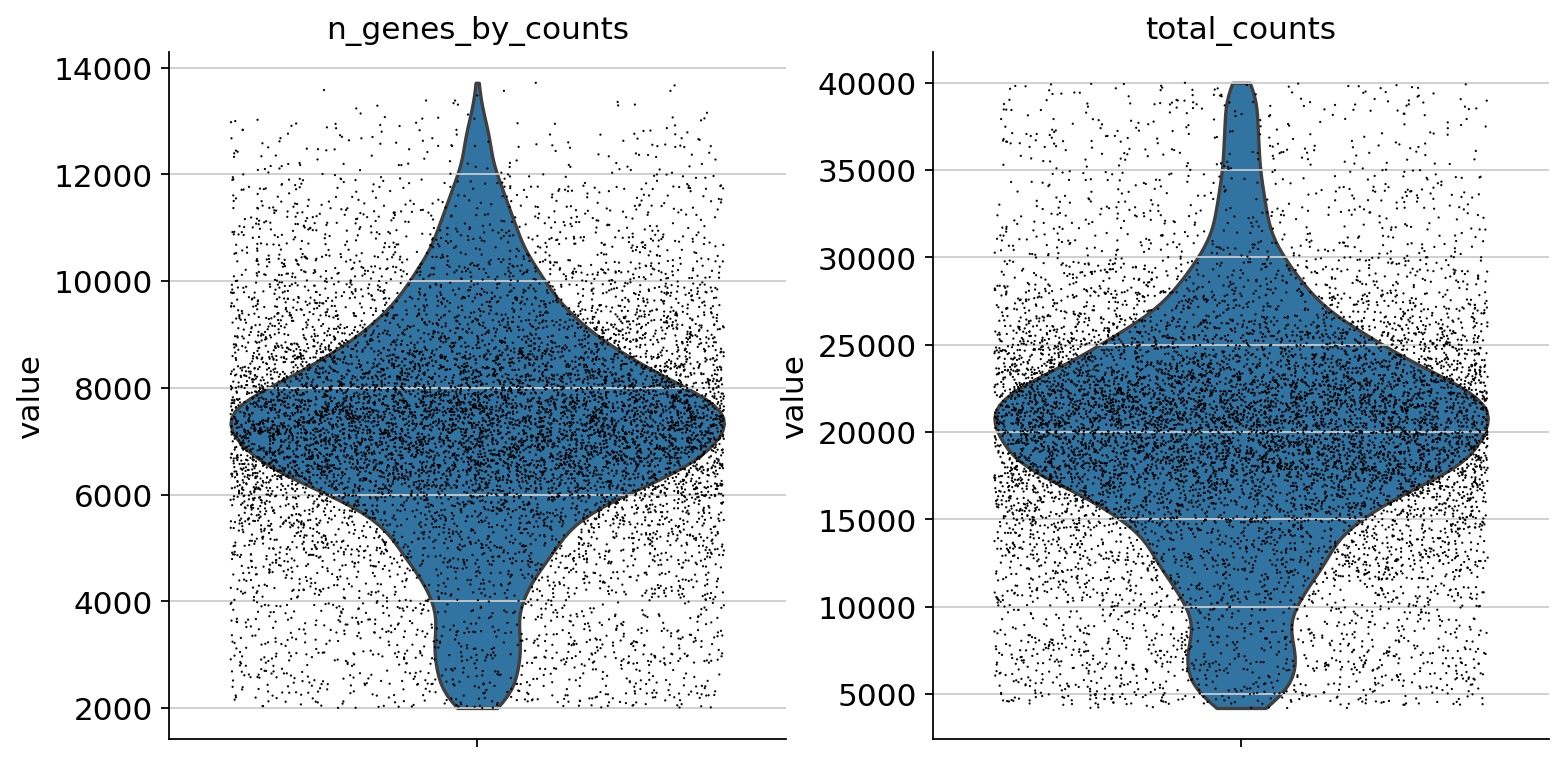
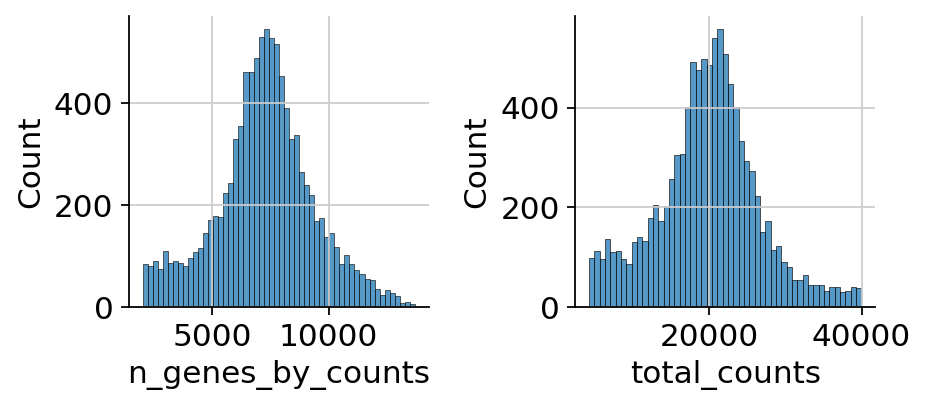
muon 还提供了直方图,可以从不同角度查看指标。
mu.pl.histogram(atac, ["n_genes_by_counts", "total_counts"])
现在我们已经初步过滤掉了具有太少峰的细胞和在太少细胞中检测到的峰,我们可以开始使用 muon 进行 ATAC 特定的质量控制。muon 在相应的模块中具有特定模态的预处理功能。我们导入 ATAC 模块以访问 ATAC 特定的预处理功能。
from muon import atac as ac
# Perform rudimentary quality control with muon's ATAC module
atac.obs["NS"] = 1
ac.tl.nucleosome_signal(atac, n=1e6)
ac.tl.get_gene_annotation_from_rna(mdata["rna"]).head(3)
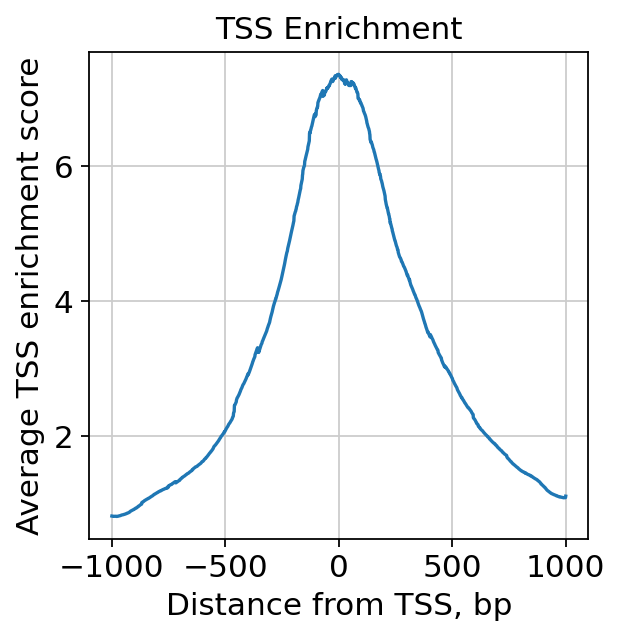
tss = ac.tl.tss_enrichment(mdata, n_tss=1000)
ac.pl.tss_enrichment(tss)
Reading Fragments: 100%|██████████| 1000000/1000000 [00:05<00:00, 185917.24it/s]
Fetching Regions...: 100%|██████████| 1000/1000 [00:19<00:00, 50.03it/s]
# Save original counts, normalize data and select highly variable genes with scanpy
atac.layers["counts"] = atac.X
sc.pp.normalize_per_cell(atac, counts_per_cell_after=1e4)
sc.pp.log1p(atac)
sc.pp.highly_variable_genes(atac, min_mean=0.05, max_mean=1.5, min_disp=0.5)
atac.raw = atac
虽然 PCA 也常用于 ATAC 数据,但潜在语义索引(LSI)是另一种流行的选择。它在 muon 的 ATAC 模块中实现。
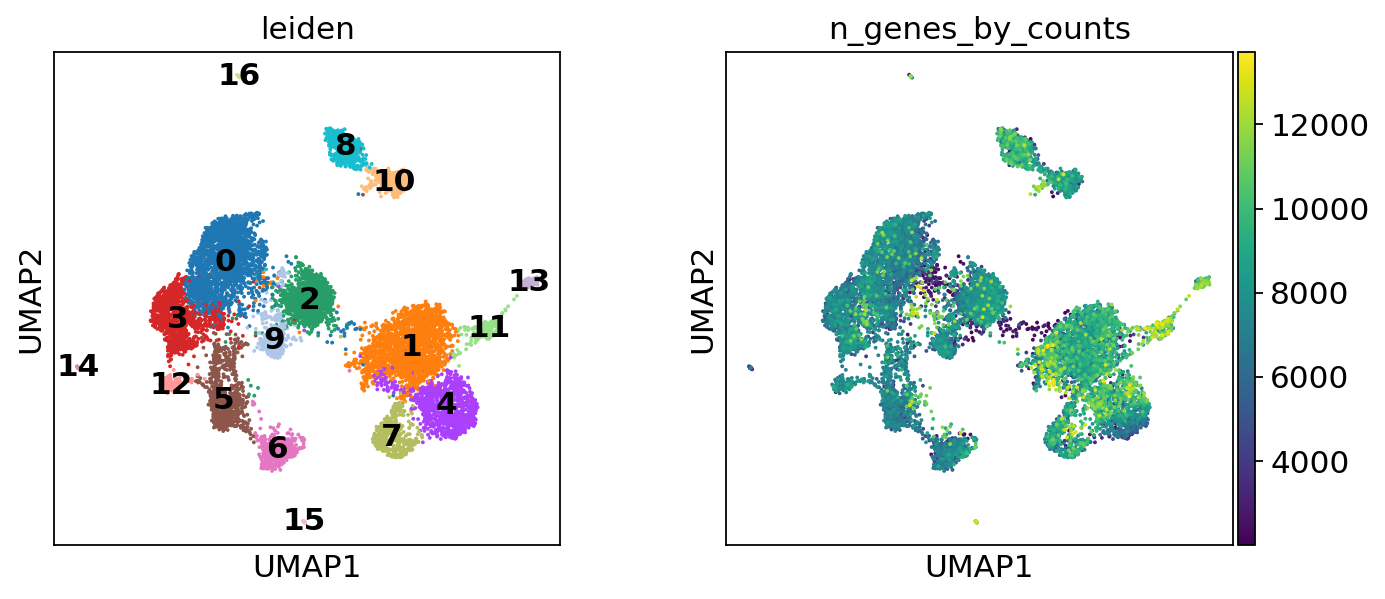
sc.pp.neighbors(atac, use_rep="X_lsi", n_neighbors=10, n_pcs=30)
sc.tl.leiden(atac, resolution=0.5)
sc.tl.umap(atac, spread=1.5, min_dist=0.5, random_state=20)
sc.pl.umap(atac, color=["leiden", "n_genes_by_counts"], legend_loc="on data")
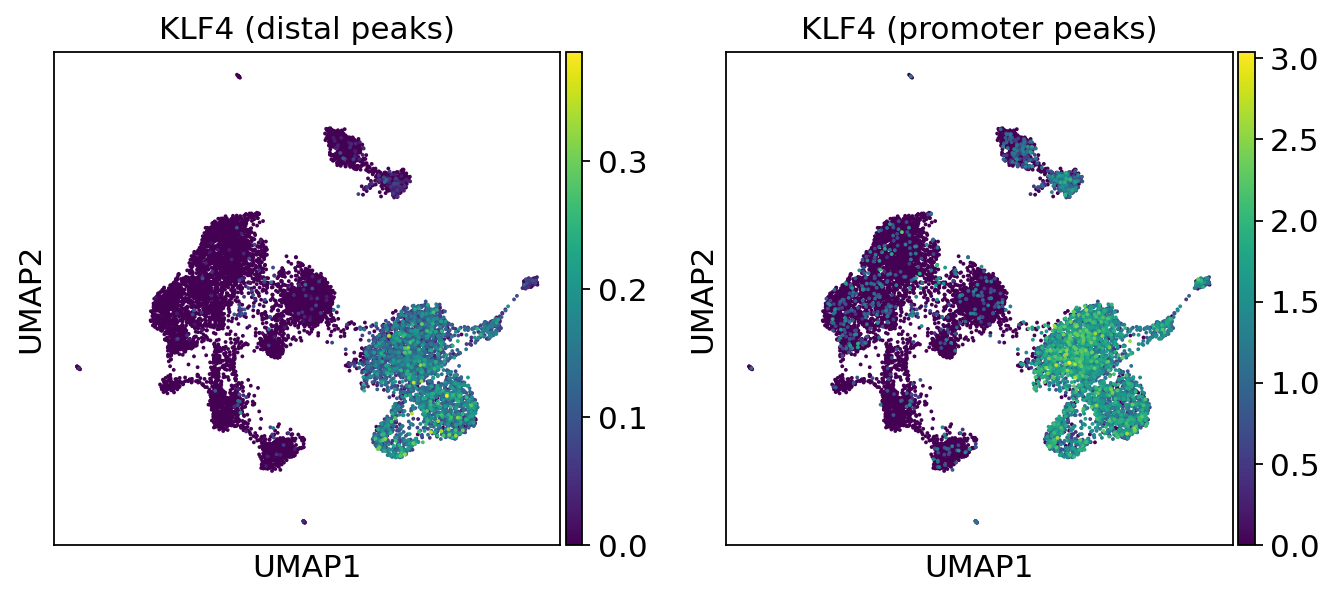
我们可以使用 muon 的 ATAC 模块的功能,根据对应于特定基因的峰值的切值来为图表着色。
ac.pl.umap(atac, color=["KLF4"], average="peak_type")
有关 muon 所有可用功能的更多详细信息,请阅读 muon API 参考文档:https://muon.readthedocs.io/en/latest/api/index.html 和 muon 教程:https://muon-tutorials.readthedocs.io/en/latest/。