2.1 Quality Control¶
Motivation¶
单细胞RNA-seq数据集在分析时需要注意两个重要特性。
首先,scRNA-seq数据存在“掉落”现象,这意味着由于mRNA的限制,数据中有过多的零值。其次,由于数据可能受到生物学因素的混淆,纠正数据和执行质量控制的潜力可能有限。因此,选择适合基础数据的预处理方法,而不过度纠正或去除生物效应至关重要。
由于新测序技术的发展和捕获细胞、测量基因和识别细胞群体数量的增加,用于单细胞RNA测序数据分析的工具集正在迅速演变【2021】。其中许多工具专注于预处理,旨在解决以下分析步骤:双重检测、质量控制、标准化、特征选择和降维。本章中选择的工具会严重影响下游分析和数据解释。
例如,如果在质量控制过程中过滤掉了太多的细胞,可能会失去稀有的细胞亚群,从而错过对有趣细胞生物学的洞察。而如果太过宽松,则在预处理过程中没有排除质量较差的细胞时,可能很难对细胞进行注释。因此,选择提供最佳实践并且在下游任务中表现优于其他工具的工具非常重要。在许多情况下,仍需要重新评估预处理分析,并例如更改过滤策略。
本笔记本的起点是经过原始处理章节中描述处理的单细胞数据。数据经过比对以获得分子计数矩阵,即所谓的计数矩阵或读取计数(读取矩阵)。计数矩阵和读取矩阵之间的区别取决于单细胞文库构建协议中是否包含唯一分子标识符(UMI)。读取和计数矩阵的维度为条形码数量 x 转录本数量。需要注意的是,这里使用“条形码”一词而不是“细胞”,因为条形码可能错误地标记了多个细胞(双重检测),或者可能没有标记任何细胞(空液滴/孔)。我们将在“双重检测”部分详细说明这一点。
环境设置和数据¶
我们使用在2021年NeurIPS会议上为单细胞数据整合挑战生成的10x Multiome数据集【2021】。该数据集捕获了来自12名健康人类供体骨髓单个核细胞的单细胞多组学数据,这些数据在四个不同的地点测量,以获得嵌套的批次效应。在本教程中,我们将使用上述数据集中的一个批次,即供体8的样本4,来展示scRNA-seq数据预处理的最佳实践。
虽然单细胞计数矩阵的预处理通常是一个相对线性的过程,其中各种质量控制和预处理步骤按明确的顺序进行,但在这里引入特定步骤时,有时需要提前进行一些后续小节中将介绍的步骤。例如,我们在环境RNA校正中使用了聚类,但会在后面的部分才介绍聚类。
第一步,我们首先使用scanpy加载托管在figshare上的数据集。
import numpy as np
import scanpy as sc
import seaborn as sns
from scipy.stats import median_abs_deviation
sc.settings.verbosity = 0
sc.settings.set_figure_params(
dpi=80,
facecolor="white",
frameon=False,
)
adata = sc.read_10x_h5(
filename="filtered_feature_bc_matrix.h5",
backup_url="https://figshare.com/ndownloader/files/39546196",
)
adata
Variable names are not unique. To make them unique, call `.var_names_make_unique`. Variable names are not unique. To make them unique, call `.var_names_make_unique`.
AnnData object with n_obs × n_vars = 16934 × 36601
var: 'gene_ids', 'feature_types', 'genome'
读取数据后,scanpy会显示一个警告,提示并非所有变量名都是唯一的。这表明某些变量(基因)出现多次,这可能导致下游分析任务的错误或意外行为。我们执行建议的函数 var_names_make_unique(),该函数通过在每个重复索引元素后附加一个数字字符串‘1’,‘2’等,使变量名唯一。
adata.var_names_make_unique()
adata
AnnData object with n_obs × n_vars = 16934 × 36601
var: 'gene_ids', 'feature_types', 'genome'
数据集的形状为 n_obs 16,934 x n_vars 36,601,这对应于 条形码 x 转录本数量。我们还检查了 .var 中的基因信息,包括基因ID(Ensembl Id)、特征类型和基因组。
大多数后续的分析任务假设数据集中的每个观测值都代表一个完整的单细胞的测量结果。在某些情况下,这一假设可能会因为低质量细胞、细胞外RNA污染或双重细胞而被违反。本教程将指导您如何纠正和删除这些问题,从而获得高质量的数据集。
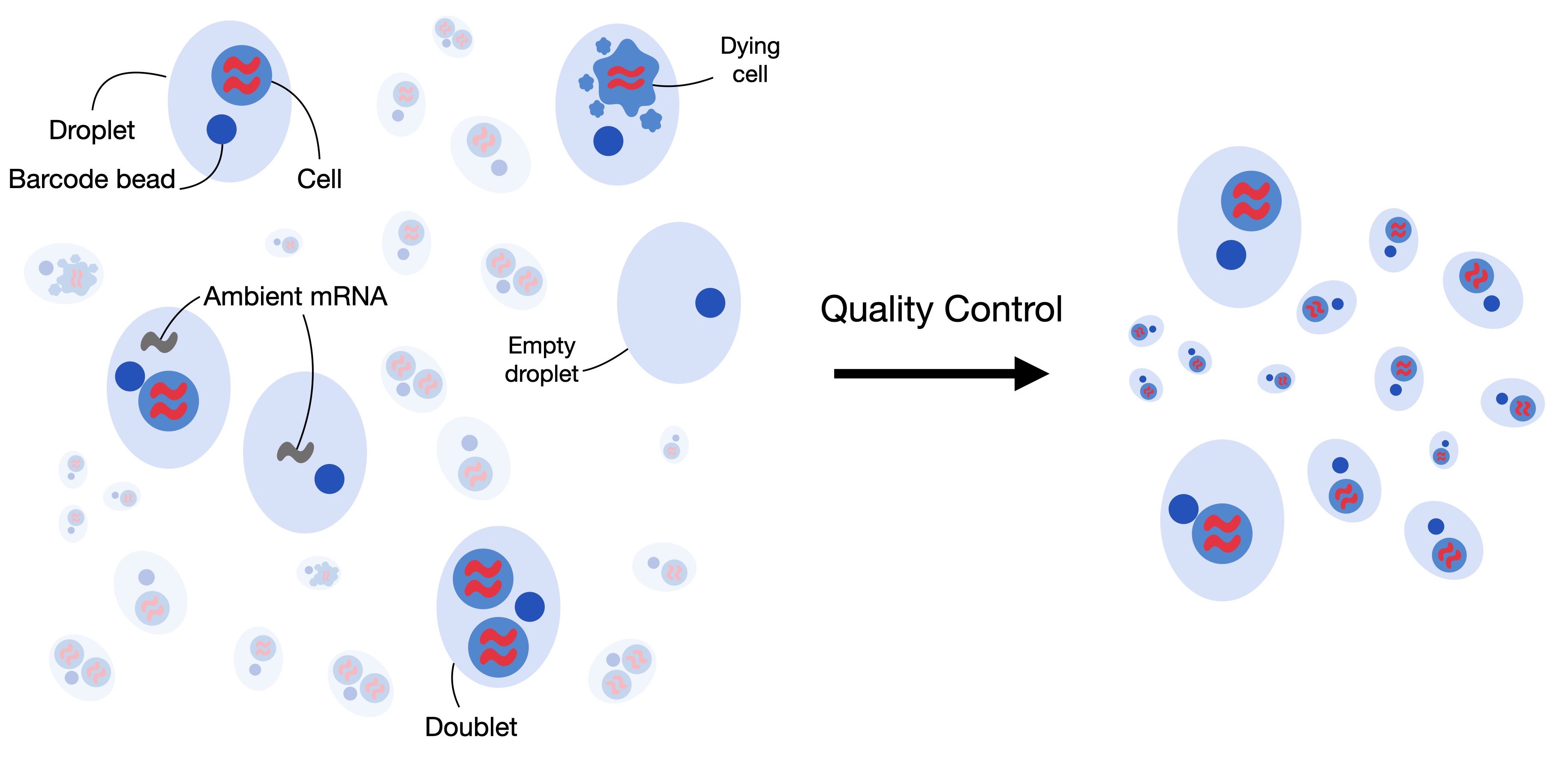
单细胞RNA-seq数据集可能包含低质量细胞、细胞外RNA和双重细胞。质量控制旨在移除和纠正这些问题,以获得每个观测值都是完整单细胞的高质量数据集。
过滤低质量细胞¶
质量控制的第一步是从数据集中移除低质量细胞。当一个细胞检测到的基因数量少、计数深度低且线粒体计数比例高时,这可能表明该细胞的膜已破裂,可能正在死亡。由于这些细胞通常不是我们分析的主要目标,并且可能会扭曲下游分析,因此我们在质量控制过程中将其移除。为识别这些细胞,我们定义细胞质量控制(QC)阈值。细胞QC通常基于以下三个QC协变量进行:
- 每个条形码的计数数目(计数深度)
- 每个条形码的基因数目
- 每个条形码中来自线粒体基因的计数比例
在细胞QC中,这些协变量通过设定阈值进行过滤,因为它们可能对应于正在死亡的细胞。如前所述,这些细胞可能反映了膜破裂的细胞,其细胞质中的mRNA已经泄漏,因此只有线粒体中的mRNA仍然存在。这些细胞可能显示出低计数深度、检测到的基因少以及线粒体读取比例高。然而,关键是要联合考虑这三个QC协变量,否则可能会导致细胞信号的误判。例如,线粒体计数比例较高的细胞可能参与呼吸过程,不应被过滤掉。而计数低或高的细胞可能对应于静息细胞群或较大的细胞。因此,在对单个协变量进行阈值决策时,最好考虑多个协变量。一般建议排除较少的细胞,并尽可能宽松,以避免过滤掉可行的细胞群或小的亚群。
在少量或小数据集上进行QC时,通常通过查看不同QC协变量的分布并手动识别异常值来进行。然而,随着数据集的增长,这一任务变得越来越耗时,因此值得考虑通过MAD(中位数绝对偏差)进行自动阈值设定。MAD的计算公式为 $MAD = median(|X_i - median(X)|)$,其中 $X_i$ 是观测值的相应QC指标,描述了该指标变异性的稳健统计量。类似于【2020】的做法,如果细胞的MAD偏差超过5个MAD,我们将其标记为异常值,这是一种相对宽松的过滤策略。我们要强调的是,在细胞注释之后重新评估过滤可能是合理的。
在QC中,第一步是计算QC协变量或指标。我们使用scanpy函数 sc.pp.calculate_qc_metrics 计算这些指标,该函数还可以计算特定基因群体的计数比例。因此,我们定义线粒体、核糖体和血红蛋白基因。需要注意的是,根据数据集中所考虑的物种,线粒体计数要么以“mt-”前缀标注,要么以“MT-”前缀标注。如前所述,本笔记本中使用的数据集是人类骨髓,因此线粒体计数以“MT-”前缀标注。对于小鼠数据集,前缀通常是小写的,即“mt-”。
# mitochondrial genes
adata.var["mt"] = adata.var_names.str.startswith("MT-")
# ribosomal genes
adata.var["ribo"] = adata.var_names.str.startswith(("RPS", "RPL"))
# hemoglobin genes.
adata.var["hb"] = adata.var_names.str.contains(("^HB[^(P)]"))
现在我们可以使用scanpy计算相应的QC指标。
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt", "ribo", "hb"], inplace=True, percent_top=[20], log1p=True
)
adata
AnnData object with n_obs × n_vars = 16934 × 36601
obs: 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb'
var: 'gene_ids', 'feature_types', 'genome', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts'
正如我们所看到的,函数向 .var 和 .obs 添加了几列额外的列。这里我们重点介绍其中的几项,更多关于不同指标的信息可以在scanpy文档中找到:
.obs中的n_genes_by_counts是细胞中具有正计数的基因数量,total_counts是细胞的总计数,这也可能被称为文库大小,pct_counts_mt是细胞中线粒体总计数的比例。
我们现在绘制每个样本的三个QC协变量 n_genes_by_counts、total_counts 和 pct_counts_mt,以评估各个细胞的捕获情况。
p1 = sns.displot(adata.obs["total_counts"], bins=100, kde=False)
# sc.pl.violin(adata, 'total_counts')
p2 = sc.pl.violin(adata, "pct_counts_mt")
p3 = sc.pl.scatter(adata, "total_counts", "n_genes_by_counts", color="pct_counts_mt")
... storing 'feature_types' as categorical ... storing 'genome' as categorical



这些图表显示,一些细胞的线粒体计数比例相对较高,这通常与细胞降解有关。但由于每个细胞的总计数足够高,并且大多数细胞的线粒体读取比例低于20%,我们仍然可以处理这些数据。基于这些图表,我们可以手动定义过滤细胞的阈值。相反,我们将展示基于MAD的自动阈值设定和过滤。
首先,我们定义一个函数,该函数接收一个 metric(即 .obs 中的一列)和在过滤策略中仍然允许的MAD数(nmad)。
def is_outlier(adata, metric: str, nmads: int):
M = adata.obs[metric]
outlier = (M < np.median(M) - nmads * median_abs_deviation(M)) | (
np.median(M) + nmads * median_abs_deviation(M) < M
)
return outlier
现在我们将此函数应用于 log1p_total_counts、log1p_n_genes_by_counts 和 pct_counts_in_top_20_genes 三个QC协变量,每个协变量的阈值设为5个MAD。
adata.obs["outlier"] = (
is_outlier(adata, "log1p_total_counts", 5)
| is_outlier(adata, "log1p_n_genes_by_counts", 5)
| is_outlier(adata, "pct_counts_in_top_20_genes", 5)
)
adata.obs.outlier.value_counts()
False 16065 True 869 Name: outlier, dtype: int64
pct_counts_Mt 使用3个MAD进行过滤。此外,线粒体计数比例超过8%的细胞也被过滤掉。
adata.obs["mt_outlier"] = is_outlier(adata, "pct_counts_mt", 3) | (
adata.obs["pct_counts_mt"] > 8
)
adata.obs.mt_outlier.value_counts()
False 15240 True 1694 Name: mt_outlier, dtype: int64
现在我们根据这两个附加列过滤我们的AnnData对象。
print(f"Total number of cells: {adata.n_obs}")
adata = adata[(~adata.obs.outlier) & (~adata.obs.mt_outlier)].copy()
print(f"Number of cells after filtering of low quality cells: {adata.n_obs}")
Total number of cells: 16934 Number of cells after filtering of low quality cells: 14814
p1 = sc.pl.scatter(adata, "total_counts", "n_genes_by_counts", color="pct_counts_mt")

校正环境RNA¶
对于基于液滴的单细胞RNA-seq实验,稀释液中存在一定量的背景mRNA,这些mRNA分布到含有细胞的液滴中并与细胞一起被测序。其净效应是产生一种背景污染,这种污染代表了液滴中所含细胞之外的表达,而是稀释液中的细胞。
基于液滴的scRNA-seq在多个细胞中生成基因的唯一分子标识符(UMIs)计数,旨在识别每个基因和每个细胞的分子数量。它假设每个液滴包含来自单个细胞的mRNA。双重细胞、空液滴和无细胞RNA可能会违反这一假设。无细胞的mRNA分子代表稀释液中存在的背景mRNA。这些分子分布在液滴中并与液滴一起被测序。输入溶液中无细胞mRNA的污染通常称为“汤”,由细胞裂解产生。
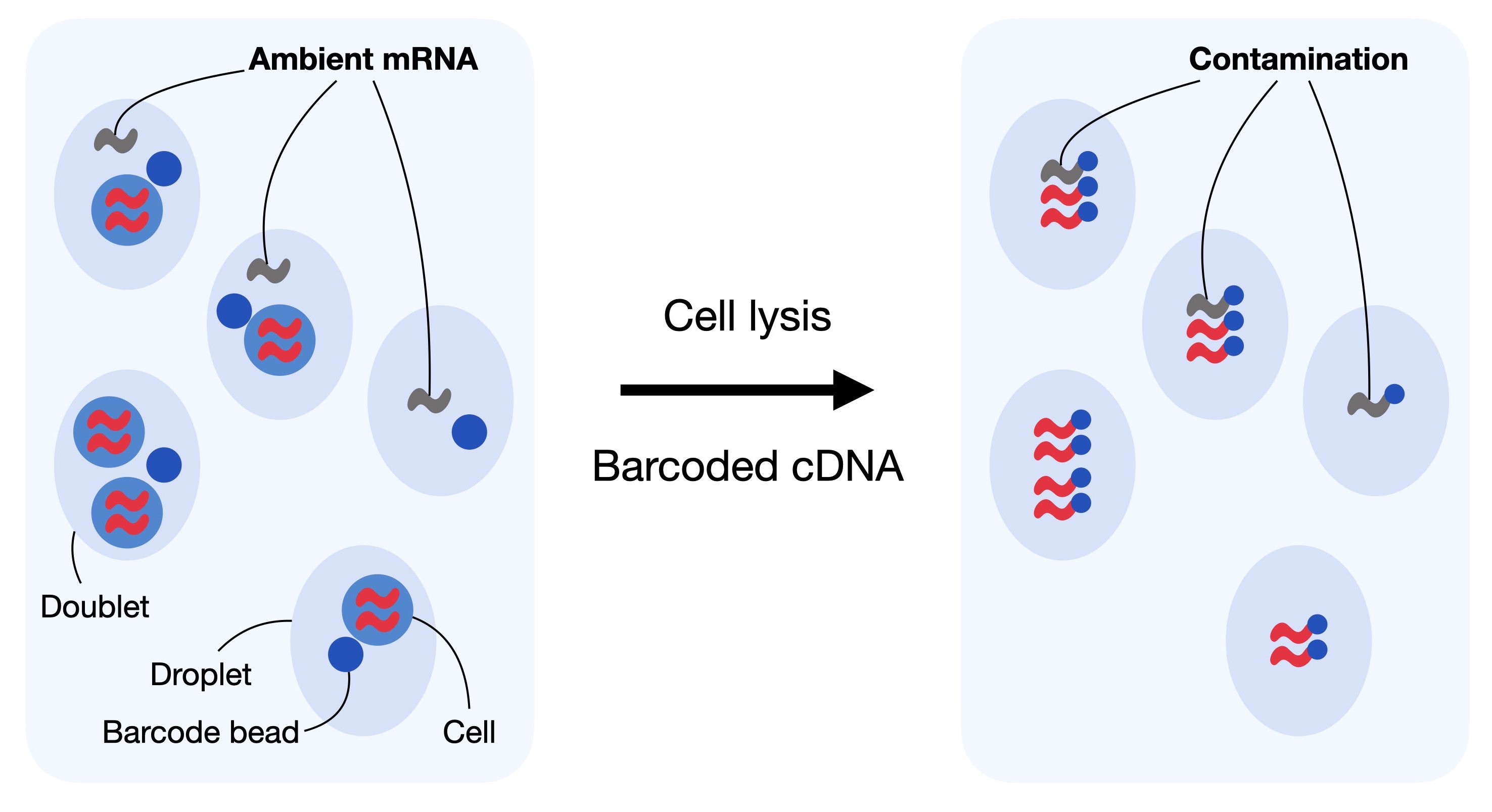
在基于液滴的测序技术中,液滴可以包含环境RNA或双重细胞(捕获多个细胞的液滴)。污染的环境RNA被条形码标记并与细胞的天然mRNA一起计数,导致计数混淆。
无细胞mRNA分子,也称为环境RNA,会混淆观察到的计数,并被视为背景污染。校正基于液滴的scRNA-seq数据集中的无细胞mRNA非常重要,因为它可能会扭曲下游分析中数据的解释。通常,每种输入溶液的汤有所不同,具体取决于数据集中各个细胞的表达模式。去除环境mRNA的方法如SoupX【2020】和DecontX【2020】旨在估计汤的成分并根据汤的表达校正计数表。
第一步,SoupX计算汤的特征。它从未过滤的Cellranger矩阵中估计空液滴的环境mRNA表达特征。接下来,SoupX估计特定细胞的污染比例。最后,它根据环境mRNA表达特征和估计的污染校正表达矩阵。
SoupX的输出是一个修改后的计数矩阵,可用于任何下游分析工具。
我们现在加载运行SoupX所需的Python和R包。
import anndata2ri
import logging
import rpy2.rinterface_lib.callbacks as rcb
import rpy2.robjects as ro
rcb.logger.setLevel(logging.ERROR)
ro.pandas2ri.activate()
anndata2ri.activate()
%load_ext rpy2.ipython
%%R
library(SoupX)
SoupX可以在没有聚类信息的情况下运行,但正如Young和Behjati【2020】所指出的,如果提供基本的聚类信息,结果会更好。SoupX可以使用cellranger生成的默认聚类,也可以手动定义聚类。我们将在本笔记本中展示后者,因为SoupX的结果对所使用的聚类并不强烈敏感。
现在,我们创建AnnData对象的副本,对其进行标准化,降维,并在处理后的副本上计算默认的Leiden聚类。后续章节将更详细地介绍聚类。目前我们只需要知道Leiden聚类给我们提供了数据集中细胞的分区(社区)。我们将获得的聚类保存为soupx_groups,并删除AnnData对象的副本以节省笔记本中的内存。
首先,我们生成AnnData对象的副本,对其进行标准化和log1p变换。此时我们使用简单的移位对数标准化。关于不同标准化技术的更多信息可以在标准化章节中找到。
adata_pp = adata.copy()
sc.pp.normalize_per_cell(adata_pp)
sc.pp.log1p(adata_pp)
接下来,我们将计算数据的主成分,以获得一个低维度的表示。 然后使用这个表示来生成数据的邻域图,并在 KNN 图上运行 Leiden 聚类。我们将聚类结果作为 soupx_groups 添加到 .obs 中并保存为向量。
sc.pp.pca(adata_pp)
sc.pp.neighbors(adata_pp)
sc.tl.leiden(adata_pp, key_added="soupx_groups")
# Preprocess variables for SoupX
soupx_groups = adata_pp.obs["soupx_groups"]
现在我们可以删除 AnnData 对象的副本了,因为我们已经生成了一个簇向量,可以直接用于 soupX 分析。
del adata_pp
接下来,我们将保存过滤后的 cellranger 输出中的细胞名称、基因名称和数据矩阵。SoupX 需要一个格式为“特征 x 条形码”的矩阵,因此我们需要对 .X 进行转置。
cells = adata.obs_names
genes = adata.var_names
data = adata.X.T
SoupX还需要原始的基因细胞矩阵,该矩阵通常在cellranger输出中称为raw_feature_bc_matrix.h5。我们同样使用scanpy加载该文件,就像之前加载filtered_feature_bc_matrix.h5一样,并在对象上运行.var_names_make_unique()使变量名称唯一,并转置相应的.X。
adata_raw = sc.read_10x_h5(
filename="raw_feature_bc_matrix.h5",
backup_url="https://figshare.com/ndownloader/files/39546217",
)
adata_raw.var_names_make_unique()
data_tod = adata_raw.X.T
Variable names are not unique. To make them unique, call `.var_names_make_unique`. Variable names are not unique. To make them unique, call `.var_names_make_unique`.
del adata_raw
我们现在已经准备好运行SoupX。所需输入包括形状为条形码 x 细胞的过滤后的cellranger矩阵、形状为条形码 x 液滴的cellranger原始液滴表、基因和细胞名称以及通过简单Leiden聚类获得的聚类。输出将是校正后的计数矩阵。
首先,我们从液滴表和细胞表构建一个所谓的SoupChannel。接下来,我们向SoupChannel对象添加元数据,这些元数据可以是任何形式的data.frame。
%%R -i data -i data_tod -i genes -i cells -i soupx_groups -o out
# specify row and column names of data
rownames(data) = genes
colnames(data) = cells
# ensure correct sparse format for table of counts and table of droplets
data <- as(data, "sparseMatrix")
data_tod <- as(data_tod, "sparseMatrix")
# Generate SoupChannel Object for SoupX
sc = SoupChannel(data_tod, data, calcSoupProfile = FALSE)
# Add extra meta data to the SoupChannel object
soupProf = data.frame(row.names = rownames(data), est = rowSums(data)/sum(data), counts = rowSums(data))
sc = setSoupProfile(sc, soupProf)
# Set cluster information in SoupChannel
sc = setClusters(sc, soupx_groups)
# Estimate contamination fraction
sc = autoEstCont(sc, doPlot=FALSE)
# Infer corrected table of counts and rount to integer
out = adjustCounts(sc, roundToInt = TRUE)
SoupX成功推断出校正后的计数,现在我们可以将其存储为一个额外的层。在所有后续的分析步骤中,我们将使用SoupX校正的计数矩阵,因此我们用SoupX校正的矩阵覆盖.X。
adata.layers["counts"] = adata.X
adata.layers["soupX_counts"] = out.T
adata.X = adata.layers["soupX_counts"]
接下来,我们还要过滤掉在至少20个细胞中未检测到的基因,因为这些基因不具有信息性。
print(f"Total number of genes: {adata.n_vars}")
# Min 20 cells - filters out 0 count genes
sc.pp.filter_genes(adata, min_cells=20)
print(f"Number of genes after cell filter: {adata.n_vars}")
Total number of genes: 36601 Number of genes after cell filter: 20171
双重细胞检测¶
双重细胞是指在相同细胞条形码下测序的两个细胞,例如它们被捕获在同一个液滴中。这就是为什么我们直到现在一直使用“条形码”而不是“细胞”这个术语。双重细胞如果由相同类型的细胞(但来自不同个体)形成,则称为同源双重细胞,否则称为异源双重细胞。同源双重细胞不一定能从计数矩阵中识别出来,通常被认为是无害的,因为它们可以通过细胞哈希或SNP识别。因此,它们的识别不是双重细胞检测方法的主要目标。
由不同细胞类型或状态形成的双重细胞称为异源双重细胞。其识别至关重要,因为它们很可能被错误分类,导致下游分析步骤的扭曲。因此,双重细胞检测和去除通常是初始预处理步骤。双重细胞可以通过其高读数和检测到的特征来识别,或者通过创建人工双重细胞并将其与数据集中的细胞进行比较的方法来识别。双重细胞检测方法在计算上是高效的,并且有几个软件包可以完成这一任务。
【2021】评估了九种不同的双重细胞检测方法,并根据计算效率和双重细胞检测准确性评估了它们的性能。此外,他们在基准测试的附录中评估了scDblFinder,该方法实现了最高的双重细胞检测准确性以及良好的计算效率和稳定性【2021】。
在本教程中,我们将展示scDblFinder R包。scDblFinder随机选择两个液滴,并通过平均它们的基因表达谱来创建人工双重细胞。然后,双重细胞得分定义为在主成分空间中每个液滴的k最近邻图中的人工双重细胞比例。
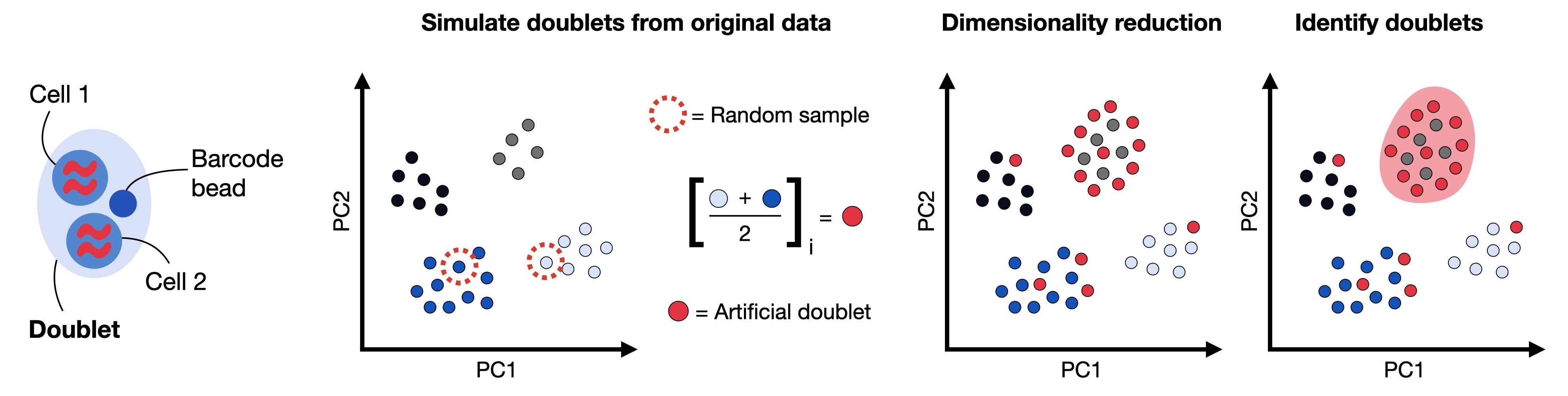
双重细胞是指包含多个细胞的液滴。常见的双重细胞检测方法通过随机抽样成对的细胞,并平均其基因表达谱以获得双重细胞计数,生成人工双重细胞。这些人工双重细胞与其余的细胞一起投射到低维的主成分空间中。双重细胞检测方法根据k最近邻图中人工双重细胞邻居的数量计算双重细胞得分。
We first load some additional python and R packages.
%%R
library(Seurat)
library(scater)
library(scDblFinder)
library(BiocParallel)
data_mat = adata.X.T
我们现在可以通过使用data_mat作为输入,将其加载到SingleCellExperiment中的scDblFinder来启动双重细胞检测。scDblFinder会向sce的colData添加几列,其中三列可能对分析很有帮助:
sce$scDblFinder.score:最终的双重细胞得分(分数越高,细胞是双重细胞的可能性越大)sce$scDblFinder.ratio:细胞邻域中人工双重细胞的比例sce$scDblFinder.class:分类结果(双重细胞或单细胞)
我们将只输出class参数,并将其存储在AnnData对象的.obs中。其他参数也可以类似地添加到AnnData对象中。
%%R -i data_mat -o doublet_score -o doublet_class
set.seed(123)
sce = scDblFinder(
SingleCellExperiment(
list(counts=data_mat),
)
)
doublet_score = sce$scDblFinder.score
doublet_class = sce$scDblFinder.class
scDblFinder输出的分类包括Singlet(1)和Doublet(2)。我们将其添加到AnnData对象的.obs中。
adata.obs["scDblFinder_score"] = doublet_score
adata.obs["scDblFinder_class"] = doublet_class
adata.obs.scDblFinder_class.value_counts()
singlet 11956 doublet 2858 Name: scDblFinder_class, dtype: int64
我们建议暂时将识别出的双重细胞保留在数据集中,并在可视化过程中检查双重细胞。
在下游聚类过程中,重新评估质量控制和选择的参数可能会有所帮助,以便可能过滤掉更多或更少的细胞。现在我们可以保存数据集,并继续进行标准化章节。
adata.write("s4d8_quality_control.h5ad")
Key Takeaways¶
- 过滤低质量细胞时应基于中位数绝对偏差(MAD)并使用宽松的截止值,以避免对较小的亚群产生偏见。
- 基于特征的过滤对下游任务没有显著好处。
- 可以使用如scDblFinder等工具高效检测双重细胞。
- 双重细胞检测方法不应在表示多个批次的聚合scRNA-seq数据上运行。