2.4 Dimensionality Reduction¶
如前所述,scRNA-seq是一种高通量测序技术,生成的数据显示在细胞和基因数量上具有高维度。这立即表明scRNA-seq数据存在“维度诅咒”问题。
{admonition}
维度诅咒最早由R. Bellman【1957】提出,描述了理论上高维数据包含更多信息,但在实践中情况并非如此。高维数据通常包含更多的噪声和冗余,因此添加更多信息不会为下游分析步骤提供好处。
并非所有基因都具有信息性并且对于基于表达谱的细胞类型聚类任务是重要的。我们已经通过特征选择来减少数据的维度,作为下一步,可以通过降维算法进一步减少单细胞RNA-seq数据的维度。这些算法是预处理中减少数据复杂性和可视化的重要步骤。已经开发并使用了几种降维技术用于单细胞数据分析。
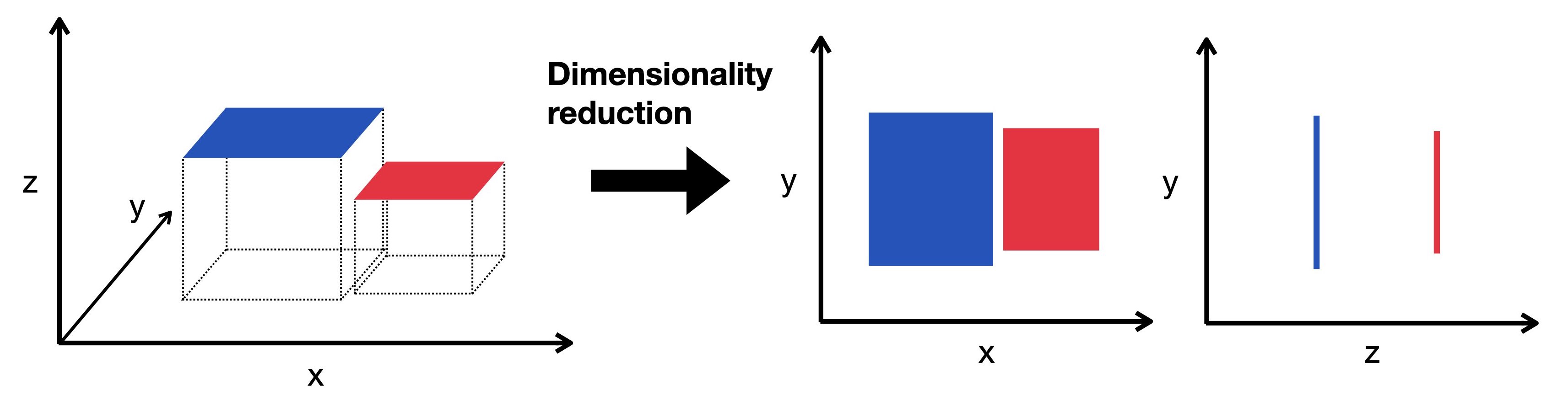
降维将高维数据嵌入到低维空间中。低维表示仍然捕捉数据的潜在结构,同时尽可能少地包含维度。这里我们将一个三维对象投影到二维空间中进行可视化。
Xing等人通过独立比较对10种不同的降维方法的稳定性、准确性和计算成本进行了比较【2021】。他们建议使用t分布随机邻嵌入(t-SNE),因为它在整体性能上表现最佳。统一流形近似和投影(UMAP)显示了最高的稳定性,并且最能分离原始细胞群体。在这种情况下,值得一提的另一个降维方法是主成分分析(PCA),它仍被广泛使用。
通常,如果选择特定的初始化选项,t-SNE和UMAP是非常稳健且大多等效的【2019】。
所有上述方法都在scanpy中得到了实现。
import scanpy as sc
sc.settings.verbosity = 0
sc.settings.set_figure_params(
dpi=80,
facecolor="white",
frameon=False,
)
adata = sc.read(
filename="s4d8_feature_selection.h5ad",
backup_url="https://figshare.com/ndownloader/files/40016014",
)
我们将使用数据集的标准化表示进行降维和可视化,特别是使用移位对数变换。
adata.X = adata.layers["log1p_norm"]
我们从以下内容开始:
PCA¶
在我们的数据集中,每个细胞是由一些正交标准基组成的 n_var 维向量空间中的一个向量。由于scRNA-seq存在“维度诅咒”,我们知道并非所有特征对于理解数据集的潜在动态都是重要的,并且存在固有的冗余【2014】。PCA通过对原始数据集进行正交变换,创建了一组新的不相关变量,即主成分(PCs)。PCs是原始数据集中特征的线性组合,并按照方差递减的顺序排列以定义变换。通过这种排序,通常第一个PC具有最大的可能方差。方差最低的PC被舍弃,以有效减少数据的维度而不丢失信息。
PCA的优势在于它具有高度的可解释性和计算效率。然而,由于scRNA-seq数据集由于掉落事件而相当稀疏,因此高度非线性,使用线性降维技术PCA进行可视化并不十分合适。PCA通常用于选择前10-50个PCs,这些PCs用于下游分析任务。
# setting highly variable as highly deviant to use scanpy 'use_highly_variable' argument in sc.pp.pca
adata.var["highly_variable"] = adata.var["highly_deviant"]
sc.pp.pca(adata, svd_solver="arpack", use_highly_variable=True)
sc.pl.pca_scatter(adata, color="total_counts")

t-SNE¶
t-SNE 是一种基于图的非线性降维技术,可以将高维数据投影到二维或三维空间。该方法基于数据点之间的高维欧几里得距离定义了一个高斯概率分布。然后,使用学生 t 分布在低维空间中重建概率分布,并使用梯度下降优化嵌入。
sc.tl.tsne(adata, use_rep="X_pca")
sc.pl.tsne(adata, color="total_counts")

UMAP¶
UMAP 也是一种基于图的非线性降维技术,在原理上与 t-SNE 类似。它通过构建数据集的高维图表示,然后优化低维图表示,使其与原始图尽可能在结构上保持相似。
降维过程通常分为两步:
- 我们首先计算数据的PCA (主成分分析)。
- 然后基于 PCA 结果构建邻域图。
sc.pp.neighbors(adata)
sc.tl.umap(adata)
sc.pl.umap(adata, color="total_counts")

检查质量控制指标¶
现在我们还可以将之前计算的质量控制指标可视化到 PCA、t-SNE 或 UMAP 图中,并有可能识别出低质量的细胞。
sc.pl.umap(
adata,
color=["total_counts", "pct_counts_mt", "scDblFinder_score", "scDblFinder_class"],
)

观察双线体评分¶
正如我们观察到的,具有高双线体评分的细胞在 UMAP 上投影到相同区域。我们暂时会将它们保留在数据集中,但可能会在以后重新审视我们的质量控制策略
adata.write("s4d8_dimensionality_reduction.h5ad")