3.1 PCA
主成分分析(PCA)是什么?¶
主成分分析是一种将多维数据降维为低维数据的方法。由于主成分分析能够提取最能代表数据性质的要素,因此在数据可视化和使用机器学习方法进行聚类分析时非常有用。
在 RNA-seq 分析中,通过使用主成分分析,可以更简单和快速地分析大量的表达数据。主成分分析对原始表达数据进行降维,提取主要的表达变化模式。通过这种方式,可以更直观地可视化大量的表达数据,并基于结果分析基因表达变化和表达模式。
使用 R 进行 RNA-seq 数据的主成分分析方法¶
要使用 R 进行 RNA-seq 数据的主成分分析,可以编写如下代码。首先保存这段代码。
install.packages("ggplot2")
library(ggplot2)
install.packages("devtools")
library(devtools)
install_github("sinhrks/ggfortify")
library(ggfortify)
library(tidyverse)
Sys.setenv("VROOM_CONNECTION_SIZE" = 131072 * 2)
mydata <- read_tsv("sample.tsv")
t_mydata <- as.data.frame(t(mydata))
gene_names <- t_mydata[1, ]
colnames(t_mydata) <- gene_names
delete_data <- c()
for (x in t_mydata) {
data5 <- sapply(x[2:length(x)], as.numeric)
data5_var <- var(data5)
if (data5_var == 0) {
delete_data <- c(delete_data, x[1])
}
}
for (x in delete_data) {
t_mydata <- select(t_mydata, -x)
}
t_mydata_as_num <- sapply(t_mydata, as.numeric)
data2 <- na.omit(t_mydata_as_num)
pca_res <- prcomp(data2, scale. = TRUE)
autoplot(pca_res)
代码保存路径:File→Save As,命名后将 Where 设为「Desktop」。
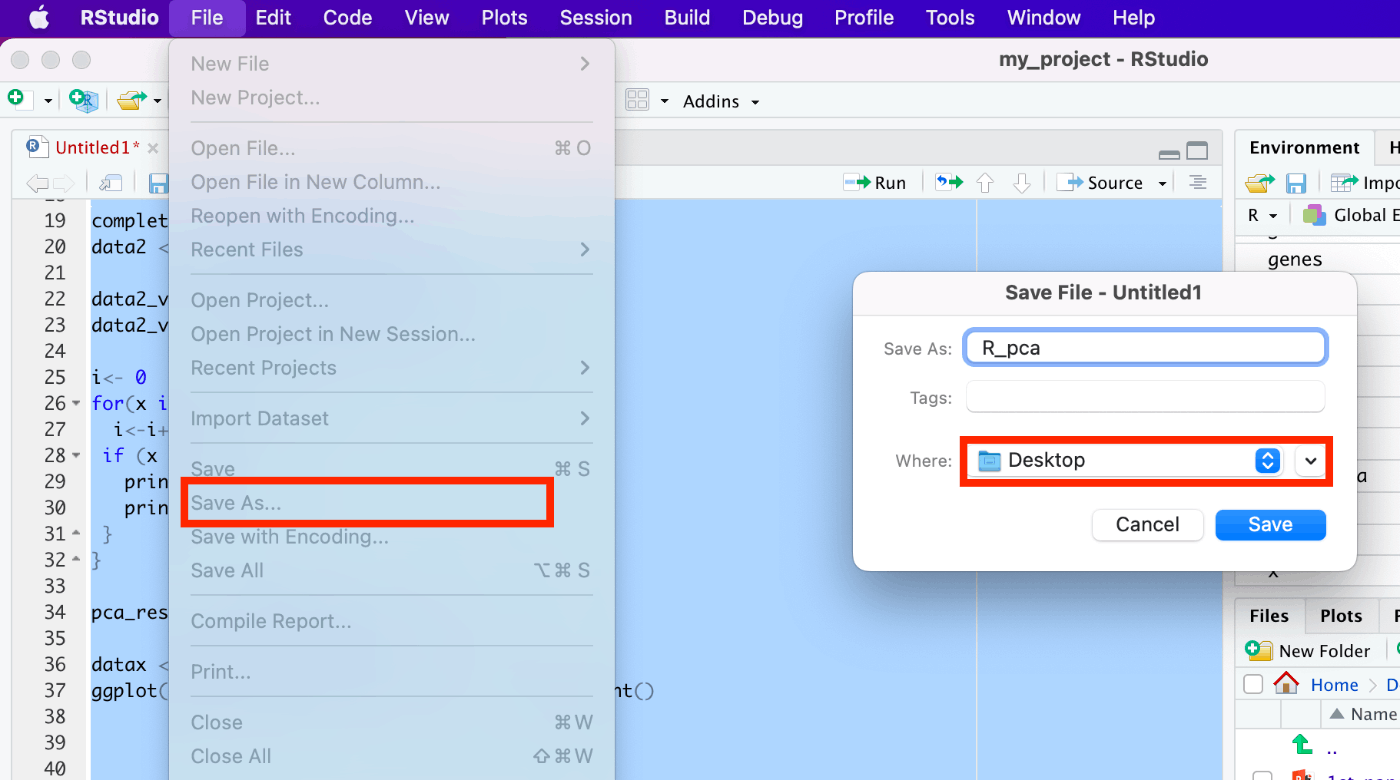
实际运行代码¶
在运行代码之前,首先需要准备解析数据。实际上用于解析的文件大致如下矩阵文件的格式:
SRR11111111 SRR11111112 SRR11111113 ……
A1BG 1.204012121 1.305946294 1.058155716
A1CF 0.8814023041 1.010914863 1.174708571
A2M 1.868867371 1 0.9556968534
A2ML1 0.6709431377 1.126605147 1
A3GALT2 1 1 1
A4GALT 0.5010093702 1.364249746 1.664916037
A4GNT 1.881158763 1 1
AAAS 0.7203903135 0.8684943431 1.474680837
AACS 1.06245034 1.256530797 1.544221892
(以下略)
这次已经准备好了示例文件,请将其下载到桌面上使用。
文件名:sample.tsv
实际读取示例文件并运行代码时,应该会得到如下分析结果。
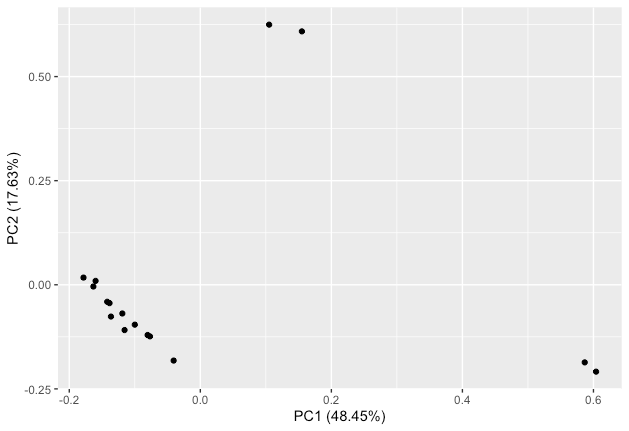
从这里开始,我会尝试给每个样品涂上不同的颜色。
我们准备了一个样本文件 sample_2.tsv,请下载到您的桌面上。
sample_2.tsv 的第 1 列新增了一列,名为 Sample。
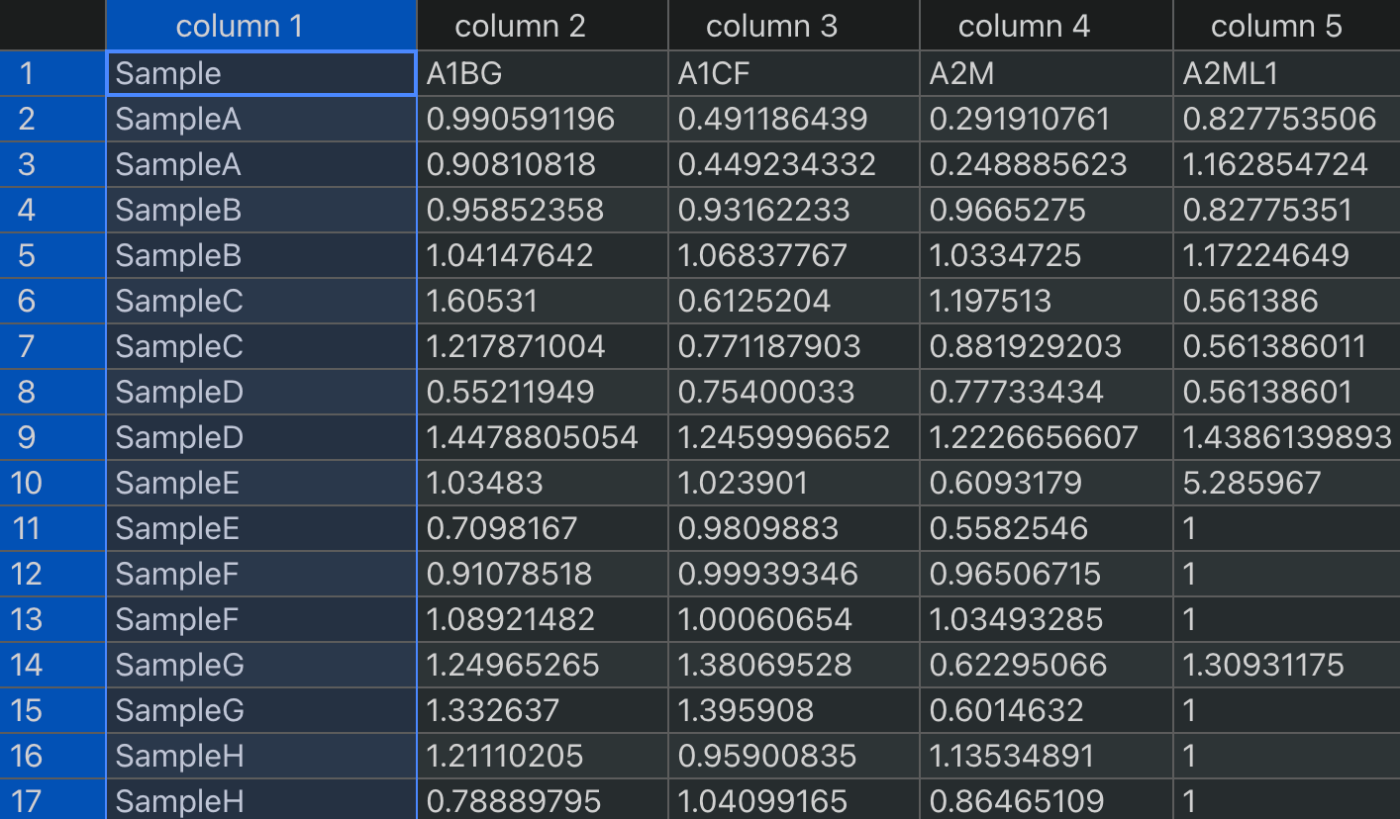
准备就绪后,运行以下代码。请注意,如果 sample_2.tsv 没有放在桌面上,就会出现错误。
mydata <- read_tsv("sample_2.tsv")
pca_res <- prcomp(mydata[2:ncol(mydata)], scale. = TRUE)
autoplot(pca_res, data = mydata, colour = "Sample")
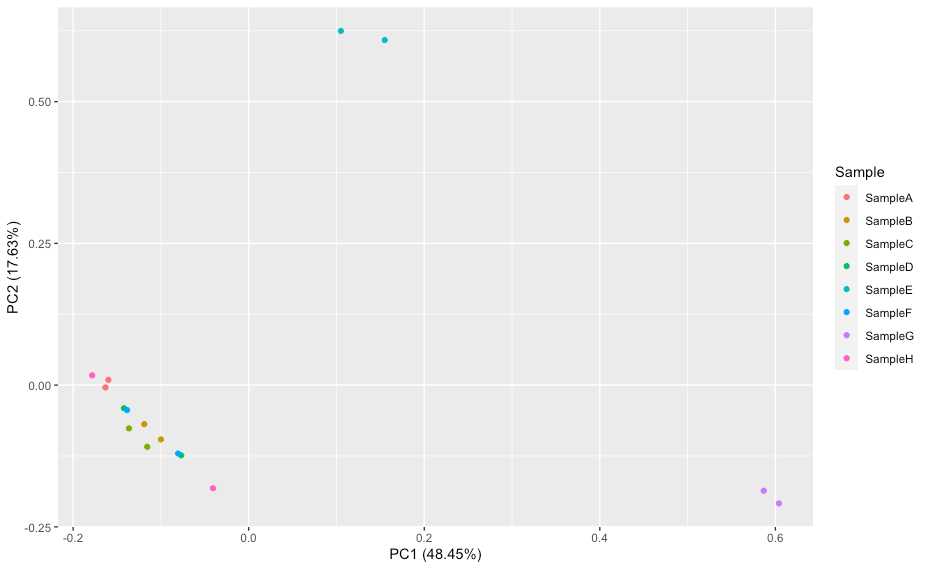
可以按样本划分。
代码说明¶
以下代码为 PCA 安装必要的软件包。
install.packages("ggplot2")
library(ggplot2)
install.packages("devtools")
library(devtools)
install_github('sinhrks/ggfortify')
library(ggfortify)
library(tidyverse)
以下是提升 Rstudio 数据加载容量的步骤。对于像这次这样的小文件来说没有问题,但如果文件较大,则无法加载,需要执行以下命令,否则会出现错误。
Sys.setenv("VROOM_CONNECTION_SIZE" = 131072 * 2)
使用 read_tsv() 函数从名为 'sample.tsv' 的 tsv 文件中读取数据,并将其存储在变量 mydata 中。接下来,使用 as.data.frame() 函数和 t() 函数将 mydata 转置为数据框 t_mydata。
mydata <- read_tsv("sample.tsv")
t_mydata <- as.data.frame(t(mydata))
将样本名称赋值给 gene_names。然后,使用 colnames() 函数将 t_mydata 的列名设置为 gene_names。
gene_names <- t_mydata[1, ]
colnames(t_mydata) <- gene_names
以下代码的详细说明省略。简而言之,在主成分分析中,需要去除单位方差(在这种情况下,所有基因表达量为 1)的成分,代码实现了这个功能。
delete_data <- c()
for (x in t_mydata) {
data5 <- sapply(x[2:length(x)], as.numeric)
data5_var <- var(data5)
if (data5_var == 0) {
delete_data <- c(delete_data, x[1])
}
}
for (x in delete_data) {
t_mydata <- select(t_mydata, -x)
}
此处从数据中删除缺失值。被删除的数据存储在 data2 中。
t_mydata_as_num <- sapply(t_mydata, as.numeric)
data2 <- na.omit(t_mydata_as_num)
最后,使用 prcomp 函数执行主成分分析。通过指定参数 scale. = TRUE 来进行标准化,然后执行主成分分析。
pca_res <- prcomp(data2, scale. = TRUE)
使用 autoplot 函数进行可视化。
autoplot(pca_res)
在 autoplot 中指定 colour 和要分组的列,可以进行分组。由于要分组的列是字符串,因此在进行 PCA 时需避开这些字符串。否则会报错,请注意。
pca_res <- prcomp(mydata[2:ncol(mydata)], scale. = TRUE)
autoplot(pca_res, data = mydata, colour = "Sample")