3.1 Clustering¶
Motivation¶
预处理和可视化使我们能够描述scRNA-seq数据集并降低其维度。到目前为止,我们已经嵌入并可视化细胞以了解数据集的潜在属性。然而,它们仍然定义得比较抽象。单细胞分析的下一个自然步骤是识别数据集中的细胞结构。
在scRNA-seq数据分析中,我们通过找到与已知细胞状态或细胞周期阶段相关的细胞身份来描述数据集中的细胞结构。这个过程通常被称为细胞身份注释。为此,我们将细胞结构化为簇,以推断相似细胞的身份。聚类本身是一个常见的无监督机器学习问题。我们可以通过最小化在降低维度的表达空间中的簇内距离来导出簇。在这种情况下,表达空间决定了细胞的基因表达相似性,其基础是降维表示。例如,通过主成分分析确定这个低维表示,相似性评分基于欧氏距离。
在KNN图中,节点反映数据集中的细胞。我们首先在PC降维表达空间中为所有细胞计算欧氏距离矩阵,然后将每个细胞连接到其最相似的K个细胞。通常,K的值设置在5到100之间,具体取决于数据集的大小。KNN图通过表示表达空间中的密集区域来反映表达数据的底层拓扑结构,这些密集区域在图中也表现为密集连接的区域 {cite}wolf_paga_2019。KNN图中的密集区域由社区检测方法如Leiden和Louvain检测到{cite}blondel_fast_2008。
Leiden算法是Louvain算法的改进版,在单细胞RNA-seq数据分析中优于其他聚类方法 ({cite}du_systematic_2018, freytag_comparison_2018, weber_comparison_2016)。由于Louvain算法不再维护,因此推荐使用Leiden算法。
我们因此建议在单细胞k最近邻(KNN)图上使用Leiden算法{cite}traag_louvain_2019来对单细胞数据集进行聚类。
Leiden算法通过考虑簇内细胞之间的连接数量与数据集中总体预期的连接数量来创建簇。
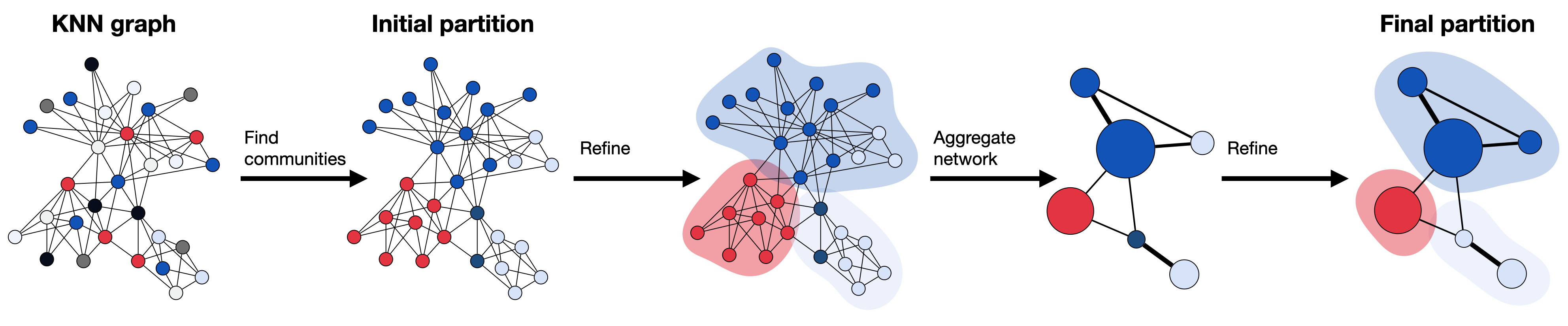
Leiden算法在从PC降维表达空间获得的KNN图上计算聚类。它从初始分区开始,其中每个节点都属于自己的社区。接下来,算法将单个节点从一个社区移动到另一个社区以找到一个分区,然后进行细化。基于细化后的分区生成一个聚合网络,再次细化直到无法进一步改进,最终达到最终分区。
起点是单节点分区,其中每个节点作为其自己的邻域(a)。接下来,算法通过将个别节点从一个社区移动到另一个社区来创建分区(b),然后进行细化以增强分区(c)。细化后的分区然后聚合成一个网络(d)。随后,算法再次在聚合网络中移动个别节点(e),直到细化不再改变分区(f)。所有步骤重复进行,直到创建最终聚类且分区不再改变。
Leiden模块有一个分辨率参数,允许确定分区簇的规模和聚类的粗细度。更高的分辨率参数会导致更多的簇。该算法还允许通过子集KNN图对数据集中的特定簇进行高效的子聚类。子聚类使用户能够识别簇内的细胞类型特定状态或更细的细胞类型标记{cite}wagner_revealing_2016,但也可能导致仅由于数据中存在的噪声而产生的模式。
如前所述,Leiden算法在scanpy中实现。
import scanpy as sc
sc.settings.verbosity = 0
sc.settings.set_figure_params(dpi=80, facecolor="white", frameon=False)
Clustering human bone marrow cells¶
首先,我们加载数据集。我们在已经预处理的NeurIPS人类骨髓数据集中对site4-donor8样本进行聚类。
这个数据集已经使用log1pPF、scran和scTransform进行了标准化。根据预处理章节的建议,为了更好地识别个体细胞的亚状态,我们将在本笔记本中重点关注scran标准化版本的数据集。
adata = sc.read("s4d8_subset_gex.h5ad")
Leiden算法利用KNN图在降维表达空间上。我们可以使用scanpy函数sc.pp.neighbors在低维基因表达表示上计算KNN图。我们在前30个主成分上调用此函数,因为它们捕捉了数据集中大部分的变异。可视化聚类可以帮助我们理解结果,因此我们将细胞嵌入到UMAP嵌入中。更多详细信息可以在{ref}pre-processing:dimensionality-reduction章节找到。
sc.pp.neighbors(adata, n_pcs=30)
sc.tl.umap(adata)
/Users/anna.schaar/opt/miniconda3/envs/bp_pp/lib/python3.8/site-packages/tqdm/auto.py:22: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html from .autonotebook import tqdm as notebook_tqdm OMP: Info #276: omp_set_nested routine deprecated, please use omp_set_max_active_levels instead.
现在我们可以调用Leiden算法进行聚类。
sc.tl.leiden(adata)
scanpy中的默认分辨率参数为1.0。然而,在许多情况下,分析人员可能希望尝试不同的分辨率参数来控制聚类的粗细度。因此,我们建议将聚类结果保存在一个指定的键下,以指示所选择的分辨率。
sc.tl.leiden(adata, key_added="leiden_res0_25", resolution=0.25)
sc.tl.leiden(adata, key_added="leiden_res0_5", resolution=0.5)
sc.tl.leiden(adata, key_added="leiden_res1", resolution=1.0)
现在我们可视化使用Leiden算法在不同分辨率下获得的不同聚类结果。正如我们所见,分辨率对聚类的粗细度有很大的影响。较高的分辨率参数导致更多的社区,即识别出更多的簇,而较低的分辨率参数导致更少的社区。因此,分辨率参数控制了算法如何将KNN嵌入中的密集聚类区域组合在一起。这对于注释簇将变得尤为重要。
sc.pl.umap(
adata,
color=["leiden_res0_25", "leiden_res0_5", "leiden_res1"],
legend_loc="on data",
)

现在我们可以清晰地检查不同分辨率对聚类结果的影响。在0.25的分辨率下,聚类结果更加粗糙,算法检测到的社区较少。此外,与在1.0分辨率下获得的聚类相比,聚类区域的密度较低。
我们再次强调,显示的簇之间的距离必须谨慎解读。由于UMAP嵌入是在二维空间中,距离不一定能很好地反映所有点之间的关系。我们建议不要解释在UMAP嵌入中可视化的簇之间的距离。
Key takeaways¶
- 在单细胞KNN图上使用Leiden社区检测。
- 使用不同分辨率参数进行子聚类,允许用户专注于数据集中更详细的子结构,以便潜在地识别更细微的细胞状态。