3.2 使用 clusterPlofiler 进行富集分析
关于富集分析¶
富集分析是用于分析生物学数据的一种方法。该分析的目的是确定特定的生物标志物或基因集与指定的生物学通路或生物学过程的相关性程度。
该分析包括评估目标基因集是否以统计学上显著高的比例包含在目标通路或生物学过程中,与预期值进行比较。通过这种分析,可以发现新的假设以更深入地理解生物学数据,或验证现有知识。
使用 R 语言进行富集分析的方法有多种,这里介绍“clusterProfiler”。使用 clusterProfiler 进行富集分析的一个优点是其丰富的可视化手段。clusterProfiler 提供了许多绘图函数,可以直观地表示富集分析的结果。
关于基因名称的表示¶
在使用 clusterProfiler 时,了解基因名称的不同表示方法是很重要的。对于大家熟悉的形式之一是 SYMBOL 型。例如,“Cyclin-Dependent Kinase 4”在 SYMBOL 型中被简写为“CDK4”。SYMBOL 由人类基因组织(HUGO)基因命名委员会管理,并被广泛采用为国际标准。
除此之外,还有以下几种表示方法,以下均表示 CDK4:
- SYMBOL: CDK4
- Entrez Gene ID: 1019
- RefSeq: NM_000075.3
- Ensembl Gene ID: ENSG00000135446
- UniProtKB/Swiss-Prot: P11802
- HGNC: 1773
- MGI: 88346
使用 clusterProfiler 进行富集分析的实现方法
接下来,我们将进行实际的富集分析。作为 R 的运行环境,我们使用 Rstudio,如果尚未准备好,请参考相关文章进行环境构建。
使用 clusterProfiler 进行富集分析的最小代码如下。我们已经为你准备了放入 geneList 中的基因名称。
library(clusterProfiler)
geneList <- c(
"UBL5", "NDUFB8", "CHMP2B", "PRPF8", "NOSTRIN", "MFAP1", "CWC22",
"PLCH2", "PRPF31", "ATP6AP1", "DSC3", "CLN5", "CHDC2", "PIP", "ZNF473",
"DHX8", "RAB5A", "NUP98", "NUPL1", "HRK", "SLC41A3", "SNRPD3", "SNRPD2",
"PCGF6", "GSK3A", "SLC2A2", "ARCN1", "ANGPTL3", "COPB1", "COPB2", "STARD5",
"CYB5R4", "DMD", "FUS", "COPS6", "NOS3", "SFXN2", "CNTN5", "IQSEC1",
"CRADD", "STAC3", "COPZ1", "SULT1C4", "EFTUD2", "SLC35A1", "B4GALT2",
"THRSP", "NHP2L1", "ZNF224", "NXF1", "PDLIM3", "SAMM50", "MTFR1",
"SART1", "PCDHGA1", "GINS2", "MPG", "GJA3", "UBE2A", "ESAM", "CLK3",
"EIF4A2", "NUTF2", "INMT", "SCYL3", "XIAP", "TRIM28", "MPZL1", "LY6G6C",
"RBM14", "TPRX1", "ATP6V0B", "NUP107", "ABCD1", "TGS1", "RPS4X", "CRNKL1",
"YTHDC1", "PPAN", "PRRT1", "KRTCAP2", "GDPD5", "ZNF132", "WDR18", "ATP5F1",
"AMOTL1", "RPS16", "CCDC74B", "CCDC74A", "SF3B1", "SF3B3", "SF3B2",
"MYOCD", "MLKL", "PRSS8", "CACTIN", "EIF2S1", "ZNF16", "PGD", "SRP54",
"AQR", "DYNC1I1", "DCLRE1B", "CALCOCO2", "EVC2", "LRP1B", "ZNF552",
"COPG1", "EPRS", "COPA", "ATP6V1G1", "PIEZO1", "FCGR2A", "CPLX1",
"SNRPB", "BACE1", "ZNF154", "RAB29", "ATP6V0E2", "AHCY", "SLC1A3"
)
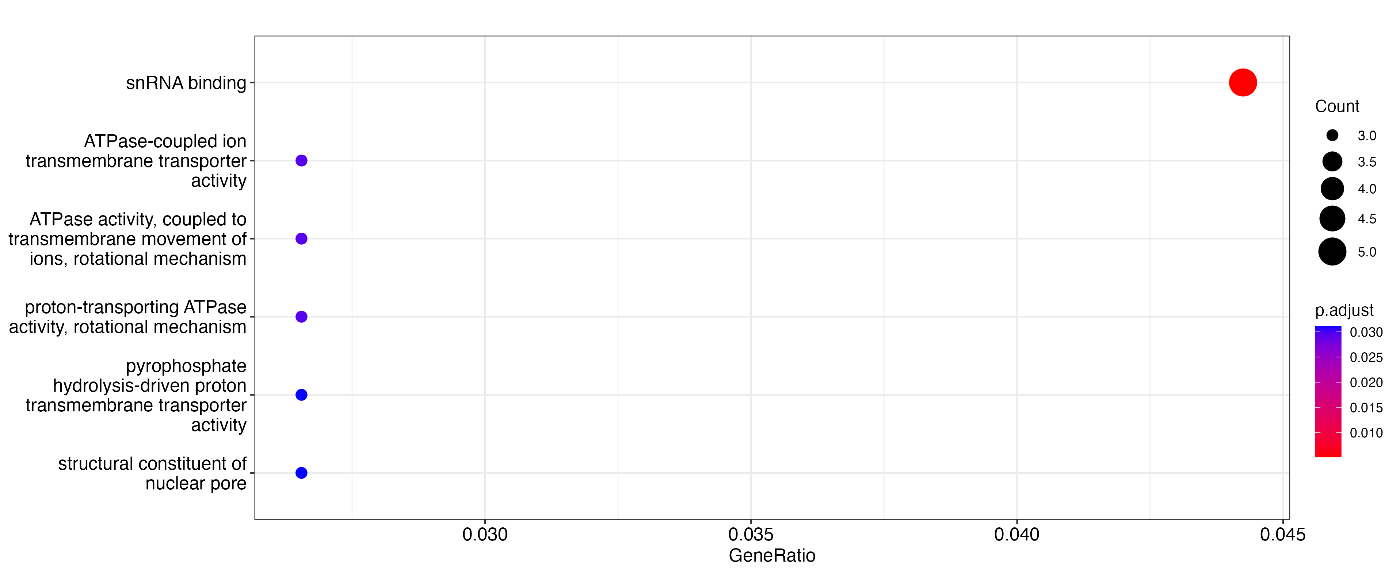
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
dotplot(result)
实际运行上述代码时,你将获得类似如下的图。默认情况下,图表是基于 GeneRatio(分析目标 ID 在类群中所占的比例)生成的。
代码内容解读如下:
library(clusterProfiler)
geneList <- c("UBL5","NDUFB8","CHMP2B"~~)
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
dotplot(result)
如果你想用自己的基因列表进行分析,只需更改 geneList 中的基因名。
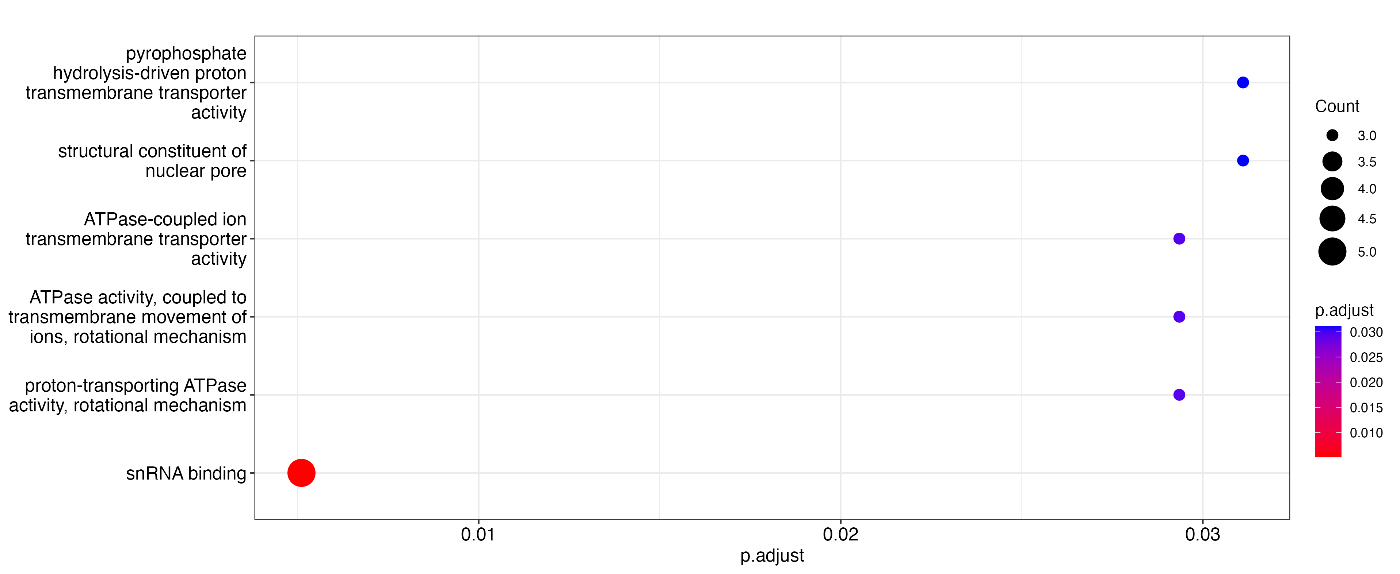
通过指定 dotplot 中的 x 参数,可以更改横轴。例如,将其更改为 p.adjust(多个检验执行后校正的 p 值):
dotplot(result, x = "p.adjust")
运行上述代码后,你将得到如下图所示的结果。
你还可以使用以下值来更改图的横轴。请尝试不同的值:
- ID: 属于集群的分析目标的 ID(例如,基因 ID 或 GO term ID 等)
- Description: 分析目标 ID 的描述
- BgRatio: 在整体背景集合中,分析目标 ID 所占的比例
- pvalue: 用于判断分析目标 ID 在集群内是否偶然集中的统计量 p 值
- qvalue: 用于控制错误发现率(FDR)的统计量 q 值
- geneID: 在集群内对应于分析目标 ID 的基因名
- Count: 集群内对应于分析目标 ID 的基因数量
以下是如何在 dotplot 中应用这些参数的示例:
dotplot(result, x = "Description")
使用 clusterProfiler 的其他可视化方法
使用 clusterProfiler,你还可以进行各种其他类型的可视化。请尝试不同的可视化方法,找到最能帮助你解释分析结果的图表。
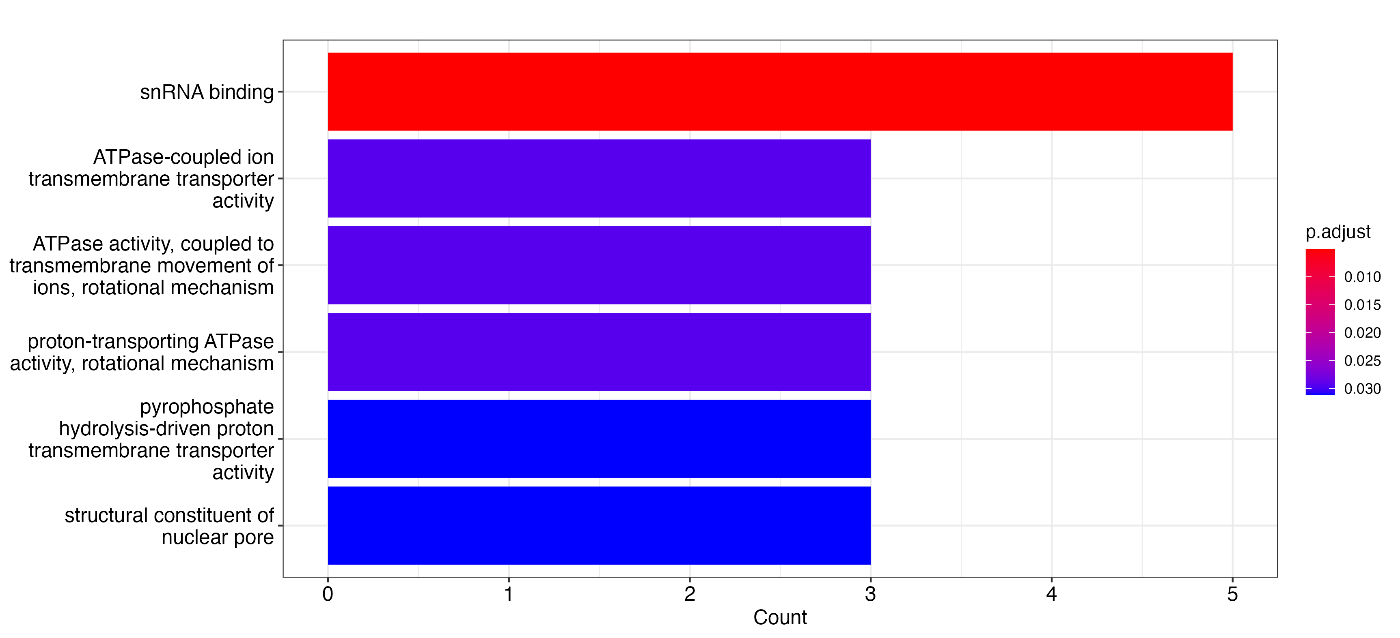
barplot¶
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
barplot(result)
ggupset¶
BiocManager::install("ggupset")
library(enrichplot)
library(ggupset)
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
upsetplot(result)

emapplot¶
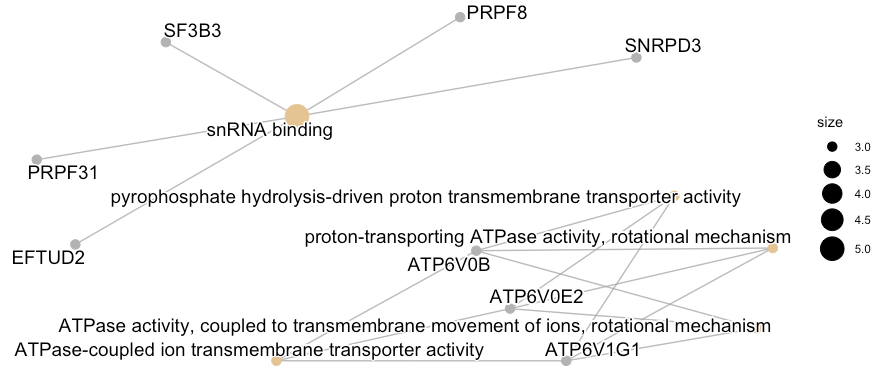
如果使用基因本体分析(如 Gene Ontology 或 KEGG Pathway)的结果,可以将其可视化为富集图。还可以绘制连接重叠基因组的网络图。
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
result2 <- pairwise_termsim(result)
emapplot(result2)
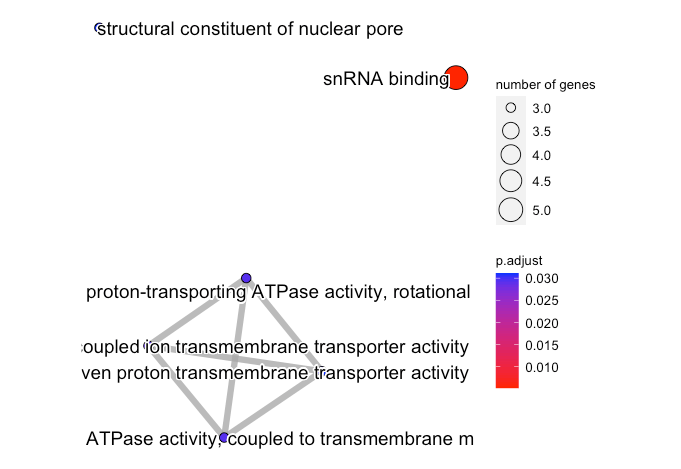
请注意,emapplotting 需要使用 pairwise_termsim 函数来计算两个或多个给定术语集之间的相似性。
pairwise_termsim 函数接收两个或多个具有共同注释项的列表,并根据注释项的共性为每个列表中的所有词对计算相似度得分。
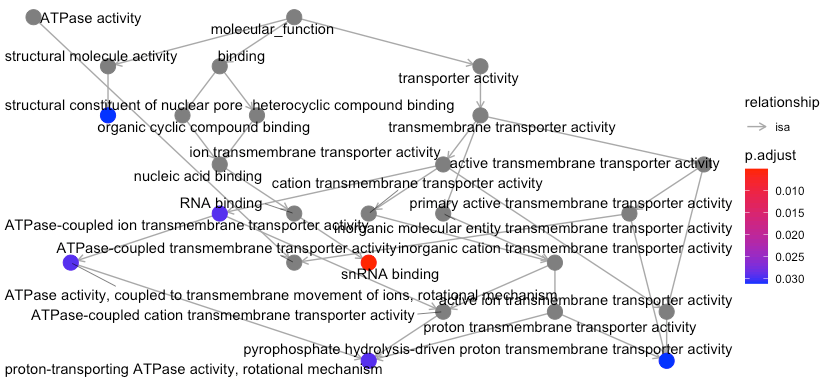
goplot¶
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
goplot(result)
cnetplot¶
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
cnetplot(result)