3.3 使用 DEseq2 提取 RNA seq 分析数据中的 DEG
DEseq2 是什么?¶
DESeq2 是一个 R 软件包,用于对 RNA 测序(RNA-seq)数据进行差异表达分析。RNA-seq 是一种量化转录组表达水平的技术,可用于研究不同条件和时间点下基因表达的变化。
DESeq2 的主要特点
- 正规化:考虑到样本间读取总数的差异,对基因的表达量进行正规化。
- 差异表达分析:统计评估两个或多个条件间基因表达的变化。
- 方差估计:通过共享基因间的方差,提高表达量变化的估计。
- 多重检验校正:为了同时评估大量基因,进行校正以减少假阳性风险。
DESeq2 差异表达分析
DESeq2 使用正规化因子(尺寸因子)考虑文库深度差异,对原始计数进行建模。接着,估计每个基因的方差,并对估计值进行缩小,以生成更准确的方差估计值,从而对计数进行建模。最后,DESeq2 拟合负二项分布模型,并使用 Wald 检验或似然比检验进行假设检验。
运行 DESeq2
本次 DESeq2 使用以下文件。请下载并放在桌面上:
- counts_data.csv
- sample_info.csv
需要加载以下库:
library(DESeq2)
library(tidyverse)
读取数据:
setwd("~/Desktop")
counts_data <- read.csv("counts_data.csv")
colData <- read.csv("sample_info.csv")
查看 counts_data 和 sample_info 的内容:
counts_data 是一个矩阵文件,行是基因名,列是样本名,每个单元格记录对应的读取计数。
> head(counts_data)
# SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517
# ENSG00000000003 679 448 873 408 1138 1047
# ENSG00000000005 0 0 0 0 0 0
# ENSG00000000419 467 515 621 365 587 799
# ENSG00000000457 260 211 263 164 245 331
# ENSG00000000460 60 55 40 35 78 63
# ENSG00000000938 0 0 2 0 1 0
# SRR1039520 SRR1039521
# ENSG00000000003 770 572
# ENSG00000000005 0 0
# ENSG00000000419 417 508
# ENSG00000000457 233 229
# ENSG00000000460 76 60
# ENSG00000000938 0 0
colData 提供了每个样本的实验变量或条件等元数据。
> head(colData)
# cellLine dexamethasone
# SRR1039508 N61311 untreated
# SRR1039509 N61311 treated
# SRR1039512 N052611 untreated
# SRR1039513 N052611 treated
# SRR1039516 N080611 untreated
# SRR1039517 N080611 treated
colData 示例输出:
- cellLine: 表示样本的细胞株,用于区分不同细胞株的样本。
- dexamethasone: 表示样本是否经过地塞米松处理。
基于这些数据,DESeq2 会按照 design = ~ dexamethasone 指定评估地塞米松处理对基因表达的影响。
使用 DESeqDataSetFromMatrix 函数创建 DESeq2 分析的数据集对象(DESeqDataSet):
dds <- DESeqDataSetFromMatrix(
countData = counts_data,
colData = colData,
design = ~dexamethasone
)
design 是解析的设计公式,指定要基于哪些变量构建统计模型。在此例中,design 指定为 dexamethasone(地塞米松处理的有无)以研究其对基因表达的影响。
在执行 DESeq2 前进行预过滤步骤。总读取数不少于 10 的基因会被保留:
keep <- rowSums(counts(dds)) >= 10
dds <- dds[keep, ]
执行 DESeq2 并保存结果:
dds <- DESeq(dds)
res <- results(dds)
write.csv(res, "deseq2_results.csv")
查看 DESeq2 结果
查看 res:
> res
# log2 fold change (MLE): dexamethasone untreated vs treated
# Wald test p-value: dexamethasone untreated vs treated
# DataFrame with 22369 rows and 6 columns
# baseMean log2FoldChange lfcSE stat pvalue padj
# <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
# ENSG00000000003 708.6022 0.3788474 0.173165 2.187781 0.0286856 0.138465
# ENSG00000000419 520.2979 -0.2037622 0.100756 -2.022329 0.0431424 0.183141
# ENSG00000000457 237.1630 -0.0340364 0.126493 -0.269078 0.7878699 0.929861
# ENSG00000000460 57.9326 0.1171825 0.301588 0.388551 0.6976080 0.894183
# ENSG00000000971 5817.3529 -0.4409590 0.258783 -1.703975 0.0883858 0.297174
# ... ... ... ... ... ... ...
# ENSG00000273483 2.68957 -0.600436 1.084493 -0.553656 0.5798145 NA
# ENSG00000273485 1.28645 -0.194034 1.346631 -0.144089 0.8854304 NA
# ENSG00000273486 15.45254 0.113349 0.426061 0.266040 0.7902086 0.930772
# ENSG00000273487 8.16323 -1.017777 0.575826 -1.767508 0.0771432 0.271676
# ENSG00000273488 8.58448 -0.218082 0.570756 -0.382094 0.7023919 0.896539
列的含义:
- baseMean: 基因的平均计数值。
- log2FoldChange: 对数 2 倍变化值。
- lfcSE: 对数 2 倍变化的标准误差。
- stat: Wald 统计量。
- pvalue: Wald 检验的 p 值。
- padj: 校正后的 p 值。
查看 res 的摘要:
> summary(res)
# out of 22369 with nonzero total read count
# adjusted p-value < 0.1
# LFC > 0 (up) : 1502, 6.7%
# LFC < 0 (down) : 1884, 8.4%
# outliers [1] : 51, 0.23%
# low counts [2] : 3903, 17%
# (mean count < 4)
# [1] see 'cooksCutoff' argument of ?results
# [2] see 'independentFiltering' argument of ?results
summary(res) 的输出解读:
- 22369 个非零总读取计数的基因。
- 调整后的 p 值< 0.1 的基因信息。
- LFC > 0(上调): 1502 个基因(占 6.7%)。
- LFC < 0(下调): 1884 个基因(占 8.4%)。
- outliers(离群值): 51 个基因(占 0.23%)。
- low counts(低计数): 3903 个基因(占 17%)。
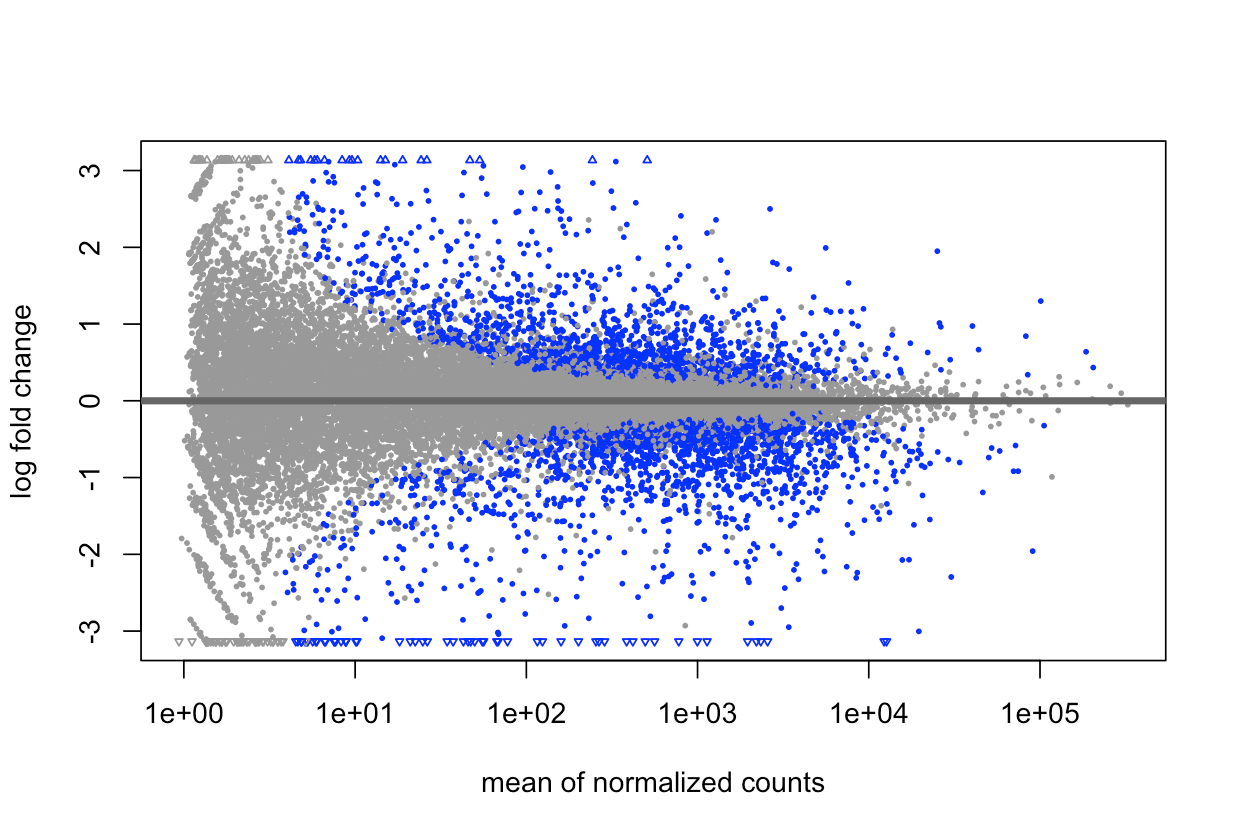
最后,使用 MAplot 可视化变化基因:
plotMA(res)
成功后会显示如下图表,蓝色点表示校正后 p 值小于指定阈值(默认为 0.1)的统计显著差异表达基因。