3.3 Data integration¶
动机¶
在大多数scRNA-seq数据分析中,一个核心挑战是批次效应。批次效应是指由于将细胞分组或分批处理而导致的测量表达水平的变化。例如,如果两个实验室从同一队列中取样,但样本的解离方式不同,就可能产生批次效应。如果实验室A优化了其解离协议,在解离样本细胞时尽量减少对细胞的压力,而实验室B则没有这样做,那么即使原始组织中的细胞具有相同的特征,来自B组的数据中的细胞可能会表达更多的与压力相关的基因(如JUN、JUNB、FOS等,见{cite}Van_den_Brink2017-si)。总的来说,批次效应的来源是多种多样且难以确定的。一些批次效应源可能是技术性的,例如样本处理、实验协议或测序深度的差异,但生物效应(如供体差异、组织或取样位置)也常常被解释为批次效应{cite}Luecken2021-jo。是否应将生物因素视为批次效应,取决于实验设计和所提出的问题。消除批次效应对于进行联合分析至关重要,这样可以专注于在不同批次的数据中找到共同结构,并使我们能够跨数据集进行查询。通常只有在消除这些效应后,才能识别出先前被批次差异掩盖的稀有细胞群。跨数据集进行查询使我们能够提出单独分析个体数据集无法回答的问题,例如_哪些细胞类型表达SARS-CoV-2入口因子,这种表达在个体之间有何不同?_ {cite}Muus2021-ti。
在去除组学数据中的批次效应时,必须做出两个核心选择:(1) 方法和参数设定,以及 (2) 批次协变量。由于批次效应可能在不同层次的细胞分组之间产生(如样本、供体、数据集等),批次协变量的选择决定了应保留哪个层次的变异,去除哪个层次的变异。批次分辨率越细,去除的效应越多。然而,细致的批次变异也更可能与具有生物学意义的信号混淆。例如,样本通常来自不同的个体或组织的不同位置。这些效应可能具有研究意义。因此,批次协变量的选择将取决于你的整合任务的目标。你是想看到个体之间的差异,还是专注于共同的细胞类型变异?一种基于定量分析的批次协变量选择方法在最近构建人类肺集成图谱的努力中首次提出,其中利用不同技术协变量的方差来进行选择{cite}Sikkema2022-tk。
整合模型的类型¶
去除scRNA-seq批次效应的方法通常包括以下(最多)三个步骤:
- 降维
- 建模并去除批次效应
- 投影回高维空间
虽然建模并去除批次效应(步骤2)是任何批次去除方法的核心部分,但许多方法首先将数据投影到低维空间(步骤1)以提高信噪比(见{ref}降维章节 <pre-processing:dimensionality-reduction>),并在该空间中进行批次校正以获得更好的性能(见{cite}Luecken2021-jo)。在第三步中,方法可能会在去除拟合的批次效应后,将数据投影回原始的高维特征空间,以输出一个经过批次校正的基因表达矩阵。
去除批次效应的方法在上述三个步骤中可能有所不同。它们可能使用各种线性或非线性降维方法、线性或非线性批次效应模型,并可能输出不同格式的批次校正数据。总体上,我们可以将批次效应去除方法分为四类。按照其发展的顺序,这些方法分别是全局模型、线性嵌入模型、基于图的方法和深度学习方法(图I1)。
全局模型 起源于整体转录组学,将批次效应建模为在所有细胞中一致的(加性和/或乘性)效应。一个常见的例子是ComBat {cite}Johnson2007-sl。
线性嵌入模型 是最早的单细胞特定批次去除方法。这些方法通常使用奇异值分解(SVD)的变体来嵌入数据,然后在嵌入中寻找跨批次的相似细胞的局部邻域,并在局部自适应(非线性)的方式下校正批次效应。方法通常使用SVD负载将数据投影回基因表达空间,但也可能仅输出校正的嵌入。这是最常见的一组方法,著名的例子包括开创性的相互最近邻(MNN)方法 {cite}Haghverdi2018-bd(不执行任何降维),Seurat集成 {cite}Butler2018-js,Stuart2019-lq,Scanorama {cite}Hie2019-er,FastMNN {cite}Haghverdi2018-bd,以及Harmony {cite}Korsunsky2019-ex。
基于图的方法 通常是运行速度最快的方法。这些方法使用最近邻图表示每个批次的数据。通过强制连接来自不同批次的细胞并允许通过修剪强制边缘来考虑细胞类型组成的差异,从而校正批次效应。最著名的例子是批次平衡_k_最近邻(BBKNN)方法 {cite}Polanski2019-zy。
深度学习(DL)方法 是最新和最复杂的批次效应去除方法,通常需要最多的数据以获得良好的性能。大多数深度学习集成方法基于自编码器网络,或在条件变分自编码器(CVAE)中基于批次协变量进行降维,或在嵌入空间中拟合局部线性校正。著名的DL方法包括scVI {cite}Lopez2018-au,scANVI {cite}Xu2021-dh,以及scGen {cite}Lotfollahi2019-cy。
有些方法可以使用细胞身份标签为方法提供参考,以便不会将生物变异视为批次效应去除。由于批次效应去除通常是预处理任务,因此此类方法在许多集成场景中可能不适用,因为在此阶段通常还没有标签。
有关批次效应去除方法的更详细概述,请参见{cite}Argelaguet2021-pb和{cite}Luecken2021-jo。
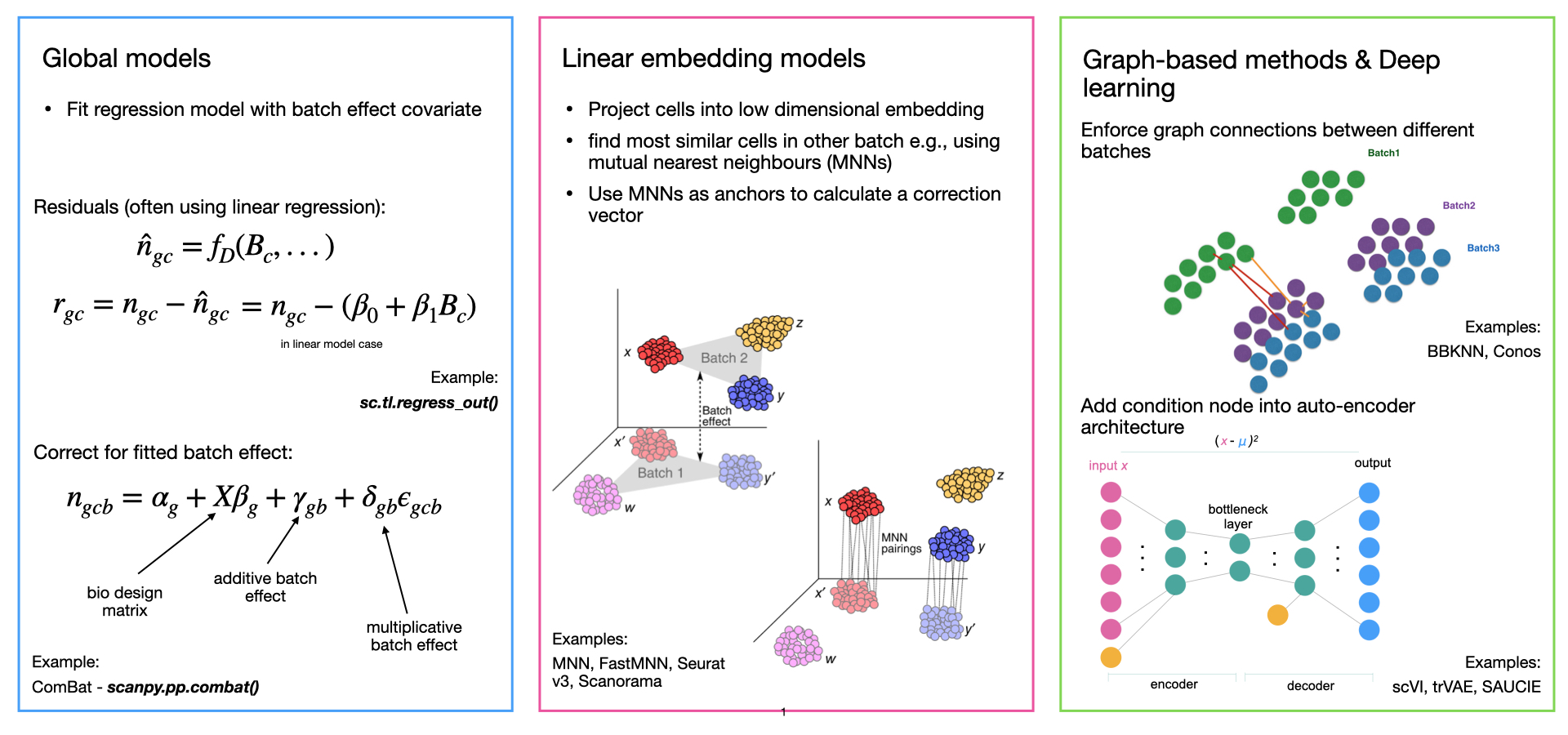
图I1:不同类型的整合方法概述及示例。
批次效应去除的复杂性¶
scRNA-seq数据中的批次效应去除以前被分为两个子任务:批次校正和数据整合 {cite}Luecken2019-og。这些子任务在需要去除的批次效应的复杂性方面有所不同。批次校正方法处理同一实验中样本之间的批次效应,其中细胞身份组成是一致的,效应通常是准线性的。相反,数据整合方法处理数据集之间的复杂、常常是嵌套的批次效应,这些数据集可能使用不同的协议生成,且不同批次之间可能不共享细胞身份。虽然我们在这里使用这种区分,但需要注意,这些术语在日常使用中经常被互换使用。鉴于复杂性的差异,不同方法在这两个子任务中被评估为最佳也是不足为奇的。
数据整合方法的比较¶
先前有多项基准测试评估了批次校正和数据整合方法的性能。在去除批次效应时,方法可能会过度校正,除了批次效应外还会去除有意义的生物变异。因此,评估整合性能时,考虑批次效应的去除和生物变异的保留都很重要。
k-最近邻批次效应测试(kBET)是第一个量化scRNA-seq数据批次校正的指标 {cite}Buttner2019-yl。利用kBET,作者发现ComBat在主要比较全局模型时,批次校正效果优于其他方法。在此基础上,最近的两项基准测试 {cite}Tran2020-ia 和 {cite}Chazarra-Gil2021-ri 还对批次较少或生物复杂性低的任务进行了线性嵌入和深度学习模型的基准测试。这些研究发现,线性嵌入模型Seurat {cite}Butler2018-js,Stuart2019-lq 和Harmony {cite}Korsunsky2019-ex 在简单批次校正任务中表现良好。
对复杂整合任务进行基准测试由于数据集的规模和数量以及场景的多样性而面临额外的挑战。最近,一项大型研究使用14个指标对16种方法在五个RNA任务和两个模拟上的整合方法类别进行了基准测试 {cite}Luecken2021-jo。虽然每个任务的最佳方法不同,但使用细胞类型标签的方法在各任务中表现更好。此外,深度学习方法scANVI(带标签)、scVI和scGen(带标签)以及线性嵌入模型Scanorama在复杂任务中表现最佳,而Harmony在不太复杂的任务中表现良好。一项专门为整合视网膜数据集以构建_眼部巨型图谱_的类似基准测试也发现scVI优于其他方法 {cite}Swamy2021-uy。
选择整合方法¶
尽管整合方法已经进行了广泛的基准测试,但并不存在适用于所有场景的最佳方法。像scIB和batchbench这样的整合性能指标包和评估管道可以用来评估您自己数据的整合性能。然而,许多指标(特别是那些测量生物变异保留的指标)需要真实的细胞身份标签。参数优化可以调整许多方法以适用于特定任务,但总体而言,可以说Harmony和Seurat在简单的批次校正任务中表现稳定,而scVI、scGen、scANVI和Scanorama在更复杂的数据整合任务中表现良好。在选择方法时,建议首先考虑这些选项。此外,整合方法的选择可能会受到所需输出数据格式的指导(例如,您是需要校正后的基因表达数据,还是整合的嵌入就足够了?)。在选择之前,最好测试多种方法,并基于定量的成功定义评估输出。有关数据整合方法选择的详细指南可以在{cite}Luecken2021-jo中找到。
在本章的其余部分,我们将展示一些表现最佳的方法,并简要演示如何评估整合性能。
# Python packages
import scanpy as sc
import scvi
import bbknn
import scib
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import matplotlib
# R interface
from rpy2.robjects import pandas2ri
from rpy2.robjects import r
import rpy2.rinterface_lib.callbacks
import anndata2ri
pandas2ri.activate()
anndata2ri.activate()
%load_ext rpy2.ipython
Global seed set to 0
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/pytorch_lightning/utilities/warnings.py:53: LightningDeprecationWarning: pytorch_lightning.utilities.warnings.rank_zero_deprecation has been deprecated in v1.6 and will be removed in v1.8. Use the equivalent function from the pytorch_lightning.utilities.rank_zero module instead.
new_rank_zero_deprecation(
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/pytorch_lightning/utilities/warnings.py:58: LightningDeprecationWarning: The `pytorch_lightning.loggers.base.rank_zero_experiment` is deprecated in v1.7 and will be removed in v1.9. Please use `pytorch_lightning.loggers.logger.rank_zero_experiment` instead.
return new_rank_zero_deprecation(*args, **kwargs)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/rpy2/robjects/pandas2ri.py:263: DeprecationWarning: The global conversion available with activate() is deprecated and will be removed in the next major release. Use a local converter.
warnings.warn('The global conversion available with activate() '
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/rpy2/robjects/numpy2ri.py:205: DeprecationWarning: The global conversion available with activate() is deprecated and will be removed in the next major release. Use a local converter.
warnings.warn('The global conversion available with activate() '
%%R
# R packages
library(Seurat)
WARNING: The R package "reticulate" does not
consider that it could be called from a Python process. This
results in a quasi-obligatory segfault when rpy2 is evaluating
R code using it. On the hand, rpy2 is accounting for the
fact that it might already be running embedded in a Python
process. This is why:
- Python -> rpy2 -> R -> reticulate: crashes
- R -> reticulate -> Python -> rpy2: works
The issue with reticulate is tracked here:
https://github.com/rstudio/reticulate/issues/208
数据集¶
我们将用来演示数据整合的数据集包含几个骨髓单个核细胞样本。这些样本最初是为单细胞分析开放问题NeurIPS 2021竞赛创建的 {cite}Luecken2022-kv, Lance2022-yy。该数据集使用10x Multiome协议测量同一细胞中的RNA表达(scRNA-seq)和染色质可及性(scATAC-seq)。我们这里使用的数据版本已经预处理过,以去除低质量细胞。
!wget https://figshare.com/ndownloader/files/45452260 -O openproblems_bmmc_multiome_genes_filtered.h5ad
adata_raw = sc.read_h5ad("openproblems_bmmc_multiome_genes_filtered.h5ad")
adata_raw.layers["logcounts"] = adata_raw.X
adata_raw
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/h5ad.py:238: OldFormatWarning: Element '/layers' was written without encoding metadata.
d[k] = read_elem(f[k])
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/GEX_pct_counts_mt' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/GEX_n_counts' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/GEX_n_genes' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/GEX_size_factors' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/GEX_phase' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/GEX_phase' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/ATAC_nCount_peaks' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/ATAC_atac_fragments' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/ATAC_reads_in_peaks_frac' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/ATAC_blacklist_fraction' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/ATAC_nucleosome_signal' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/cell_type' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/cell_type' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/batch' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/batch' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/ATAC_pseudotime_order' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/GEX_pseudotime_order' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/Samplename' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/Samplename' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/Site' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/Site' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/DonorNumber' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/DonorNumber' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/Modality' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/Modality' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/VendorLot' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/DonorID' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/DonorAge' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/DonorBMI' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/DonorBloodType' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/DonorBloodType' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/DonorRace' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/DonorRace' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/Ethnicity' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/Ethnicity' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/DonorGender' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/DonorGender' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/QCMeds' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/QCMeds' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/obs/__categories/DonorSmoker' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/obs/DonorSmoker' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/obs/_index' was written without encoding metadata.
return read_elem(dataset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/h5ad.py:238: OldFormatWarning: Element '/obsm' was written without encoding metadata.
d[k] = read_elem(f[k])
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:92: OldFormatWarning: Element '/obsm/ATAC_lsi_full' was written without encoding metadata.
return {k: read_elem(v) for k, v in elem.items()}
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:92: OldFormatWarning: Element '/obsm/ATAC_lsi_red' was written without encoding metadata.
return {k: read_elem(v) for k, v in elem.items()}
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:92: OldFormatWarning: Element '/obsm/ATAC_umap' was written without encoding metadata.
return {k: read_elem(v) for k, v in elem.items()}
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:92: OldFormatWarning: Element '/obsm/GEX_X_pca' was written without encoding metadata.
return {k: read_elem(v) for k, v in elem.items()}
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:92: OldFormatWarning: Element '/obsm/GEX_X_umap' was written without encoding metadata.
return {k: read_elem(v) for k, v in elem.items()}
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/h5ad.py:238: OldFormatWarning: Element '/uns' was written without encoding metadata.
d[k] = read_elem(f[k])
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:92: OldFormatWarning: Element '/uns/ATAC_gene_activity_var_names' was written without encoding metadata.
return {k: read_elem(v) for k, v in elem.items()}
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:92: OldFormatWarning: Element '/uns/dataset_id' was written without encoding metadata.
return {k: read_elem(v) for k, v in elem.items()}
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:92: OldFormatWarning: Element '/uns/genome' was written without encoding metadata.
return {k: read_elem(v) for k, v in elem.items()}
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:92: OldFormatWarning: Element '/uns/organism' was written without encoding metadata.
return {k: read_elem(v) for k, v in elem.items()}
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/var/__categories/feature_types' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/var/feature_types' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:584: OldFormatWarning: Element '/var/__categories/gene_id' was written without encoding metadata.
categories = read_elem(categories_dset)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:587: OldFormatWarning: Element '/var/gene_id' was written without encoding metadata.
read_elem(dataset), categories, ordered=ordered
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata/_io/specs/methods.py:590: OldFormatWarning: Element '/var/_index' was written without encoding metadata.
return read_elem(dataset)
AnnData object with n_obs × n_vars = 69249 × 129921
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker'
var: 'feature_types', 'gene_id'
uns: 'ATAC_gene_activity_var_names', 'dataset_id', 'genome', 'organism'
obsm: 'ATAC_gene_activity', 'ATAC_lsi_full', 'ATAC_lsi_red', 'ATAC_umap', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts', 'logcounts'
整个数据集包含69,249个细胞和129,921个特征的测量值。表达矩阵有两个版本,counts 包含原始计数值,logcounts 包含标准化的对数计数值(这些值也存储在 adata.X 中)。
obs 插槽包含若干变量,其中一些是在预处理中计算的(用于质量控制),另一些则包含样本的元数据。我们在这里感兴趣的变量是:
cell_type- 每个细胞的注释标签batch- 每个细胞的测序批次
在实际分析中,考虑更多的变量是很重要的,但为了简化,这里我们只看这些变量。
我们定义变量来保存这些名称,以明确我们在代码中如何使用它们。这也有助于重现性,因为如果我们出于任何原因决定更改其中一个变量,我们可以确保它在整个笔记本中都已更改。
label_key = "cell_type"
batch_key = "batch"
{admonition}
As mentioned above, deciding what to use as a "batch" for data integration is one of the central choices when integrating your data. The most common approach is to define each sample as a batch (as we have here) which generally produces the strongest batch correction. However, samples are usually confounded with biological factors that you may want to preserve. For example, imagine an experiment that took samples from two locations in a tissue. If samples are considered as batches then data integration methods will attempt to remove differences between them and therefore differences between the locations. In this case, it may be more appropriate to use the donor as the batch to remove differences between individuals but not between locations. The planned analysis should also be considered. In cases where you are integrating many datasets and want to capture differences between individuals, the dataset may be a useful batch covariate. In our example, it may be better to have consistent cell type labels for the two locations and then test for differences between them than to have separate clusters for each location which need to be annotated separately and then matched.
The issue of confounding between samples and biological factors can be mitigated through careful experimental design that minimizes the batch effect overall by using multiplexing techniques that allow biological samples to be combined into a single sequencing sample. However, this is not always possible and requires both extra processing in the lab and as well as extra computational steps.
Let's have a look at the different batches and how many cells we have for each.
adata_raw.obs[batch_key].value_counts()
s4d8 9876 s4d1 8023 s3d10 6781 s1d2 6740 s1d1 6224 s2d4 6111 s2d5 4895 s3d3 4325 s4d9 4325 s1d3 4279 s2d1 4220 s3d7 1771 s3d6 1679 Name: batch, dtype: int64
数据集中有13个不同的批次。在这次实验中,从一组供体中采集了多个样本,并在不同的设施中进行了测序,因此这里的名称是样本编号(例如 "s1")和供体(例如 "d2")的组合。为了简化和减少计算时间,我们将选择三个样本来使用。
keep_batches = ["s1d3", "s2d1", "s3d7"]
adata = adata_raw[adata_raw.obs[batch_key].isin(keep_batches)].copy()
adata
AnnData object with n_obs × n_vars = 10270 × 129921
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker'
var: 'feature_types', 'gene_id'
uns: 'ATAC_gene_activity_var_names', 'dataset_id', 'genome', 'organism'
obsm: 'ATAC_gene_activity', 'ATAC_lsi_full', 'ATAC_lsi_red', 'ATAC_umap', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts', 'logcounts'
在子集中选择这些批次后,我们剩下10,270个细胞。
对于存储在var中的特征,我们有两个注释:
feature_types- 每个特征的类型(RNA或ATAC)gene_id- 与每个特征相关联的基因
让我们来看一下特征类型。
adata.var["feature_types"].value_counts()
ATAC 116490 GEX 13431 Name: feature_types, dtype: int64
我们可以看到有超过100,000个ATAC特征,但只有大约13,000个基因表达("GEX")特征。多模态整合是一个复杂的问题,将在{ref}多模态整合章节<multimodal-integration:advanced-integration>中详细描述,因此现在我们将仅选择基因表达特征。我们还进行简单的过滤,以确保没有计数为零的特征(这是必要的,因为通过选择一个样本子集,我们可能已经移除了表达特定特征的所有细胞)。
adata = adata[:, adata.var["feature_types"] == "GEX"].copy()
sc.pp.filter_genes(adata, min_cells=1)
adata
AnnData object with n_obs × n_vars = 10270 × 13431
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker'
var: 'feature_types', 'gene_id', 'n_cells'
uns: 'ATAC_gene_activity_var_names', 'dataset_id', 'genome', 'organism'
obsm: 'ATAC_gene_activity', 'ATAC_lsi_full', 'ATAC_lsi_red', 'ATAC_umap', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts', 'logcounts'
由于进行了子集选择,我们还需要重新标准化数据。这里我们仅通过每个细胞的总计数使用全局缩放进行标准化。
adata.X = adata.layers["counts"].copy()
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
adata.layers["logcounts"] = adata.X.copy()
我们将使用这个数据集来演示整合。
大多数整合方法需要一个包含所有样本和一个批次变量的单一对象(如我们这里所示)。如果你有单独的对象分别表示每个样本,可以使用anndata的concat()函数将它们连接起来。更多详细信息请参见合并教程。其他生态系统中也有类似的功能。
!!! note {整合UMI和全长数据}
整合来自UMI和全长协议的样本可能会带来额外的挑战。这是因为全长协议受到基因长度偏差的影响(较长的基因会更高表达),而UMI数据则不会 {cite}Phipson2017-qt。因此,一般建议在尝试整合之前,将全长样本的计数转换为一种校正基因长度的单位(如每百万转录本数(TPM){cite}Wagner2012-qf)。然而,如果所有整合的样本都使用全长协议,则不需要进行此转换。
未整合的数据¶
在进行任何整合之前,建议先查看原始数据。这可以大致了解批次效应的大小及其可能的原因(从而确定哪些变量应作为批次标签考虑)。对于某些实验,如果样本已经有重叠,甚至可能表明不需要进行整合。这在单个实验室的鼠类或细胞系研究中并不少见,因为批次效应的许多变量可以得到控制(即批次校正设置)。
我们将进行高度可变基因(HVG)选择、PCA和UMAP降维,如前几章所述。
sc.pp.highly_variable_genes(adata)
sc.tl.pca(adata)
sc.pp.neighbors(adata)
sc.tl.umap(adata)
adata
AnnData object with n_obs × n_vars = 10270 × 13431
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker'
var: 'feature_types', 'gene_id', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'ATAC_gene_activity_var_names', 'dataset_id', 'genome', 'organism', 'log1p', 'hvg', 'pca', 'neighbors', 'umap'
obsm: 'ATAC_gene_activity', 'ATAC_lsi_full', 'ATAC_lsi_red', 'ATAC_umap', 'GEX_X_pca', 'GEX_X_umap', 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts', 'logcounts'
obsp: 'distances', 'connectivities'
这为我们的AnnData对象添加了几个新项。var插槽现在包括均值、离散度和选择的可变基因。在obsp插槽中,我们有KNN图的距离和连接性,在obsm中是PCA和UMAP嵌入。
让我们绘制UMAP图,用细胞身份和批次标签对点进行着色。如果数据集尚未被标记(这通常是实际情况),我们只能考虑批次标签。
adata.uns[batch_key + "_colors"] = [
"#1b9e77",
"#d95f02",
"#7570b3",
] # Set custom colours for batches
sc.pl.umap(adata, color=[label_key, batch_key], wspace=1)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:163: PendingDeprecationWarning: The get_cmap function will be deprecated in a future version. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap(obj)`` instead. cmap = copy(get_cmap(cmap)) /Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored cax = scatter( /Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored cax = scatter(

通常,当查看这些图时,您会看到批次之间有明显的分离。在这种情况下,我们看到的是更微妙的情况,虽然来自相同标签的细胞通常彼此靠近,但批次之间存在一定的偏移。如果我们使用这些原始数据进行聚类分析,可能会得到一些仅包含单一批次的聚类,这在注释阶段将难以解释。同时,我们也可能忽略一些稀有细胞类型,因为它们在任何单个样本中都不够常见,无法形成自己的聚类。虽然UMAP图通常可以显示批次效应,但在考虑这些二维表示时,重要的是不要过度解读它们。对于实际分析,您应通过其他方式确认整合效果,例如检查标记基因的分布。在下面的“评估您自己的整合”部分中,我们将讨论量化整合质量的指标。
现在我们已经确认需要纠正批次效应,我们可以继续讨论不同的整合方法。如果批次完美重叠,或者我们可以在不进行校正的情况下发现有意义的细胞聚类,那么就没有必要进行整合。
Batch-aware feature selection¶
如{ref}前几章所示<pre-processing:feature-selection>,我们通常选择一部分基因进行分析,以减少噪声和处理时间。当我们有多个样本时,也需要进行相同的操作,但是重要的是要以批次感知的方式进行基因选择。这是因为在整个数据集中可变的基因可能捕捉的是批次效应,而不是我们感兴趣的生物信号。基因选择还应有助于识别稀有细胞类型,例如,如果一种细胞类型仅存在于一个样本中,那么它的标记基因可能在所有样本中都不具有变异性,但在那个样本中应该是有的。
我们可以通过在scanpy的highly_variable_genes()函数中设置batch_key参数来执行批次感知的高度可变基因选择。scanpy将分别为每个批次计算HVGs,并通过选择在最多批次中高度可变的基因来合并结果。我们在这里使用scanpy函数是因为它内置了这种批次感知功能。对于其他方法,我们需要分别对每个批次运行,然后手动合并结果。
sc.pp.highly_variable_genes(
adata, n_top_genes=2000, flavor="cell_ranger", batch_key=batch_key
)
adata
adata.var
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/preprocessing/_highly_variable_genes.py:478: FutureWarning: The frame.append method is deprecated and will be removed from pandas in a future version. Use pandas.concat instead. hvg = hvg.append(missing_hvg, ignore_index=True) /Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/preprocessing/_highly_variable_genes.py:478: FutureWarning: The frame.append method is deprecated and will be removed from pandas in a future version. Use pandas.concat instead. hvg = hvg.append(missing_hvg, ignore_index=True) /Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/preprocessing/_highly_variable_genes.py:478: FutureWarning: The frame.append method is deprecated and will be removed from pandas in a future version. Use pandas.concat instead. hvg = hvg.append(missing_hvg, ignore_index=True)
| feature_types | gene_id | n_cells | highly_variable | means | dispersions | dispersions_norm | highly_variable_nbatches | highly_variable_intersection | |
|---|---|---|---|---|---|---|---|---|---|
| AL627309.5 | GEX | ENSG00000241860 | 112 | False | 0.006533 | 0.775289 | 0.357973 | 1 | False |
| LINC01409 | GEX | ENSG00000237491 | 422 | False | 0.024462 | 0.716935 | -0.126119 | 0 | False |
| LINC01128 | GEX | ENSG00000228794 | 569 | False | 0.030714 | 0.709340 | -0.296701 | 0 | False |
| NOC2L | GEX | ENSG00000188976 | 675 | False | 0.037059 | 0.704363 | -0.494025 | 0 | False |
| KLHL17 | GEX | ENSG00000187961 | 88 | False | 0.005295 | 0.721757 | -0.028456 | 0 | False |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| MT-ND5 | GEX | ENSG00000198786 | 4056 | False | 0.269375 | 0.645837 | -0.457491 | 0 | False |
| MT-ND6 | GEX | ENSG00000198695 | 1102 | False | 0.051506 | 0.697710 | -0.248421 | 0 | False |
| MT-CYB | GEX | ENSG00000198727 | 5647 | False | 0.520368 | 0.613233 | -0.441362 | 0 | False |
| AL592183.1 | GEX | ENSG00000273748 | 732 | False | 0.047486 | 0.753417 | 0.413699 | 0 | False |
| AC240274.1 | GEX | ENSG00000271254 | 43 | False | 0.001871 | 0.413772 | -0.386635 | 0 | False |
13431 rows × 9 columns
我们可以看到,现在var中有一些新增的列:
highly_variable_nbatches- 每个基因在多少个批次中被发现是高度可变的highly_variable_intersection- 每个基因是否在每个批次中都是高度可变的highly_variable- 在合并每个批次的结果后,每个基因是否被选择为高度可变的
让我们检查每个基因在多少个批次中是可变的:
n_batches = adata.var["highly_variable_nbatches"].value_counts()
ax = n_batches.plot(kind="bar")
n_batches
0 9931 1 1824 2 852 3 824 Name: highly_variable_nbatches, dtype: int64

首先,我们注意到大多数基因并不是高度可变的。这通常是这种情况,但这也取决于我们试图整合的样本之间的差异。随着我们增加更多的样本,重叠部分会减少,只有相对较少的基因在所有三个批次中都是高度可变的。通过选择前2000个基因,我们选择了在两个或三个批次中存在的所有HVGs,以及大多数在一个批次中存在的HVGs。
!!! note {使用多少基因?}
这个问题没有明确的答案。我们在下文中使用的**scvi-tools**包的作者建议使用1000到10000个基因,但具体数量取决于上下文,包括数据集的复杂性和批次数量。以前的一篇最佳实践论文中的调查 {cite}`Luecken2019-og` 表明人们在标准分析中通常使用1000到6000个HVGs。虽然选择较少的基因可以帮助去除批次效应 {cite}`Luecken2021-jo`(最高度可变的基因通常仅描述主要的生物变异),但我们建议选择稍多的基因,而不是选择太少并冒去除对稀有细胞类型或感兴趣的通路重要的基因的风险。然而,需要注意的是,更多的基因也会增加运行整合方法所需的时间。我们将创建一个仅包含所选基因的对象以用于整合。
adata_hvg = adata[:, adata.var["highly_variable"]].copy()
adata_hvg
AnnData object with n_obs × n_vars = 10270 × 2000
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker'
var: 'feature_types', 'gene_id', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'ATAC_gene_activity_var_names', 'dataset_id', 'genome', 'organism', 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'batch_colors', 'cell_type_colors'
obsm: 'ATAC_gene_activity', 'ATAC_lsi_full', 'ATAC_lsi_red', 'ATAC_umap', 'GEX_X_pca', 'GEX_X_umap', 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts', 'logcounts'
obsp: 'distances', 'connectivities'
基于变分自编码器(VAE)的整合¶
我们将使用的第一个整合方法是scVI(单细胞变分推断),这是一种基于条件变分自编码器的方法 {cite}Lopez2018-au,可在scvi-tools包中找到 {cite}Gayoso2022-ar。变分自编码器(VAE)是一种试图降低数据集维度的人工神经网络。条件部分指的是将这一降维过程条件化于特定协变量(在这种情况下是批次),使得该协变量不影响低维表示。在基准测试研究中,scVI在多个数据集中表现良好,能够在批次校正和平衡保留生物变异性之间取得良好平衡 {cite}Luecken2021-jo。scVI直接对原始计数进行建模,因此提供给它的是计数矩阵而不是标准化表达矩阵非常重要。
首先,让我们复制一份数据集用于此整合。通常不需要这样做,但由于我们将演示多种整合方法,复制一份数据集使得更容易展示每种方法添加了什么。
adata_scvi = adata_hvg.copy()
Data preparation¶
使用scVI的第一步是准备我们的AnnData对象。这一步会存储一些scVI所需的信息,例如使用哪个表达矩阵和批次 key 是什么。
scvi.model.SCVI.setup_anndata(adata_scvi, layer="counts", batch_key=batch_key)
adata_scvi
AnnData object with n_obs × n_vars = 10270 × 2000
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', '_scvi_batch', '_scvi_labels'
var: 'feature_types', 'gene_id', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'ATAC_gene_activity_var_names', 'dataset_id', 'genome', 'organism', 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'batch_colors', 'cell_type_colors', '_scvi_uuid', '_scvi_manager_uuid'
obsm: 'ATAC_gene_activity', 'ATAC_lsi_full', 'ATAC_lsi_red', 'ATAC_umap', 'GEX_X_pca', 'GEX_X_umap', 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts', 'logcounts'
obsp: 'distances', 'connectivities'
scVI创建的字段以_scvi为前缀。这些字段是为内部使用设计的,不应手动修改。scvi-tools作者的一般建议是在模型训练完成之前,不要对对象进行任何更改。在其他数据集上,您可能会看到关于输入表达矩阵包含未标准化计数数据的警告。这通常意味着您应该检查提供给设置函数的层确实包含计数值,但如果您对全长协议的数据进行基因长度校正或使用其他不生成整数计数的量化方法,也可能会发生这种情况。
Building the model¶
现在我们可以构建一个scVI模型对象。除了我们在这里使用的scVI模型外,scvi-tools包中还包含其他各种模型(我们将在下文中使用scANVI模型)。
model_scvi = scvi.model.SCVI(adata_scvi)
model_scvi
SCVI Model with the following params: n_hidden: 128, n_latent: 10, n_layers: 1, dropout_rate: 0.1, dispersion: gene, gene_likelihood: zinb, latent_distribution: normal Training status: Not Trained
scVI模型对象包含提供的AnnData对象以及模型本身的神经网络。您可以看到当前模型尚未训练。如果我们想修改网络的结构,可以向模型构造函数提供其他参数,但这里我们仅使用默认设置。
我们还可以打印模型的详细描述,以显示关联的AnnData对象中存储信息的位置。
model_scvi.view_anndata_setup()
Anndata setup with scvi-tools version 0.18.0.
Setup via `SCVI.setup_anndata` with arguments:
{ │ 'layer': 'counts', │ 'batch_key': 'batch', │ 'labels_key': None, │ 'size_factor_key': None, │ 'categorical_covariate_keys': None, │ 'continuous_covariate_keys': None }
Summary Statistics ┏━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━┓ ┃ Summary Stat Key ┃ Value ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━┩ │ n_batch │ 3 │ │ n_cells │ 10270 │ │ n_extra_categorical_covs │ 0 │ │ n_extra_continuous_covs │ 0 │ │ n_labels │ 1 │ │ n_vars │ 2000 │ └──────────────────────────┴───────┘
Data Registry ┏━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━┓ ┃ Registry Key ┃ scvi-tools Location ┃ ┡━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━┩ │ X │ adata.layers['counts'] │ │ batch │ adata.obs['_scvi_batch'] │ │ labels │ adata.obs['_scvi_labels'] │ └──────────────┴───────────────────────────┘
batch State Registry ┏━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━┓ ┃ Source Location ┃ Categories ┃ scvi-tools Encoding ┃ ┡━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━┩ │ adata.obs['batch'] │ s1d3 │ 0 │ │ │ s2d1 │ 1 │ │ │ s3d7 │ 2 │ └────────────────────┴────────────┴─────────────────────┘
labels State Registry ┏━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━┓ ┃ Source Location ┃ Categories ┃ scvi-tools Encoding ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━┩ │ adata.obs['_scvi_labels'] │ 0 │ 0 │ └───────────────────────────┴────────────┴─────────────────────┘
在这里,我们可以准确看到scVI分配了哪些信息,包括每个不同批次在模型中的编码方式等详细信息。
Training the model¶
模型将根据给定的训练轮数(epochs)进行训练,每轮训练都会将每个细胞通过网络一次。默认情况下,scVI使用以下启发式方法来设置轮数。对于少于20,000个细胞的数据集,使用400轮训练;随着细胞数量超过20,000,轮数会逐渐减少。这背后的原理是,由于网络在每轮训练中看到的细胞更多,它可以从较少轮数中学习到与较多轮数相同的信息。
max_epochs_scvi = np.min([round((20000 / adata.n_obs) * 400), 400])
max_epochs_scvi
400
现在我们开始训练模型,使用选定的轮数(这将根据您使用的计算机大约需要20到40分钟)。
model_scvi.train()
GPU available: False, used: False TPU available: False, using: 0 TPU cores IPU available: False, using: 0 IPUs HPU available: False, using: 0 HPUs
Epoch 400/400: 100%|██████████| 400/400 [15:49<00:00, 2.27s/it, loss=648, v_num=1]
`Trainer.fit` stopped: `max_epochs=400` reached.
Epoch 400/400: 100%|██████████| 400/400 [15:49<00:00, 2.37s/it, loss=648, v_num=1]
!!! note {提前停止}
除了设置目标训练轮数外,还可以在训练函数中设置`early_stopping=True`。这将让**scVI**根据模型的收敛情况决定提前停止训练。提前停止的具体条件可以通过其他参数来控制。Extracting the embedding¶
我们想从训练好的模型中提取的主要结果是每个细胞的潜在表示。这是一个多维嵌入,其中批次效应已被去除,可以类似于在分析单个数据集时使用PCA维度的方式使用。我们将其存储在obsm中,键为X_scvi。
adata_scvi.obsm["X_scVI"] = model_scvi.get_latent_representation()
Calculate a batch-corrected UMAP¶
现在我们将像整合前一样可视化数据。我们计算一个新的UMAP嵌入,但不是在PCA空间中找到最近邻,而是从scVI的校正表示开始。
sc.pp.neighbors(adata_scvi, use_rep="X_scVI")
sc.tl.umap(adata_scvi)
adata_scvi
AnnData object with n_obs × n_vars = 10270 × 2000
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', '_scvi_batch', '_scvi_labels'
var: 'feature_types', 'gene_id', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'ATAC_gene_activity_var_names', 'dataset_id', 'genome', 'organism', 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'batch_colors', 'cell_type_colors', '_scvi_uuid', '_scvi_manager_uuid'
obsm: 'ATAC_gene_activity', 'ATAC_lsi_full', 'ATAC_lsi_red', 'ATAC_umap', 'GEX_X_pca', 'GEX_X_umap', 'X_pca', 'X_umap', 'X_scVI'
varm: 'PCs'
layers: 'counts', 'logcounts'
obsp: 'distances', 'connectivities'
一旦我们有了新的UMAP表示,我们就可以像之前一样绘制图表,并按批次和身份标签进行着色。
sc.pl.umap(adata_scvi, color=[label_key, batch_key], wspace=1)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:163: PendingDeprecationWarning: The get_cmap function will be deprecated in a future version. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap(obj)`` instead. cmap = copy(get_cmap(cmap)) /Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored cax = scatter( /Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored cax = scatter(

这看起来好多了!之前,各个批次之间有所偏移。现在,批次之间更多地重叠,每个细胞身份标签都有一个单一的聚集。
在许多情况下,我们不会事先有身份标签,因此从这个阶段开始,我们会继续进行聚类、注释和进一步分析,如其他章节所述。
使用细胞标签的VAE整合¶
在使用scVI进行整合时,我们假装没有任何细胞标签(尽管我们在图中显示了它们)。这种情况很常见,但在某些情况下,我们确实事先知道一些关于细胞身份的信息。通常,这种情况发生在我们想将一个或多个公开可用的数据集与新研究的数据结合时。当我们至少对部分细胞有标签时,可以使用scANVI(使用变分推断的单细胞注释) {cite}Xu2021-dh。这是scVI模型的扩展版本,能够结合细胞身份标签信息和批次信息。因为它具有额外的信息,所以可以在去除批次效应的同时保留细胞标签之间的差异。基准测试表明,与scVI相比,scANVI在保留生物信号方面更好,但有时在去除批次效应方面不如scVI {cite}Luecken2021-jo。尽管我们这里对所有细胞都有标签,scANVI也可以在半监督的情况下使用,仅为部分细胞提供标签。
!!! note {标签一致性} 如果您使用scANVI来整合多个已有标签的数据集,首先进行标签一致性检查很重要。这指的是检查要整合的数据集中标签的一致性。例如,一个数据集中可能将细胞注释为“T细胞”,但在另一个数据集中,相同类型的细胞可能被标记为“CD8+ T细胞”。如何最好地统一标签是一个开放性问题,通常需要专家的输入。
我们首先创建一个scANVI模型对象。注意,因为scANVI是在已经训练好的scVI模型基础上进行优化的,所以我们提供scVI模型而不是AnnData对象。如果我们还没有训练scVI模型,需要先进行这一步。我们还提供adata.obs中的一个列的键,其中包含我们的细胞标签以及未标记细胞对应的标签。在这种情况下,我们所有的细胞都有标签,所以我们只提供一个虚拟值。在大多数情况下,重要的是检查此设置是否正确,以便scANVI在训练过程中知道忽略哪个标签。
# Normally we would need to run scVI first but we have already done that here
# model_scvi = scvi.model.SCVI(adata_scvi) etc.
model_scanvi = scvi.model.SCANVI.from_scvi_model(
model_scvi, labels_key=label_key, unlabeled_category="unlabelled"
)
print(model_scanvi)
model_scanvi.view_anndata_setup()
ScanVI Model with the following params: unlabeled_category: unlabelled, n_hidden: 128, n_latent: 10, n_layers: 1, dropout_rate: 0.1, dispersion: gene, gene_likelihood: zinb Training status: Not Trained
Anndata setup with scvi-tools version 0.18.0.
Setup via `SCANVI.setup_anndata` with arguments:
{ │ 'labels_key': 'cell_type', │ 'unlabeled_category': 'unlabelled', │ 'layer': 'counts', │ 'batch_key': 'batch', │ 'size_factor_key': None, │ 'categorical_covariate_keys': None, │ 'continuous_covariate_keys': None }
Summary Statistics ┏━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━┓ ┃ Summary Stat Key ┃ Value ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━┩ │ n_batch │ 3 │ │ n_cells │ 10270 │ │ n_extra_categorical_covs │ 0 │ │ n_extra_continuous_covs │ 0 │ │ n_labels │ 22 │ │ n_vars │ 2000 │ └──────────────────────────┴───────┘
Data Registry ┏━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━┓ ┃ Registry Key ┃ scvi-tools Location ┃ ┡━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━┩ │ X │ adata.layers['counts'] │ │ batch │ adata.obs['_scvi_batch'] │ │ labels │ adata.obs['_scvi_labels'] │ └──────────────┴───────────────────────────┘
batch State Registry ┏━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━┓ ┃ Source Location ┃ Categories ┃ scvi-tools Encoding ┃ ┡━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━┩ │ adata.obs['batch'] │ s1d3 │ 0 │ │ │ s2d1 │ 1 │ │ │ s3d7 │ 2 │ └────────────────────┴────────────┴─────────────────────┘
labels State Registry ┏━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━┓ ┃ Source Location ┃ Categories ┃ scvi-tools Encoding ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━┩ │ adata.obs['cell_type'] │ B1 B │ 0 │ │ │ CD4+ T activated │ 1 │ │ │ CD4+ T naive │ 2 │ │ │ CD8+ T │ 3 │ │ │ CD8+ T naive │ 4 │ │ │ CD14+ Mono │ 5 │ │ │ CD16+ Mono │ 6 │ │ │ Erythroblast │ 7 │ │ │ G/M prog │ 8 │ │ │ HSC │ 9 │ │ │ ILC │ 10 │ │ │ Lymph prog │ 11 │ │ │ MK/E prog │ 12 │ │ │ NK │ 13 │ │ │ Naive CD20+ B │ 14 │ │ │ Normoblast │ 15 │ │ │ Plasma cell │ 16 │ │ │ Proerythroblast │ 17 │ │ │ Transitional B │ 18 │ │ │ cDC2 │ 19 │ │ │ pDC │ 20 │ │ │ unlabelled │ 21 │ └────────────────────────┴──────────────────┴─────────────────────┘
这个scANVI模型对象与之前看到的scVI模型非常相似。如前所述,我们可以修改模型网络的结构,但这里我们只使用默认参数。
同样,我们有一个启发式方法来选择训练轮数。注意,这次的训练轮数要少得多,因为我们只是对scVI模型进行优化,而不是从头开始训练整个网络。
max_epochs_scanvi = int(np.min([10, np.max([2, round(max_epochs_scvi / 3.0)])]))
model_scanvi.train(max_epochs=max_epochs_scanvi)
INFO Training for 10 epochs.
GPU available: False, used: False TPU available: False, using: 0 TPU cores IPU available: False, using: 0 IPUs HPU available: False, using: 0 HPUs
Epoch 10/10: 100%|██████████| 10/10 [00:35<00:00, 3.69s/it, loss=747, v_num=1]
`Trainer.fit` stopped: `max_epochs=10` reached.
Epoch 10/10: 100%|██████████| 10/10 [00:35<00:00, 3.56s/it, loss=747, v_num=1]
我们可以从模型中提取新的潜在表示,并像对scVI所做的那样创建一个新的UMAP嵌入。
adata_scanvi = adata_scvi.copy()
adata_scanvi.obsm["X_scANVI"] = model_scanvi.get_latent_representation()
sc.pp.neighbors(adata_scanvi, use_rep="X_scANVI")
sc.tl.umap(adata_scanvi)
sc.pl.umap(adata_scanvi, color=[label_key, batch_key], wspace=1)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:163: PendingDeprecationWarning: The get_cmap function will be deprecated in a future version. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap(obj)`` instead. cmap = copy(get_cmap(cmap)) /Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored cax = scatter( /Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored cax = scatter(

通过查看UMAP表示,很难分辨scANVI和scVI之间的区别,但正如我们将在下文中看到的,在量化整合质量的指标得分上存在差异。这提醒我们,不应过度解读这些二维表示,尤其是在比较方法时。
Graph-based integration¶
我们将要讨论的下一种方法是BBKNN或“批次平衡KNN” {cite}Polanski2019-zy。这是与scVI非常不同的方法。BBKNN不是使用神经网络将细胞嵌入批次校正的空间,而是修改了用于聚类和嵌入的_k_最近邻(KNN)图的构建方式。正如我们在{ref}前几章<cellular-structure:clustering>中看到的,正常的KNN程序将细胞连接到整个数据集中最相似的细胞。BBKNN的改进在于强制细胞与其他批次的细胞连接。虽然这是一个简单的修改,但在批次效应非常强烈时,它可以非常有效。然而,由于输出是一个整合的图,它的下游应用可能有限,因为很少有包会接受这个作为输入。
BBKNN的一个重要参数是每个批次的邻居数量。建议的启发式方法是,如果细胞数量超过100,000,则使用25个邻居;如果少于100,000,则使用默认的3个邻居。
neighbors_within_batch = 25 if adata_hvg.n_obs > 100000 else 3
neighbors_within_batch
3
在使用BBKNN之前,我们首先执行PCA,就像在构建正常的KNN图之前一样。与scVI建模原始计数不同,这里我们从对数标准化的表达矩阵开始。
adata_bbknn = adata_hvg.copy()
adata_bbknn.X = adata_bbknn.layers["logcounts"].copy()
sc.pp.pca(adata_bbknn)
现在我们可以运行BBKNN,在标准工作流程中用它替换对scanpy neighbors()函数的调用。一个重要的不同之处是要确保设置batch_key参数,该参数指定adata_hvg.obs中的一个列,该列包含批次标签。
接下来,我们可以运行BBKNN并计算UMAP嵌入,然后绘制UMAP图,用批次和细胞身份标签对点进行着色。
bbknn.bbknn(
adata_bbknn, batch_key=batch_key, neighbors_within_batch=neighbors_within_batch
)
adata_bbknn
AnnData object with n_obs × n_vars = 10270 × 2000
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker'
var: 'feature_types', 'gene_id', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'ATAC_gene_activity_var_names', 'dataset_id', 'genome', 'organism', 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'batch_colors', 'cell_type_colors'
obsm: 'ATAC_gene_activity', 'ATAC_lsi_full', 'ATAC_lsi_red', 'ATAC_umap', 'GEX_X_pca', 'GEX_X_umap', 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts', 'logcounts'
obsp: 'distances', 'connectivities'
与默认的scanpy函数不同,BBKNN不允许指定用于存储结果的键,因此结果总是存储在默认的“neighbors”键下。
我们可以像使用普通KNN图一样使用这个新的整合图来构建UMAP嵌入。
sc.tl.umap(adata_bbknn)
sc.pl.umap(adata_bbknn, color=[label_key, batch_key], wspace=1)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:163: PendingDeprecationWarning: The get_cmap function will be deprecated in a future version. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap(obj)`` instead. cmap = copy(get_cmap(cmap)) /Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored cax = scatter( /Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored cax = scatter(

与未整合的数据相比,这种整合也得到了改进,细胞身份聚集在一起,但我们仍然看到批次之间存在一些偏移。
Linear embedding integration using Mutual Nearest Neighbors (MNN)¶
有些下游应用不能接受整合的嵌入或邻域图,需要校正后的表达矩阵。一个能够生成这种输出的方法是Seurat的整合方法 {cite}Satija2015-or, Butler2018-js, Stuart2019-lq。Seurat整合方法属于一类_线性嵌入模型_,利用_相互最近邻_(Seurat称之为_anchors_)的思想来校正批次效应 {cite}Haghverdi2018-bd。相互最近邻是指当两个数据集置于相同的(潜在)空间时,相互在对方邻域中的细胞对。找到这些细胞后,可以用它们来对齐两个数据集并校正它们之间的差异。在一些评估中,Seurat也被发现是顶级混合方法之一 {cite}Tran2020-ia。
由于Seurat是一个R包,我们必须将数据从Python转移到R。这里我们准备AnnData对象以便用rpy2和anndata2ri进行转换。
adata_seurat = adata_hvg.copy()
# Convert categorical columns to strings
adata_seurat.obs[batch_key] = adata_seurat.obs[batch_key].astype(str)
adata_seurat.obs[label_key] = adata_seurat.obs[label_key].astype(str)
# Delete uns as this can contain arbitrary objects which are difficult to convert
del adata_seurat.uns
adata_seurat
AnnData object with n_obs × n_vars = 10270 × 2000
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker'
var: 'feature_types', 'gene_id', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
obsm: 'ATAC_gene_activity', 'ATAC_lsi_full', 'ATAC_lsi_red', 'ATAC_umap', 'GEX_X_pca', 'GEX_X_umap', 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts', 'logcounts'
obsp: 'distances', 'connectivities'
经过准备的AnnData现在可以通过anndata2ri在R中作为SingleCellExperiment对象使用。请注意,与AnnData对象相比,这是转置的,因此我们的观测值(细胞)现在是列,而变量(基因)现在是行。
%%R -i adata_seurat
adata_seurat
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/rpy2/robjects/pandas2ri.py:263: DeprecationWarning: The global conversion available with activate() is deprecated and will be removed in the next major release. Use a local converter.
warnings.warn('The global conversion available with activate() '
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/rpy2/robjects/numpy2ri.py:205: DeprecationWarning: The global conversion available with activate() is deprecated and will be removed in the next major release. Use a local converter.
warnings.warn('The global conversion available with activate() '
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/rpy2/robjects/pandas2ri.py:263: DeprecationWarning: The global conversion available with activate() is deprecated and will be removed in the next major release. Use a local converter.
warnings.warn('The global conversion available with activate() '
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/rpy2/robjects/numpy2ri.py:205: DeprecationWarning: The global conversion available with activate() is deprecated and will be removed in the next major release. Use a local converter.
warnings.warn('The global conversion available with activate() '
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata2ri/r2py.py:106: FutureWarning: X.dtype being converted to np.float32 from float64. In the next version of anndata (0.9) conversion will not be automatic. Pass dtype explicitly to avoid this warning. Pass `AnnData(X, dtype=X.dtype, ...)` to get the future behavour.
return AnnData(exprs, obs, var, uns, obsm or None, layers=layers)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/rpy2/robjects/pandas2ri.py:263: DeprecationWarning: The global conversion available with activate() is deprecated and will be removed in the next major release. Use a local converter.
warnings.warn('The global conversion available with activate() '
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/rpy2/robjects/numpy2ri.py:205: DeprecationWarning: The global conversion available with activate() is deprecated and will be removed in the next major release. Use a local converter.
warnings.warn('The global conversion available with activate() '
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata2ri/r2py.py:106: FutureWarning: X.dtype being converted to np.float32 from float64. In the next version of anndata (0.9) conversion will not be automatic. Pass dtype explicitly to avoid this warning. Pass `AnnData(X, dtype=X.dtype, ...)` to get the future behavour.
return AnnData(exprs, obs, var, uns, obsm or None, layers=layers)
class: SingleCellExperiment dim: 2000 10270 metadata(0): assays(3): X counts logcounts rownames(2000): GPR153 TNFRSF25 ... TMLHE-AS1 MT-ND3 rowData names(9): feature_types gene_id ... highly_variable_nbatches highly_variable_intersection colnames(10270): TCACCTGGTTAGGTTG-3-s1d3 CGTTAACAGGTGTCCA-3-s1d3 ... AGCAGGTAGGCTATGT-12-s3d7 GCCATGATCCCTTGCG-12-s3d7 colData names(28): GEX_pct_counts_mt GEX_n_counts ... QCMeds DonorSmoker reducedDimNames(8): ATAC_gene_activity ATAC_lsi_full ... PCA UMAP mainExpName: NULL altExpNames(0):
Seurat使用其自有对象来存储数据。值得庆幸的是,作者提供了一个函数用于从SingleCellExperiment进行转换。我们只需提供SingleCellExperiment对象,并告诉Seurat哪些检测(我们AnnData对象中的层)包含原始计数和标准化表达(Seurat将其存储在一个名为“data”的槽中)。
%%R -i adata_seurat
seurat <- as.Seurat(adata_seurat, counts = "counts", data = "logcounts")
seurat
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/rpy2/robjects/pandas2ri.py:263: DeprecationWarning: The global conversion available with activate() is deprecated and will be removed in the next major release. Use a local converter.
warnings.warn('The global conversion available with activate() '
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/rpy2/robjects/numpy2ri.py:205: DeprecationWarning: The global conversion available with activate() is deprecated and will be removed in the next major release. Use a local converter.
warnings.warn('The global conversion available with activate() '
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/anndata2ri/r2py.py:106: FutureWarning: X.dtype being converted to np.float32 from float64. In the next version of anndata (0.9) conversion will not be automatic. Pass dtype explicitly to avoid this warning. Pass `AnnData(X, dtype=X.dtype, ...)` to get the future behavour.
return AnnData(exprs, obs, var, uns, obsm or None, layers=layers)
An object of class Seurat 2000 features across 10270 samples within 1 assay Active assay: originalexp (2000 features, 0 variable features) 8 dimensional reductions calculated: ATAC_gene_activity, ATAC_lsi_full, ATAC_lsi_red, ATAC_umap, GEX_X_pca, GEX_X_umap, PCA, UMAP
与我们之前看到的一些仅需一个对象和一个批次键的方法不同,Seurat的整合函数需要一个对象列表。我们使用SplitObject()函数创建这个列表。
%%R -i batch_key
batch_list <- SplitObject(seurat, split.by = batch_key)
batch_list
$s1d3 An object of class Seurat 2000 features across 4279 samples within 1 assay Active assay: originalexp (2000 features, 0 variable features) 8 dimensional reductions calculated: ATAC_gene_activity, ATAC_lsi_full, ATAC_lsi_red, ATAC_umap, GEX_X_pca, GEX_X_umap, PCA, UMAP $s2d1 An object of class Seurat 2000 features across 4220 samples within 1 assay Active assay: originalexp (2000 features, 0 variable features) 8 dimensional reductions calculated: ATAC_gene_activity, ATAC_lsi_full, ATAC_lsi_red, ATAC_umap, GEX_X_pca, GEX_X_umap, PCA, UMAP $s3d7 An object of class Seurat 2000 features across 1771 samples within 1 assay Active assay: originalexp (2000 features, 0 variable features) 8 dimensional reductions calculated: ATAC_gene_activity, ATAC_lsi_full, ATAC_lsi_red, ATAC_umap, GEX_X_pca, GEX_X_umap, PCA, UMAP
现在我们可以使用这个列表为每对数据集找到锚点。通常,您首先会识别批次感知的高度可变基因(使用FindVariableFeatures()和SelectIntegrationFeatures()函数),但因为我们已经做过这一步,所以我们告诉Seurat使用对象中的所有特征。
%%R
anchors <- FindIntegrationAnchors(batch_list, anchor.features = rownames(seurat))
anchors
| | 0 % ~calculating |+++++++++++++++++ | 33% ~03s |++++++++++++++++++++++++++++++++++ | 67% ~01s |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=02s | | 0 % ~calculating |+++++++++++++++++ | 33% ~01m 39s |++++++++++++++++++++++++++++++++++ | 67% ~41s |++++++++++++++++++++++++++++++++++++++++++++++++++| 100% elapsed=01m 56s An AnchorSet object containing 25352 anchors between 3 Seurat objects This can be used as input to IntegrateData.
然后,Seurat可以使用这些锚点来计算将一个数据集映射到另一个数据集的变换。这是以成对方式进行的,直到所有数据集都被合并。默认情况下,Seurat会确定一个合并顺序,使得更相似的数据集首先合并,但也可以自行定义这个顺序。
%%R
integrated <- IntegrateData(anchors)
integrated
An object of class Seurat 4000 features across 10270 samples within 2 assays Active assay: integrated (2000 features, 2000 variable features) 1 other assay present: originalexp
结果是另一个Seurat对象,但请注意,现在的活动检测被称为“integrated”。这包含校正后的表达矩阵,是整合的最终输出。
现在我们提取该矩阵并准备将其转移回Python。
%%R -o integrated_expr
# Extract the integrated expression matrix
integrated_expr <- GetAssayData(integrated)
# Make sure the rows and columns are in the same order as the original object
integrated_expr <- integrated_expr[rownames(seurat), colnames(seurat)]
# Transpose the matrix to AnnData format
integrated_expr <- t(integrated_expr)
print(integrated_expr[1:10, 1:10])
10 x 10 sparse Matrix of class "dgCMatrix"
TCACCTGGTTAGGTTG-3-s1d3 . -0.0005365199 1.032812e-02 -2.653187e-02
CGTTAACAGGTGTCCA-3-s1d3 0.0001382038 -0.1809919733 -1.454901e-02 3.608087e-03
ATTCGTTTCAGTATTG-3-s1d3 -0.0121073040 -0.0634131457 . 2.144075e-02
GGACCGAAGTGAGGTA-3-s1d3 . . 2.972292e-04 .
ATGAAGCCAGGGAGCT-3-s1d3 -0.0139047071 -0.0313151274 . 2.239855e-02
AGTGCGGAGTAAGGGC-3-s1d3 -0.0004299227 -0.0002657828 . -1.871410e-03
CTACCTCAGACACCGC-3-s1d3 -0.0055208620 -0.0398862297 7.182253e-06 8.240406e-03
CTTCAATTCACGAATC-3-s1d3 . -0.0109928448 . 1.935677e-04
CCATTGTGTAGACAAA-3-s1d3 . 0.0171909615 . 5.711311e-05
CCGTTACTCAATGTGC-3-s1d3 0.0139905515 0.0007981117 2.303346e-03 1.356206e-02
TCACCTGGTTAGGTTG-3-s1d3 -0.023237586 0.031938502 -0.003196878 0.01777767
CGTTAACAGGTGTCCA-3-s1d3 0.114149766 -0.013183394 0.038076743 0.80491293
ATTCGTTTCAGTATTG-3-s1d3 -0.054419895 0.010955783 -0.005951634 0.37223306
GGACCGAAGTGAGGTA-3-s1d3 0.002305526 0.011544715 0.011133475 0.02366670
ATGAAGCCAGGGAGCT-3-s1d3 -0.123505733 -0.009382408 0.002153635 -0.07013584
AGTGCGGAGTAAGGGC-3-s1d3 0.035848768 0.013858992 -0.000379393 0.05617137
CTACCTCAGACACCGC-3-s1d3 -0.003837952 0.082027580 -0.001109391 -0.06307772
CTTCAATTCACGAATC-3-s1d3 0.052970708 0.153601547 0.247920321 -0.01143158
CCATTGTGTAGACAAA-3-s1d3 -0.015445186 0.025763467 -0.003632830 0.02040172
CCGTTACTCAATGTGC-3-s1d3 -0.018025385 0.022560136 0.005755797 0.61496228
TCACCTGGTTAGGTTG-3-s1d3 -6.661643e-03 -0.0183198213
CGTTAACAGGTGTCCA-3-s1d3 5.079864e-02 0.0394717101
ATTCGTTTCAGTATTG-3-s1d3 6.600434e-02 0.0009021682
GGACCGAAGTGAGGTA-3-s1d3 7.172740e-04 0.0095352521
ATGAAGCCAGGGAGCT-3-s1d3 1.226039e-01 0.0063816143
AGTGCGGAGTAAGGGC-3-s1d3 -1.830964e-03 0.0008381943
CTACCTCAGACACCGC-3-s1d3 8.559358e-01 0.0102285085
CTTCAATTCACGAATC-3-s1d3 -5.318167e-06 -0.0341661907
CCATTGTGTAGACAAA-3-s1d3 -6.362298e-03 0.0232357011
CCGTTACTCAATGTGC-3-s1d3 -4.910886e-02 0.0317359576
现在我们将校正后的表达矩阵存储为我们AnnData对象中的一个层。我们还将adata.X设置为使用此矩阵。
adata_seurat.X = integrated_expr
adata_seurat.layers["seurat"] = integrated_expr
print(adata_seurat)
adata.X
AnnData object with n_obs × n_vars = 10270 × 2000
obs: 'GEX_pct_counts_mt', 'GEX_n_counts', 'GEX_n_genes', 'GEX_size_factors', 'GEX_phase', 'ATAC_nCount_peaks', 'ATAC_atac_fragments', 'ATAC_reads_in_peaks_frac', 'ATAC_blacklist_fraction', 'ATAC_nucleosome_signal', 'cell_type', 'batch', 'ATAC_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker'
var: 'feature_types', 'gene_id', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
obsm: 'ATAC_gene_activity', 'ATAC_lsi_full', 'ATAC_lsi_red', 'ATAC_umap', 'GEX_X_pca', 'GEX_X_umap', 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts', 'logcounts', 'seurat'
obsp: 'distances', 'connectivities'
<10270x13431 sparse matrix of type '<class 'numpy.float32'>' with 14348115 stored elements in Compressed Sparse Row format>
现在我们已经得到了整合的结果,可以计算UMAP并绘制它,就像我们对其他方法所做的一样(我们也可以在R中完成这一步)。
# Reset the batch colours because we deleted them earlier
adata_seurat.uns[batch_key + "_colors"] = [
"#1b9e77",
"#d95f02",
"#7570b3",
]
sc.tl.pca(adata_seurat)
sc.pp.neighbors(adata_seurat)
sc.tl.umap(adata_seurat)
sc.pl.umap(adata_seurat, color=[label_key, batch_key], wspace=1)
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:163: PendingDeprecationWarning: The get_cmap function will be deprecated in a future version. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap(obj)`` instead. cmap = copy(get_cmap(cmap)) /Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored cax = scatter( /Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored cax = scatter(

正如我们之前看到的,批次是混合的,而标签是分开的。尽管根据UMAP选择整合方法很有吸引力,但这并不能完全代表整合的质量。在下一部分中,我们将介绍一些更严格评估整合方法的方式。
!!! note {关于可扩展性的一点说明} 在运行不同的整合方法时,您可能注意到scVI花费的时间最多。虽然这对于像这里示例的小型数据集是正确的,但基准测试显示 scVI在处理大型数据集时扩展性良好。这主要是因为训练轮数会根据数据集的大小进行调整。MNN方法通常扩展性不如其他方法,部分原因是它们执行多个成对整合,所以如果有20个批次,就要进行20次整合,而其他方法可以同时考虑所有批次。
Benchmarking your own integration¶
这里展示的方法是根据基准测试实验的结果选择的,包括单细胞整合基准测试项目 {cite}Luecken2021-jo。该项目还产生了一个名为scib的软件包,可以用于运行多种整合方法以及用于评估的指标。在本节中,我们展示如何使用这个包来评估整合的质量。
!!! note {什么是真实情况?} 其中一些指标,特别是那些评估生物变异保留的指标,需要一个已知的真实情况进行比较。通常,这是真实的细胞身份标签,但有时也可能是其他信息,如已知的轨迹。由于这种要求,评估完全新的数据集的整合是困难的,因为不清楚应该保留哪些生物信号。
scib的指标可以单独运行,但也有一次运行多个指标的包装器。在这里,我们使用metrics_fast()函数运行一部分计算快速的指标。这个函数需要一些参数:原始未整合的数据集、整合后的数据集、批次键和标签键。根据整合方法的输出,我们可能还需要提供额外的参数,例如在这里我们使用embed参数指定scVI和scANVI的嵌入。你还可以通过附加参数控制一些指标的运行方式。此外,还需要检查对象的格式是否正确,以确保scIB可以找到所需信息。
让我们为我们上面执行的每个整合方法,以及未整合的数据(在选择高度可变基因之后)运行这些指标。
metrics_scvi = scib.metrics.metrics_fast(
adata, adata_scvi, batch_key, label_key, embed="X_scVI"
)
metrics_scanvi = scib.metrics.metrics_fast(
adata, adata_scanvi, batch_key, label_key, embed="X_scANVI"
)
metrics_bbknn = scib.metrics.metrics_fast(adata, adata_bbknn, batch_key, label_key)
metrics_seurat = scib.metrics.metrics_fast(adata, adata_seurat, batch_key, label_key)
metrics_hvg = scib.metrics.metrics_fast(adata, adata_hvg, batch_key, label_key)
Silhouette score... PC regression... Isolated labels ASW... Graph connectivity... Silhouette score... PC regression... Isolated labels ASW... Graph connectivity... Silhouette score... PC regression... Isolated labels ASW... Graph connectivity... Silhouette score... PC regression... Isolated labels ASW... Graph connectivity... Silhouette score... PC regression... Isolated labels ASW... Graph connectivity...
以下是单个整合方法某个指标结果的示例:
metrics_hvg
| 0 | |
|---|---|
| NMI_cluster/label | NaN |
| ARI_cluster/label | NaN |
| ASW_label | 0.555775 |
| ASW_label/batch | 0.833656 |
| PCR_batch | 0.537850 |
| cell_cycle_conservation | NaN |
| isolated_label_F1 | NaN |
| isolated_label_silhouette | 0.487975 |
| graph_conn | 0.985533 |
| kBET | NaN |
| iLISI | NaN |
| cLISI | NaN |
| hvg_overlap | 1.000000 |
| trajectory | NaN |
每行代表一个不同的指标,值显示了该指标的得分。得分介于0和1之间,其中1表示表现良好,0表示表现较差(scib也可以返回某些指标的未缩放得分,如果需要的话)。因为我们这里只运行了快速指标,所以有些指标的得分为NaN。另外,注意有些指标不能用于某些输出格式,这也可能导致返回NaN值。
为了比较这些方法,将所有指标结果放在一个表格中是很有用的。这段代码将它们组合起来,并整理成更方便的格式。
# Concatenate metrics results
metrics = pd.concat(
[metrics_scvi, metrics_scanvi, metrics_bbknn, metrics_seurat, metrics_hvg],
axis="columns",
)
# Set methods as column names
metrics = metrics.set_axis(
["scVI", "scANVI", "BBKNN", "Seurat", "Unintegrated"], axis="columns"
)
# Select only the fast metrics
metrics = metrics.loc[
[
"ASW_label",
"ASW_label/batch",
"PCR_batch",
"isolated_label_silhouette",
"graph_conn",
"hvg_overlap",
],
:,
]
# Transpose so that metrics are columns and methods are rows
metrics = metrics.T
# Remove the HVG overlap metric because it's not relevant to embedding outputs
metrics = metrics.drop(columns=["hvg_overlap"])
metrics
| ASW_label | ASW_label/batch | PCR_batch | isolated_label_silhouette | graph_conn | |
|---|---|---|---|---|---|
| scVI | 0.570953 | 0.907165 | 0.913474 | 0.560875 | 0.982940 |
| scANVI | 0.587934 | 0.905402 | 0.887735 | 0.556232 | 0.983729 |
| BBKNN | 0.555469 | 0.847772 | 0.537850 | 0.481913 | 0.969514 |
| Seurat | 0.571231 | 0.915237 | 0.788894 | 0.488585 | 0.989366 |
| Unintegrated | 0.555775 | 0.833656 | 0.537850 | 0.487975 | 0.985533 |
现在,我们将所有得分汇总到一个表格中,指标作为列,方法作为行。通过使用渐变样式来美化表格,可以更容易看出得分之间的差异。
metrics.style.background_gradient(cmap="Blues")
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/pandas/io/formats/style.py:3930: PendingDeprecationWarning: The get_cmap function will be deprecated in a future version. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap(obj)`` instead. rgbas = plt.cm.get_cmap(cmap)(norm(gmap))
| ASW_label | ASW_label/batch | PCR_batch | isolated_label_silhouette | graph_conn | |
|---|---|---|---|---|---|
| scVI | 0.570953 | 0.907165 | 0.913474 | 0.560875 | 0.982940 |
| scANVI | 0.587934 | 0.905402 | 0.887735 | 0.556232 | 0.983729 |
| BBKNN | 0.555469 | 0.847772 | 0.537850 | 0.481913 | 0.969514 |
| Seurat | 0.571231 | 0.915237 | 0.788894 | 0.488585 | 0.989366 |
| Unintegrated | 0.555775 | 0.833656 | 0.537850 | 0.487975 | 0.985533 |
对于某些指标,得分往往在相对较小的范围内。为了强调不同方法之间的差异,并将每个指标放在相同的尺度上,我们对它们进行缩放,使得表现最差的得分为0,表现最好的得分为1,其他方法的得分介于两者之间。
metrics_scaled = (metrics - metrics.min()) / (metrics.max() - metrics.min())
metrics_scaled.style.background_gradient(cmap="Blues")
| ASW_label | ASW_label/batch | PCR_batch | isolated_label_silhouette | graph_conn | |
|---|---|---|---|---|---|
| scVI | 0.476942 | 0.901060 | 1.000000 | 1.000000 | 0.676270 |
| scANVI | 1.000000 | 0.879454 | 0.931475 | 0.941194 | 0.716018 |
| BBKNN | 0.000000 | 0.173036 | 0.000000 | 0.000000 | 0.000000 |
| Seurat | 0.485521 | 1.000000 | 0.668338 | 0.084500 | 1.000000 |
| Unintegrated | 0.009437 | 0.000000 | 0.000000 | 0.076776 | 0.806917 |
现在这些值更好地表示了方法之间的差异(并且更符合颜色比例)。然而,需要注意的是,缩放后的得分只能用于比较这一特定整合方法集的相对性能。如果我们想添加另一种方法,则需要重新进行缩放。我们也不能断言某种整合方法绝对“好”,只能说它比我们尝试的其他方法更好。这个缩放强调了方法之间的差异。例如,如果我们有0.92、0.94和0.96的指标得分,这些分数将被缩放为0、0.5和1.0。这使得第一种方法看起来得分要差很多,即使它仅比其他两种稍低,而且仍然得到了非常高的分数。当比较的方法较少且它们的原始得分相似时,这种效果更明显。是否查看原始得分或缩放得分取决于您是否想关注绝对性能还是方法之间性能的差异。
评估指标可以分为两类:衡量批次效应去除的指标和衡量生物变异保留的指标。我们可以通过取每组缩放值的平均值来计算每一类的总结分数。这种总结分数对于原始值来说没有意义,因为一些指标始终产生较高的分数(因此对平均值的影响更大)。
metrics_scaled["Batch"] = metrics_scaled[
["ASW_label/batch", "PCR_batch", "graph_conn"]
].mean(axis=1)
metrics_scaled["Bio"] = metrics_scaled[["ASW_label", "isolated_label_silhouette"]].mean(
axis=1
)
metrics_scaled.style.background_gradient(cmap="Blues")
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/pandas/io/formats/style.py:3930: PendingDeprecationWarning: The get_cmap function will be deprecated in a future version. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap(obj)`` instead. rgbas = plt.cm.get_cmap(cmap)(norm(gmap))
| ASW_label | ASW_label/batch | PCR_batch | isolated_label_silhouette | graph_conn | Batch | Bio | |
|---|---|---|---|---|---|---|---|
| scVI | 0.476942 | 0.901060 | 1.000000 | 1.000000 | 0.676270 | 0.859110 | 0.738471 |
| scANVI | 1.000000 | 0.879454 | 0.931475 | 0.941194 | 0.716018 | 0.842316 | 0.970597 |
| BBKNN | 0.000000 | 0.173036 | 0.000000 | 0.000000 | 0.000000 | 0.057679 | 0.000000 |
| Seurat | 0.485521 | 1.000000 | 0.668338 | 0.084500 | 1.000000 | 0.889446 | 0.285010 |
| Unintegrated | 0.009437 | 0.000000 | 0.000000 | 0.076776 | 0.806917 | 0.268972 | 0.043106 |
将两个总结分数绘制在一起,可以指示每种方法的侧重点。有些方法偏向于批次校正,而其他方法则倾向于保留生物变异。
fig, ax = plt.subplots()
ax.set_xlim(0, 1)
ax.set_ylim(0, 1)
metrics_scaled.plot.scatter(
x="Batch",
y="Bio",
c=range(len(metrics_scaled)),
ax=ax,
)
for k, v in metrics_scaled[["Batch", "Bio"]].iterrows():
ax.annotate(
k,
v,
xytext=(6, -3),
textcoords="offset points",
family="sans-serif",
fontsize=12,
)

在我们的小示例场景中,BBKNN显然表现最差,在批次去除和生物保留方面得分最低。其他三种方法的批次校正得分相似,其中scANVI在生物保留方面得分最高,其次是scVI和Seurat。
为了获得每种方法的总体得分,我们可以将两个总结分数结合起来。scIB论文建议将批次校正权重设为40%,生物保留权重设为60%,但您可以根据数据集的优先级调整权重。
metrics_scaled["Overall"] = 0.4 * metrics_scaled["Batch"] + 0.6 * metrics_scaled["Bio"]
metrics_scaled.style.background_gradient(cmap="Blues")
/Users/luke.zappia/miniconda/envs/bp-integration/lib/python3.9/site-packages/pandas/io/formats/style.py:3930: PendingDeprecationWarning: The get_cmap function will be deprecated in a future version. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap(obj)`` instead. rgbas = plt.cm.get_cmap(cmap)(norm(gmap))
| ASW_label | ASW_label/batch | PCR_batch | isolated_label_silhouette | graph_conn | Batch | Bio | Overall | |
|---|---|---|---|---|---|---|---|---|
| scVI | 0.476942 | 0.901060 | 1.000000 | 1.000000 | 0.676270 | 0.859110 | 0.738471 | 0.786726 |
| scANVI | 1.000000 | 0.879454 | 0.931475 | 0.941194 | 0.716018 | 0.842316 | 0.970597 | 0.919285 |
| BBKNN | 0.000000 | 0.173036 | 0.000000 | 0.000000 | 0.000000 | 0.057679 | 0.000000 | 0.023071 |
| Seurat | 0.485521 | 1.000000 | 0.668338 | 0.084500 | 1.000000 | 0.889446 | 0.285010 | 0.526785 |
| Unintegrated | 0.009437 | 0.000000 | 0.000000 | 0.076776 | 0.806917 | 0.268972 | 0.043106 | 0.133453 |
让我们制作一个快速的柱状图来可视化整体性能。这样的图表可以清晰地展示每种方法的综合得分,便于比较各方法的优劣。
metrics_scaled.plot.bar(y="Overall")
<AxesSubplot: >

正如我们已经看到的,scVI和scANVI是表现最好的方法,scANVI得分略高一些。需要注意的是,这只是展示如何为这个特定数据集运行这些指标的一个示例,并不是对这些方法的正式评估。对于正式评估,您应参考现有的基准测试出版物。特别是,我们只运行了一小部分高性能的方法和一部分指标。此外,请记住,得分是相对于所使用的方法的,因此即使方法表现几乎相同,小差异也会被放大。
现有的基准测试已经建议了一些通常表现良好的方法,但在不同的情境中,性能也可能有很大差异。对于某些分析,进行自己的整合评估可能是值得的。scib包简化了这个过程,但它仍然可能是一项重要的任务,依赖于对真实情况的良好了解和对指标的解释。
Key Takeaways¶
- 在尝试校正批次效应之前,可视化数据以评估问题的严重程度。批次效应校正并非总是必要的,有时可能会掩盖感兴趣的生物变异。
- 如果有细胞标签且生物变异是最重要的,建议使用可以利用这些标签的方法(如scANVI)。
- 考虑在数据集上运行多种整合方法,并使用scIB指标进行评估,以选择最适合您的使用情况的整合方法。
Quiz¶
- What are the sources of batch effects?
- What is the difference between technical and biological variation?
- How does one evaluate whether the integration worked well or not? What are useful metrics for this purpose?
Session information¶
Python¶
import session_info
session_info.show()
Click to view session information
----- anndata 0.8.0 anndata2ri 1.1 bbknn NA matplotlib 3.6.1 numpy 1.23.4 pandas 1.5.1 rpy2 3.5.1 scanpy 1.9.1 scib 1.0.4 scipy 1.9.3 scvi 0.18.0 session_info 1.0.0 -----
Click to view modules imported as dependencies
PIL 9.2.0 absl NA annoy NA appnope 0.1.3 asttokens NA attr 22.1.0 backcall 0.2.0 beta_ufunc NA binom_ufunc NA cffi 1.15.1 chex 0.1.5 colorama 0.4.6 contextlib2 NA cycler 0.10.0 cython_runtime NA dateutil 2.8.2 debugpy 1.6.3 decorator 5.1.1 defusedxml 0.7.1 deprecate 0.3.2 deprecated 1.2.13 docrep 0.3.2 entrypoints 0.4 etils 0.8.0 executing 1.1.1 flax 0.5.0 fsspec 2022.10.0 google NA h5py 3.7.0 hypergeom_ufunc NA igraph 0.10.2 importlib_metadata NA ipykernel 6.16.2 ipython_genutils 0.2.0 ipywidgets 8.0.2 jax 0.3.23 jaxlib 0.3.22 jedi 0.18.1 jinja2 3.1.2 joblib 1.2.0 jupyter_server 1.21.0 kiwisolver 1.4.4 leidenalg 0.9.0 llvmlite 0.39.1 louvain 0.8.0 markupsafe 2.1.1 matplotlib_inline 0.1.6 ml_collections NA mpl_toolkits NA msgpack 1.0.4 mudata 0.2.0 multipledispatch 0.6.0 natsort 8.2.0 nbinom_ufunc NA ncf_ufunc NA numba 0.56.3 numpyro 0.10.1 opt_einsum v3.3.0 optax 0.1.3 packaging 21.3 parso 0.8.3 pexpect 4.8.0 pickleshare 0.7.5 pkg_resources NA prompt_toolkit 3.0.31 psutil 5.9.3 ptyprocess 0.7.0 pure_eval 0.2.2 pycparser 2.21 pydev_ipython NA pydevconsole NA pydevd 2.8.0 pydevd_file_utils NA pydevd_plugins NA pydevd_tracing NA pygments 2.13.0 pynndescent 0.5.7 pyparsing 3.0.9 pyro 1.8.2+2e3bd02 pytorch_lightning 1.7.7 pytz 2022.5 pytz_deprecation_shim NA rich NA seaborn 0.12.0 setuptools 65.5.0 six 1.16.0 sklearn 1.1.3 sphinxcontrib NA stack_data 0.5.1 statsmodels 0.13.2 tensorboard 2.10.1 texttable 1.6.4 threadpoolctl 3.1.0 toolz 0.12.0 torch 1.12.1 torchmetrics 0.10.1 tornado 6.2 tqdm 4.64.1 traitlets 5.5.0 tree 0.1.7 typing_extensions NA tzlocal NA umap 0.5.3 wcwidth 0.2.5 wrapt 1.14.1 yaml 6.0 zipp NA zmq 24.0.1 zoneinfo NA
----- IPython 8.5.0 jupyter_client 7.4.4 jupyter_core 4.11.1 jupyterlab 3.5.0 notebook 6.5.1 ----- Python 3.9.13 | packaged by conda-forge | (main, May 27 2022, 17:01:00) [Clang 13.0.1 ] macOS-10.15.7-x86_64-i386-64bit ----- Session information updated at 2022-11-10 12:53
R¶
%%R
sessioninfo::session_info()
─ Session info ─────────────────────────────────────────────────────────────── setting value version R version 4.1.3 (2022-03-10) os macOS Catalina 10.15.7 system x86_64, darwin13.4.0 ui unknown language (EN) collate C ctype UTF-8 tz Europe/Berlin date 2022-11-10 pandoc 2.19.2 @ /Users/luke.zappia/miniconda/envs/bp-integration/bin/pandoc ─ Packages ─────────────────────────────────────────────────────────────────── package * version date (UTC) lib source abind 1.4-5 2016-07-21 [1] CRAN (R 4.1.3) Biobase * 2.54.0 2021-10-26 [1] Bioconductor BiocGenerics * 0.40.0 2021-10-26 [1] Bioconductor bitops 1.0-7 2021-04-24 [1] CRAN (R 4.1.3) cli 3.4.1 2022-09-23 [1] CRAN (R 4.1.3) cluster 2.1.4 2022-08-22 [1] CRAN (R 4.1.3) codetools 0.2-18 2020-11-04 [1] CRAN (R 4.1.3) colorspace 2.0-3 2022-02-21 [1] CRAN (R 4.1.3) cowplot 1.1.1 2020-12-30 [1] CRAN (R 4.1.3) data.table 1.14.4 2022-10-17 [1] CRAN (R 4.1.3) DelayedArray 0.20.0 2021-10-26 [1] Bioconductor deldir 1.0-6 2021-10-23 [1] CRAN (R 4.1.3) digest 0.6.30 2022-10-18 [1] CRAN (R 4.1.3) dplyr 1.0.10 2022-09-01 [1] CRAN (R 4.1.3) ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.1.3) fansi 1.0.3 2022-03-24 [1] CRAN (R 4.1.3) fastmap 1.1.0 2021-01-25 [1] CRAN (R 4.1.3) fitdistrplus 1.1-8 2022-03-10 [1] CRAN (R 4.1.3) future 1.28.0 2022-09-02 [1] CRAN (R 4.1.3) future.apply 1.9.1 2022-09-07 [1] CRAN (R 4.1.3) generics 0.1.3 2022-07-05 [1] CRAN (R 4.1.3) GenomeInfoDb * 1.30.1 2022-01-30 [1] Bioconductor GenomeInfoDbData 1.2.7 2022-10-28 [1] Bioconductor GenomicRanges * 1.46.1 2021-11-18 [1] Bioconductor ggplot2 3.3.6 2022-05-03 [1] CRAN (R 4.1.3) ggrepel 0.9.1 2021-01-15 [1] CRAN (R 4.1.3) ggridges 0.5.4 2022-09-26 [1] CRAN (R 4.1.3) globals 0.16.1 2022-08-28 [1] CRAN (R 4.1.3) glue 1.6.2 2022-02-24 [1] CRAN (R 4.1.3) goftest 1.2-3 2021-10-07 [1] CRAN (R 4.1.3) gridExtra 2.3 2017-09-09 [1] CRAN (R 4.1.3) gtable 0.3.1 2022-09-01 [1] CRAN (R 4.1.3) htmltools 0.5.3 2022-07-18 [1] CRAN (R 4.1.3) htmlwidgets 1.5.4 2021-09-08 [1] CRAN (R 4.1.3) httpuv 1.6.6 2022-09-08 [1] CRAN (R 4.1.3) httr 1.4.4 2022-08-17 [1] CRAN (R 4.1.3) ica 1.0-3 2022-07-08 [1] CRAN (R 4.1.3) igraph 1.3.5 2022-09-22 [1] CRAN (R 4.1.3) IRanges * 2.28.0 2021-10-26 [1] Bioconductor irlba 2.3.5.1 2022-10-03 [1] CRAN (R 4.1.3) jsonlite 1.8.3 2022-10-21 [1] CRAN (R 4.1.3) KernSmooth 2.23-20 2021-05-03 [1] CRAN (R 4.1.3) later 1.2.0 2021-04-23 [1] CRAN (R 4.1.3) lattice 0.20-45 2021-09-22 [1] CRAN (R 4.1.3) lazyeval 0.2.2 2019-03-15 [1] CRAN (R 4.1.3) leiden 0.4.3 2022-09-10 [1] CRAN (R 4.1.3) lifecycle 1.0.3 2022-10-07 [1] CRAN (R 4.1.3) listenv 0.8.0 2019-12-05 [1] CRAN (R 4.1.3) lmtest 0.9-40 2022-03-21 [1] CRAN (R 4.1.3) magrittr 2.0.3 2022-03-30 [1] CRAN (R 4.1.3) MASS 7.3-58.1 2022-08-03 [1] CRAN (R 4.1.3) Matrix * 1.5-1 2022-09-13 [1] CRAN (R 4.1.3) MatrixGenerics * 1.6.0 2021-10-26 [1] Bioconductor matrixStats * 0.62.0 2022-04-19 [1] CRAN (R 4.1.3) mgcv 1.8-41 2022-10-21 [1] CRAN (R 4.1.3) mime 0.12 2021-09-28 [1] CRAN (R 4.1.3) miniUI 0.1.1.1 2018-05-18 [1] CRAN (R 4.1.3) munsell 0.5.0 2018-06-12 [1] CRAN (R 4.1.3) nlme 3.1-160 2022-10-10 [1] CRAN (R 4.1.3) parallelly 1.32.1 2022-07-21 [1] CRAN (R 4.1.3) patchwork 1.1.2 2022-08-19 [1] CRAN (R 4.1.3) pbapply 1.5-0 2021-09-16 [1] CRAN (R 4.1.3) pillar 1.8.1 2022-08-19 [1] CRAN (R 4.1.3) pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.1.3) plotly 4.10.0 2021-10-09 [1] CRAN (R 4.1.3) plyr 1.8.7 2022-03-24 [1] CRAN (R 4.1.3) png 0.1-7 2013-12-03 [1] CRAN (R 4.1.3) polyclip 1.10-4 2022-10-20 [1] CRAN (R 4.1.3) progressr 0.11.0 2022-09-02 [1] CRAN (R 4.1.3) promises 1.2.0.1 2021-02-11 [1] CRAN (R 4.1.3) purrr 0.3.5 2022-10-06 [1] CRAN (R 4.1.3) R6 2.5.1 2021-08-19 [1] CRAN (R 4.1.3) RANN 2.6.1 2019-01-08 [1] CRAN (R 4.1.3) RColorBrewer 1.1-3 2022-04-03 [1] CRAN (R 4.1.3) Rcpp 1.0.9 2022-07-08 [1] CRAN (R 4.1.3) RcppAnnoy 0.0.19 2021-07-30 [1] CRAN (R 4.1.3) RCurl 1.98-1.9 2022-10-03 [1] CRAN (R 4.1.3) reshape2 1.4.4 2020-04-09 [1] CRAN (R 4.1.3) reticulate 1.26 2022-08-31 [1] CRAN (R 4.1.3) rgeos 0.5-9 2021-12-15 [1] CRAN (R 4.1.3) rlang 1.0.6 2022-09-24 [1] CRAN (R 4.1.3) ROCR 1.0-11 2020-05-02 [1] CRAN (R 4.1.3) rpart 4.1.19 2022-10-21 [1] CRAN (R 4.1.3) Rtsne 0.16 2022-04-17 [1] CRAN (R 4.1.3) S4Vectors * 0.32.4 2022-03-24 [1] Bioconductor scales 1.2.1 2022-08-20 [1] CRAN (R 4.1.3) scattermore 0.8 2022-02-14 [1] CRAN (R 4.1.3) sctransform 0.3.5 2022-09-21 [1] CRAN (R 4.1.3) sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.1.3) Seurat * 4.2.0 2022-09-21 [1] CRAN (R 4.1.3) SeuratObject * 4.1.2 2022-09-20 [1] CRAN (R 4.1.3) shiny 1.7.3 2022-10-25 [1] CRAN (R 4.1.3) SingleCellExperiment * 1.16.0 2021-10-26 [1] Bioconductor sp * 1.5-0 2022-06-05 [1] CRAN (R 4.1.3) spatstat.core 2.4-4 2022-05-18 [1] CRAN (R 4.1.3) spatstat.data 3.0-0 2022-10-21 [1] CRAN (R 4.1.3) spatstat.geom 3.0-3 2022-10-25 [1] CRAN (R 4.1.3) spatstat.random 2.2-0 2022-03-30 [1] CRAN (R 4.1.3) spatstat.sparse 3.0-0 2022-10-21 [1] CRAN (R 4.1.3) spatstat.utils 3.0-1 2022-10-19 [1] CRAN (R 4.1.3) stringi 1.7.8 2022-07-11 [1] CRAN (R 4.1.3) stringr 1.4.1 2022-08-20 [1] CRAN (R 4.1.3) SummarizedExperiment * 1.24.0 2021-10-26 [1] Bioconductor survival 3.4-0 2022-08-09 [1] CRAN (R 4.1.3) tensor 1.5 2012-05-05 [1] CRAN (R 4.1.3) tibble 3.1.8 2022-07-22 [1] CRAN (R 4.1.3) tidyr 1.2.1 2022-09-08 [1] CRAN (R 4.1.3) tidyselect 1.2.0 2022-10-10 [1] CRAN (R 4.1.3) utf8 1.2.2 2021-07-24 [1] CRAN (R 4.1.3) uwot 0.1.14 2022-08-22 [1] CRAN (R 4.1.3) vctrs 0.5.0 2022-10-22 [1] CRAN (R 4.1.3) viridisLite 0.4.1 2022-08-22 [1] CRAN (R 4.1.3) xtable 1.8-4 2019-04-21 [1] CRAN (R 4.1.3) XVector 0.34.0 2021-10-26 [1] Bioconductor zlibbioc 1.40.0 2021-10-26 [1] Bioconductor zoo 1.8-11 2022-09-17 [1] CRAN (R 4.1.3) [1] /Users/luke.zappia/miniconda/envs/bp-integration/lib/R/library ──────────────────────────────────────────────────────────────────────────────