3.4 使用其他数据库(KEGG、Reactome 等)进行富集分析
clusterProfiler 可使用的数据库¶
在之前的章节中,我们使用 enrichGO 函数进行了 GO 数据库的富集分析。实际上,clusterProfiler 还支持其他数据库,用于调查特定基因集或蛋白质集在各种生物学过程或路径中的参与情况。
以下是 clusterProfiler 支持的一些主要数据库及其对应的富集分析函数:
1. groupGO¶
- 使用数据库: Gene Ontology (GO)
- 功能: 将基因群映射到 GO 类别(生物学过程、分子功能、细胞成分)。
2. enrichGO¶
- 使用数据库: Gene Ontology (GO)
- 功能: 调查给定基因集是否在特定 GO 类别中过度表达。
3. enrichKEGG¶
- 使用数据库: KEGG (Kyoto Encyclopedia of Genes and Genomes)
- 功能: 评估基因集是否在 KEGG 路径中过度表达。
4. enrichMKEGG¶
- 使用数据库: KEGG
- 功能: 提供多变量 KEGG 富集分析。
5. enrichWP¶
- 使用数据库: WikiPathways
- 功能: 基于 WikiPathways 进行富集分析。
6. enricher¶
- 使用数据库: 多种
- 功能: 使用多个数据库进行富集分析。
7. enrichPathway¶
- 使用数据库: Reactome
- 功能: 基于 Reactome 进行富集分析。
8. enrichDO¶
- 使用数据库: Disease Ontology
- 功能: 调查基因集是否与特定疾病类别相关。
9. enrichNCG¶
- 使用数据库: Network of Cancer Genes (NCG)
- 功能: 提供癌症相关基因的富集分析。
10. enrichDGN¶
- 使用数据库: DGN (Depression Gene Nexus)
- 功能: 进行抑郁相关基因的富集分析。
11. enrichMeSH¶
- 使用数据库: MeSH (Medical Subject Headings)
- 功能: 评估基因集是否与特定 MeSH 项目相关。
以下是如何使用这些数据库进行富集分析的示例,使用 deseq2_results.csv 作为输入数据。
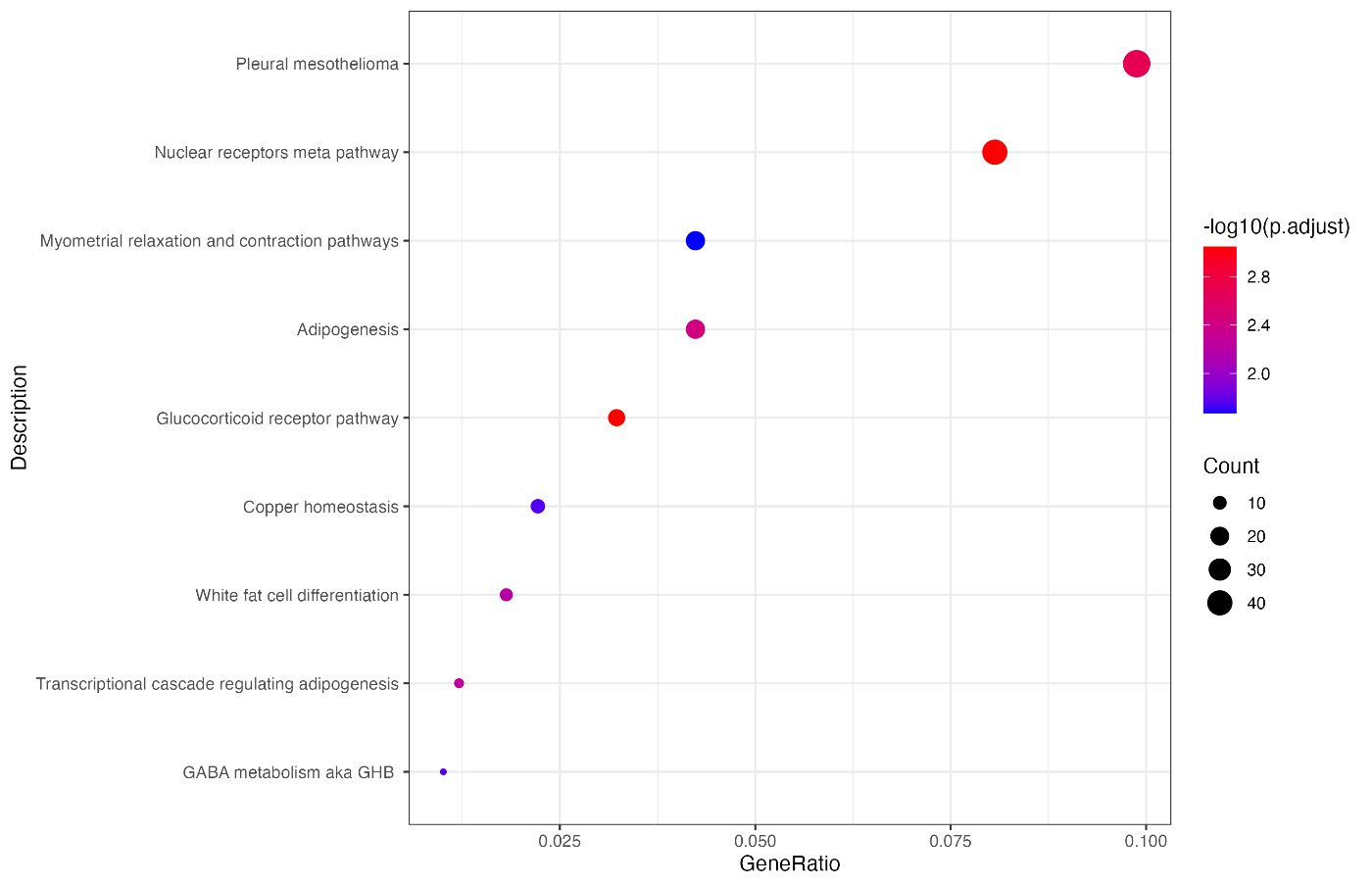
使用 KEGG 数据库的富集分析¶
# 必需的库
library(readr)
library(ggplot2)
library(clusterProfiler)
library(AnnotationDbi)
library(org.Hs.eg.db)
gene_list <- read_csv("deseq2_results.csv")
# 将基因符号转换为Entrez ID
convertToEntrez <- function(genes) {
mapIds(
org.Hs.eg.db,
keys = genes,
column = "ENTREZID",
keytype = "ENSEMBL",
multiVals = "first"
)
}
# 执行KEGG路径的富集分析
performKeggEnrichment <- function(entrez_ids) {
enrichKEGG(
gene = entrez_ids,
organism = "hsa",
pAdjustMethod = "BH",
qvalueCutoff = 0.05
)
}
differentially_expressed_genes <- subset(
gene_list, abs(log2FoldChange) > 1 & pvalue < 0.05
)
# 将基因符号转换为Entrez ID
entrez_ids_deg <- convertToEntrez(differentially_expressed_genes$...1)
# KEGG路径富集分析
ekegg_deg <- performKeggEnrichment(entrez_ids_deg)
# 转换为数据框
ekegg_deg_df <- as.data.frame(ekegg_deg)
ekegg_deg_df$Regulation <- "Deg"
visualizeKeggEnrichment <- function(ekegg_deg_df) {
# 转换GeneRatio为小数
parts <- strsplit(as.character(ekegg_deg_df$GeneRatio), "/")
ekegg_deg_df$GeneRatio <- as.numeric(sapply(parts, `[`, 1)) /
as.numeric(sapply(parts, `[`, 2))
# 根据GeneRatio降序重新排序Description
ekegg_deg_df$Description <- reorder(
ekegg_deg_df$Description, ekegg_deg_df$GeneRatio
)
# 绘图
p <- ggplot(ekegg_deg_df, aes(x = GeneRatio, y = Description)) +
geom_point(aes(size = Count, color = -log10(p.adjust))) +
scale_color_continuous(low = "blue", high = "red") +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
theme_bw()
return(p)
}
# 绘图
p <- visualizeKeggEnrichment(ekegg_deg_df)
print(p)
使用 WikiPathways 数据库的富集分析¶
# 必需的库
library(readr)
library(ggplot2)
library(clusterProfiler)
library(org.Hs.eg.db)
gene_list <- read_csv("deseq2_results.csv")
# 将基因符号转换为Entrez ID
convertToEntrez <- function(genes) {
mapIds(
org.Hs.eg.db,
keys = genes,
column = "ENTREZID",
keytype = "ENSEMBL",
multiVals = "first"
)
}
# 执行WikiPathways富集分析
performWPEnrichment <- function(entrez_ids) {
enrichWP(gene = entrez_ids, organism = "Homo sapiens")
}
differentially_expressed_genes <- subset(
gene_list, abs(log2FoldChange) > 1 & pvalue < 0.05
)
# 将基因符号转换为Entrez ID
entrez_ids_deg <- convertToEntrez(differentially_expressed_genes$...1)
# 富集分析
ego_deg <- performWPEnrichment(entrez_ids_deg)
visualizeWPEnrichment <- function(enrichment_result) {
# 转换为数据框
df <- as.data.frame(enrichment_result)
# 转换GeneRatio为小数
parts <- strsplit(as.character(df$GeneRatio), "/")
df$GeneRatio <- as.numeric(sapply(parts, `[`, 1)) /
as.numeric(sapply(parts, `[`, 2))
# 根据GeneRatio降序重新排序Description
df$Description <- reorder(df$Description, df$GeneRatio)
# 绘图
p <- ggplot(df, aes(x = GeneRatio, y = Description)) +
geom_point(aes(size = Count, color = -log10(p.adjust))) +
scale_color_continuous(low = "blue", high = "red") +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
theme_bw()
return(p)
}
# 绘图
p <- visualizeWPEnrichment(ego_deg)
print(p)
请根据需要修改代码,运行并检查输出结果。
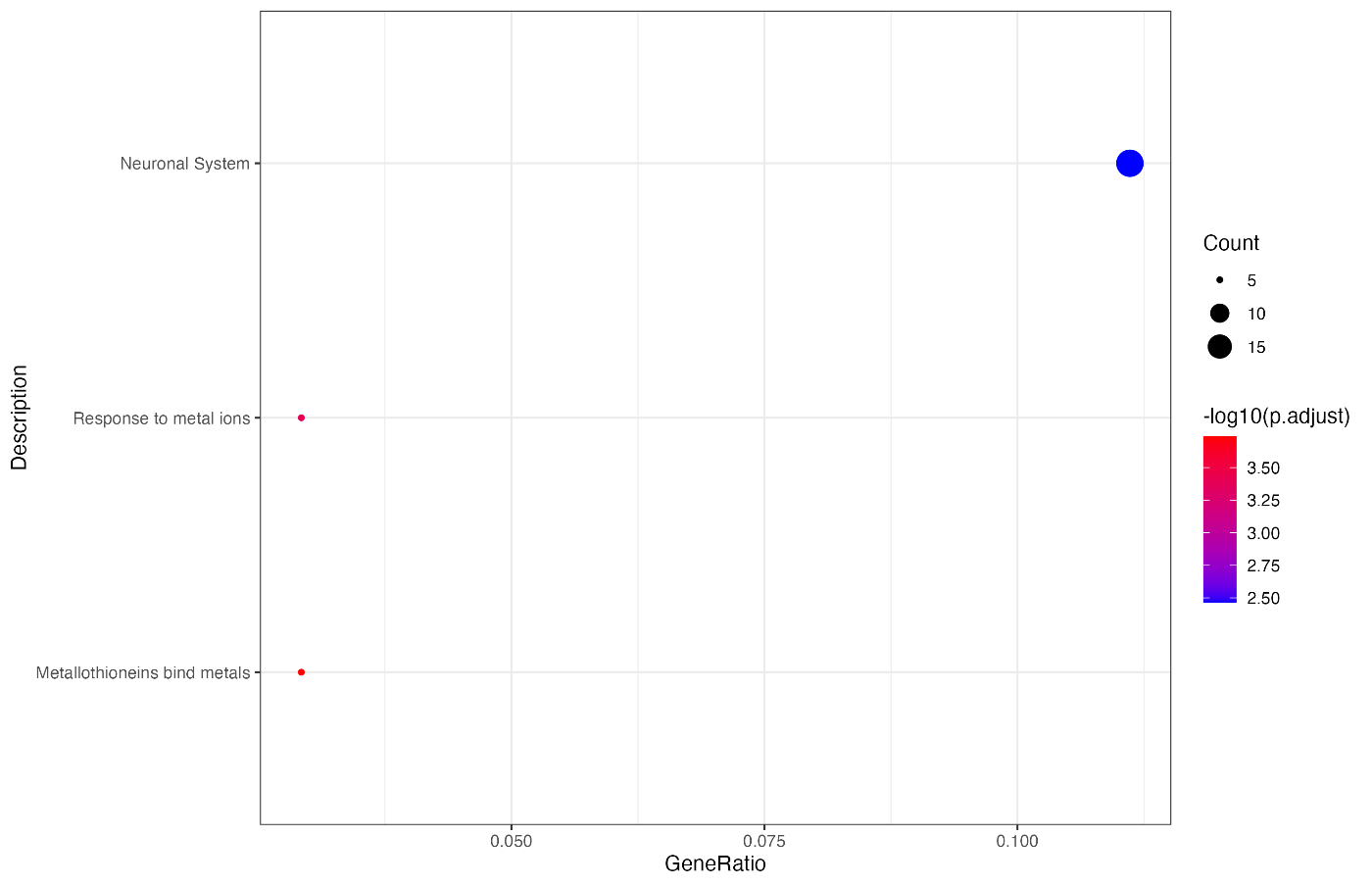
以下是使用 Reactome 数据库进行富集分析的详细步骤和代码:
1. 加载必要的库¶
library(readr)
library(patchwork)
library(ggplot2)
library(clusterProfiler)
library(ReactomePA)
library(org.Hs.eg.db)
这些库包括用于数据读取、数据处理和可视化的功能。
2. 读取数据¶
gene_list <- read_csv("deseq2_results.csv")
从 deseq2_results.csv 文件中读取 DESeq2 的结果。
3. 将基因符号转换为 Entrez ID¶
convertToEntrez <- function(genes) {
mapIds(
org.Hs.eg.db,
keys = genes,
column = "ENTREZID",
keytype = "ENSEMBL",
multiVals = "first"
)
}
定义一个函数,将基因符号转换为 Entrez ID。
4. 执行 Reactome 路径富集分析¶
performReactomePAEnrichment <- function(entrez_ids) {
enrichPathway(gene = entrez_ids, pvalueCutoff = 0.05, readable = TRUE)
}
定义一个函数,使用 enrichPathway 函数对给定的 Entrez ID 列表进行 Reactome 路径富集分析。
5. 选择差异表达基因¶
differentially_expressed_genes <- subset(
gene_list, abs(log2FoldChange) > 1 & pvalue < 0.05
)
根据 log2 倍变化和 p 值标准从基因列表中选择差异表达基因。
6. 转换为 Entrez ID¶
entrez_ids_deg <- convertToEntrez(differentially_expressed_genes$...1)
将选择的差异表达基因列表转换为 Entrez ID。
7. 富集分析¶
ego_deg <- performReactomePAEnrichment(entrez_ids_deg)
使用之前定义的 performReactomePAEnrichment 函数进行富集分析。
8. 可视化富集结果¶
visualizeReactomePAEnrichment <- function(enrichment_result) {
# 将enrichResult对象转换为数据框
df <- as.data.frame(enrichment_result)
# 将GeneRatio转换为小数
parts <- strsplit(as.character(df$GeneRatio), "/")
df$GeneRatio <- as.numeric(sapply(parts, `[`, 1)) /
as.numeric(sapply(parts, `[`, 2))
# 根据GeneRatio降序重新排序Description
df$Description <- reorder(df$Description, df$GeneRatio)
# 绘图
p <- ggplot(df, aes(x = GeneRatio, y = Description)) +
geom_point(aes(size = Count, color = -log10(p.adjust))) +
scale_color_continuous(low = "blue", high = "red") +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
theme_bw()
return(p)
}
# 绘图
p <- visualizeReactomePAEnrichment(ego_deg)
print(p)
定义一个函数来可视化富集分析的结果,然后生成并显示图表。
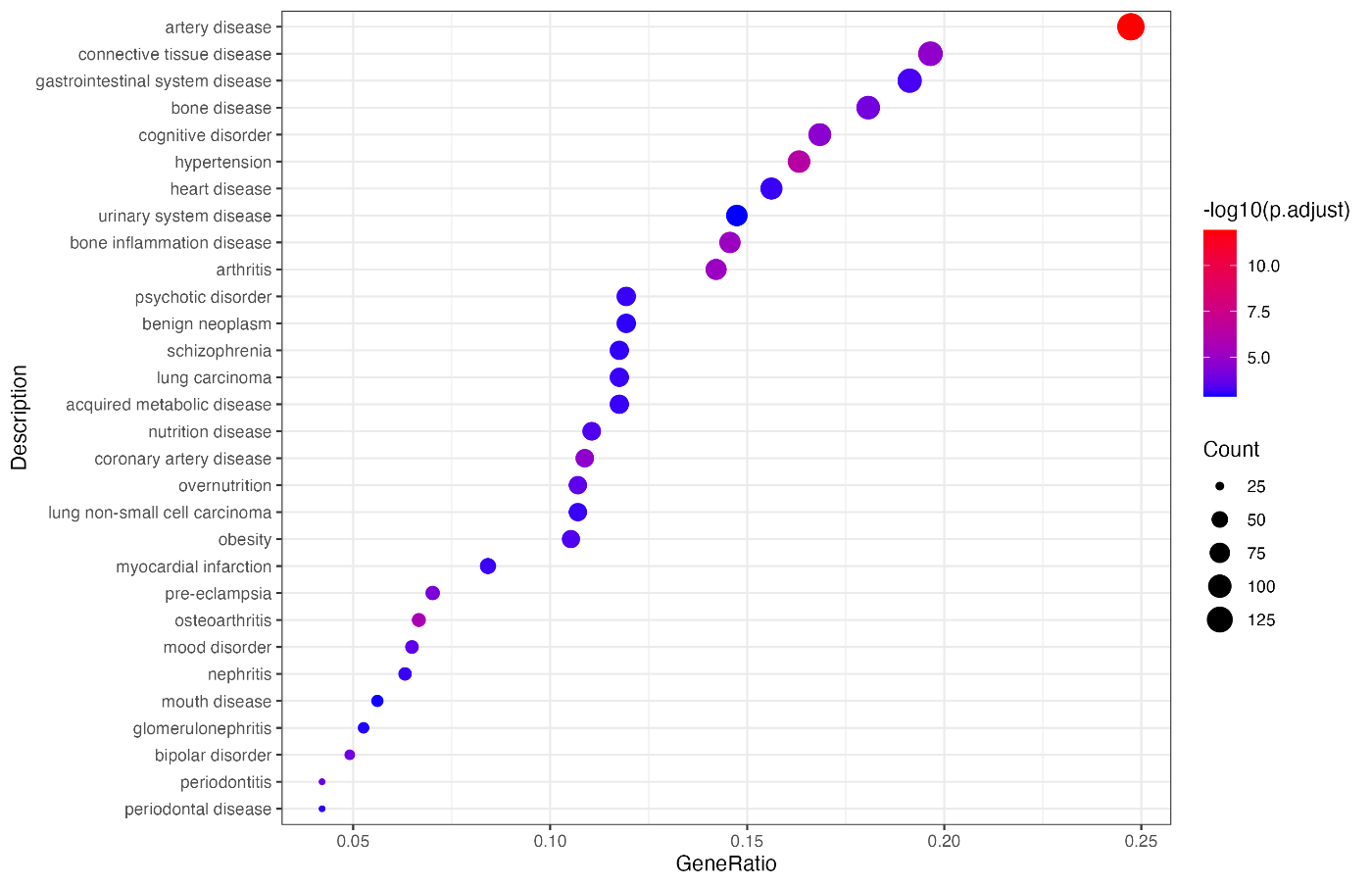
以下是使用 DOSE 数据库进行富集分析的详细步骤和代码:
1. 加载必要的库¶
library(readr)
library(patchwork)
library(ggplot2)
library(clusterProfiler)
library(DOSE)
library(org.Hs.eg.db)
这些库包括用于数据读取、数据处理和可视化的功能。
2. 读取数据¶
gene_list <- read_csv("deseq2_results.csv")
从 deseq2_results.csv 文件中读取 DESeq2 的结果。
3. 将基因符号转换为 Entrez ID¶
convertToEntrez <- function(genes) {
mapIds(
org.Hs.eg.db,
keys = genes,
column = "ENTREZID",
keytype = "ENSEMBL",
multiVals = "first"
)
}
定义一个函数,将基因符号转换为 Entrez ID。
4. 执行 DO 富集分析¶
performDOEnrichment <- function(entrez_ids) {
enrichDO(
gene = entrez_ids,
ont = "DO",
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
minGSSize = 5,
maxGSSize = 1500,
qvalueCutoff = 0.05,
readable = FALSE
)
}
定义一个函数,对给定的 Entrez ID 列表进行 DO 富集分析。
5. 选择差异表达基因¶
differentially_expressed_genes <- subset(
gene_list, abs(log2FoldChange) > 1 & pvalue < 0.05
)
使用 log2 倍变化和 p 值标准,从基因列表中选择差异表达基因。
6. 转换为 Entrez ID¶
entrez_ids_deg <- convertToEntrez(differentially_expressed_genes$...1)
将选择的差异表达基因列表转换为 Entrez ID。
7. 执行富集分析¶
ego_deg <- performDOEnrichment(entrez_ids_deg)
使用之前定义的 performDOEnrichment 函数进行富集分析。
8. 可视化富集结果¶
visualizeDOEnrichment <- function(enrichment_result) {
# 将enrichResult对象转换为数据框
df <- as.data.frame(enrichment_result)
# 将GeneRatio转换为小数
parts <- strsplit(as.character(df$GeneRatio), "/")
df$GeneRatio <- as.numeric(sapply(parts, `[`, 1)) /
as.numeric(sapply(parts, `[`, 2))
# 按p.adjust升序排序并选择前30个条目
df <- df[order(df$p.adjust)[1:30], ]
# 根据GeneRatio降序重新排序Description
df$Description <- reorder(df$Description, df$GeneRatio)
# 绘图
p <- ggplot(df, aes(x = GeneRatio, y = Description)) +
geom_point(aes(size = Count, color = -log10(p.adjust))) +
scale_color_continuous(low = "blue", high = "red") +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
theme_bw()
return(p)
}
# 绘图
p <- visualizeDOEnrichment(ego_deg)
print(p)
代码解读¶
- 加载必要的库:加载了用于数据读取、数据处理和可视化的 R 包。
- 读取数据:从 CSV 文件中读取 DESeq2 结果。
- 将基因符号转换为 Entrez ID:定义了一个函数
convertToEntrez,将基因符号转换为 Entrez ID。 - 执行 DO 富集分析:定义了一个函数
performDOEnrichment,对给定的 Entrez ID 列表进行疾病本体富集分析。 - 选择差异表达基因:从基因列表中选择符合条件的差异表达基因。
- 转换为 Entrez ID:将选择的差异表达基因列表转换为 Entrez ID。
- 执行富集分析:使用
performDOEnrichment函数对差异表达基因进行富集分析。 - 可视化富集结果:定义并使用
visualizeDOEnrichment函数将富集分析结果进行可视化,并生成一个图表。
运行上述代码后,你将获得一张图表,展示了基于 DOSE 数据库的差异表达基因的富集分析结果。