4.2 准备使用 Seurat 进行分析
什么是 Seurat?¶
Seurat 是一个用于单细胞 RNA 测序(scRNA-seq)数据分析的 R 包。它可以对 scRNA-seq 数据进行规范化处理和可视化分析。Seurat 提供了丰富的工具和功能,帮助研究人员深入分析单细胞水平的基因表达数据,从而揭示细胞异质性和生物学过程。
Seurat的介绍与安装
Seurat是一个用于单细胞RNA测序(scRNA-seq)数据分析的R包。通过Seurat,研究人员可以对scRNA-seq数据进行规范化处理、聚类分析、降维和可视化等操作。Seurat的制作者提供了详细的教程,帮助用户更好地理解和使用这个工具。以下是关于Seurat安装和环境设置的详细步骤和说明。
Seurat的安装和环境设置¶
在开始Seurat的教程之前,首先需要安装必要的R包。如果使用Docker镜像,运行install.packages("Seurat")时会安装v5版本,因此建议跳过这一步。
- 安装必要的R包:
install.packages("dplyr")
install.packages("Seurat") # 使用Docker镜像的用户建议跳过此步骤
install.packages("patchwork")
- 加载安装好的R包:
library(dplyr)
library(Seurat)
library(patchwork)
获取生数据¶
从Seurat的官方网站下载生数据。访问以下链接,并从页面中的"here"下载数据:
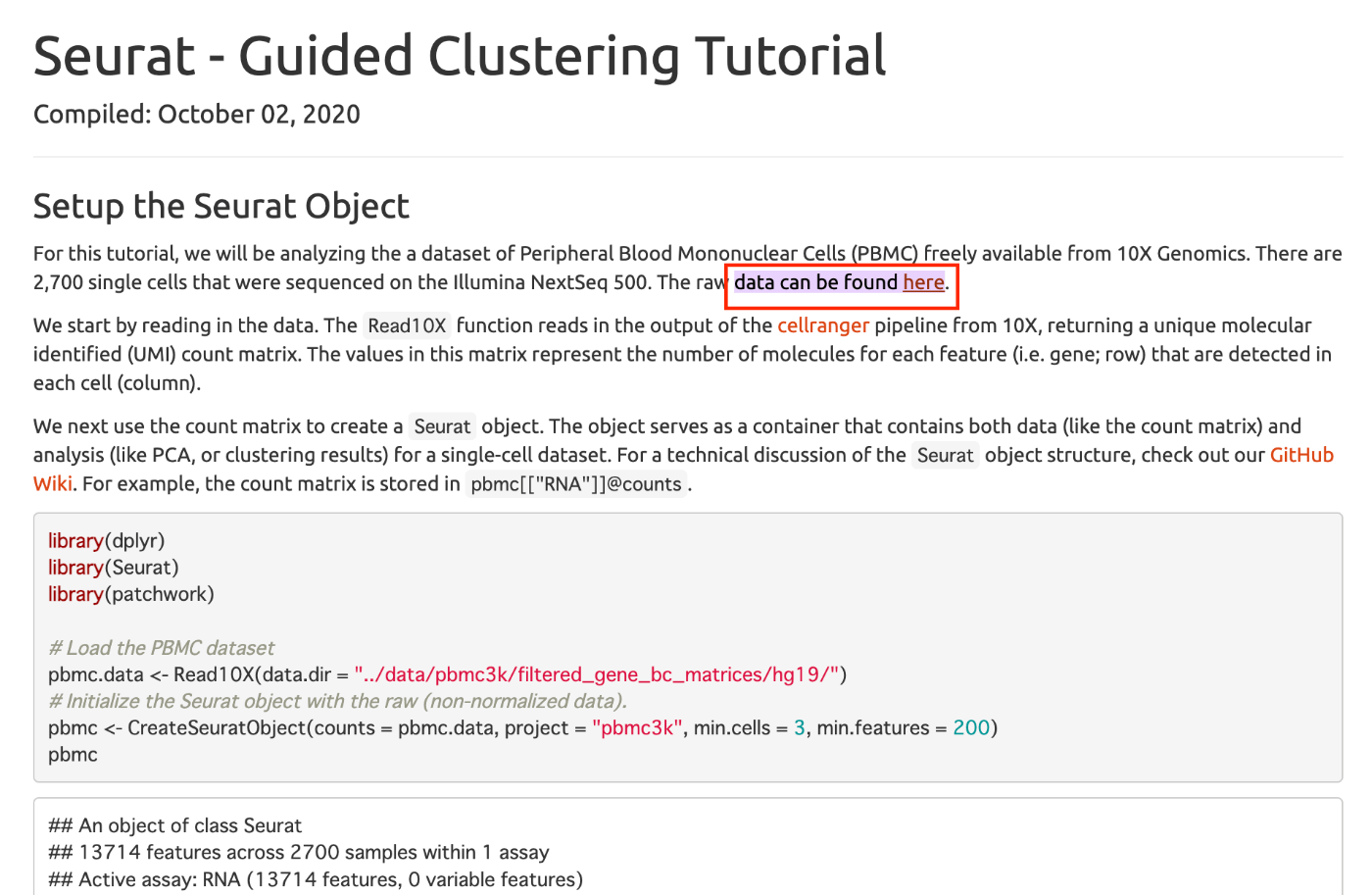
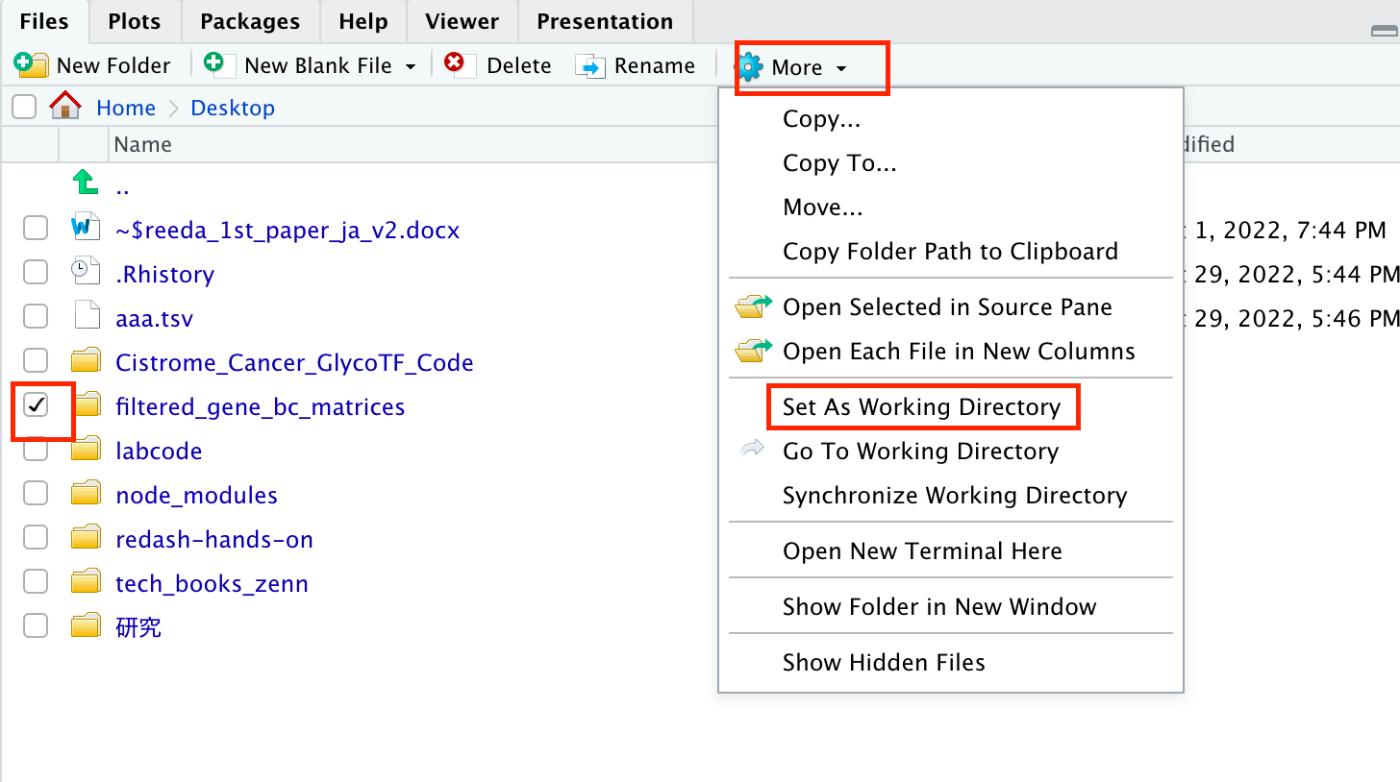
请将下载的文件解压到桌面上。然后在RStudio的右下角窗口中导航到桌面,按照以下步骤操作:
- 勾选下载的文件
- 点击"More"
- 选择"Set As Working Directory"
请注意,如果操作错误,后续代码将无法运行。
执行以下命令,如果输出的路径以Desktop结尾,则说明设置正确(仅适用于Mac)。
> getwd()
[1] "/Users/[用户名]/Desktop"