4.3 使用 Seurat 进行数据预处理
创建 Seurat 对象和导入数据¶
首先执行以下命令,创建 Seurat 对象。这将完成数据的导入。
pbmc.data <- Read10X(data.dir = "./filtered_gene_bc_matrices/hg19/")
pbmc <- CreateSeuratObject(
counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200
)
pbmc
质量控制 (QC) 和过滤¶
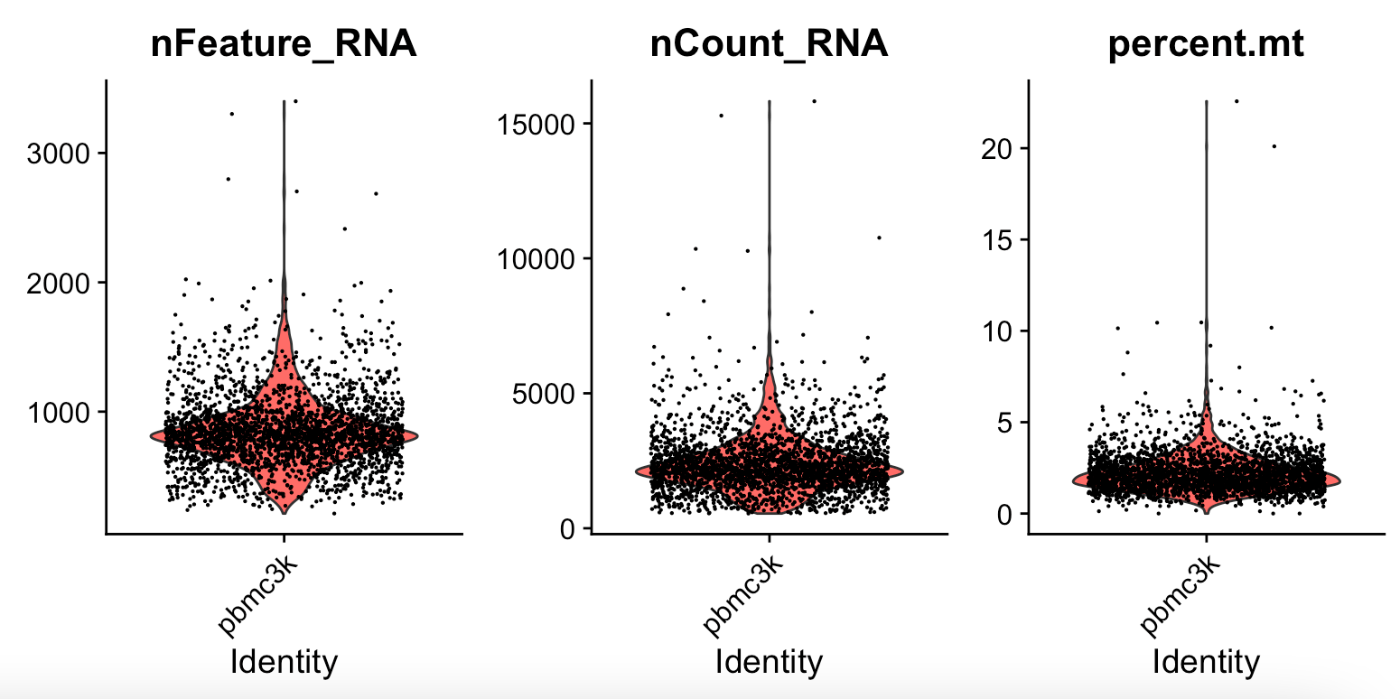
在 scRNA-seq 中,使用小提琴图进行质量控制,以检查是否存在异常的细胞。
低质量的细胞(如死细胞)通常会表现出极低的基因表达量或存在线粒体来源的污染。此外,如果在计数基因数时细胞发生双胞现象(即不是单细胞),则会导致异常的基因数计数。
# 使用以MT-开头的所有基因作为线粒体基因集合
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
运行上述代码后,RStudio 的右下角应会出现如下图所示的小提琴图。
通过查看小提琴图的膨胀部分,可以确定细胞群体的合理基因计数。以下是此次样本的质量控制参考值:
- 基于
nFeature_RNA,过滤掉唯一特征数大于 2,500 或小于 200 的细胞。 - 基于
percent.mt,过滤掉线粒体基因数超过 5% 的细胞。
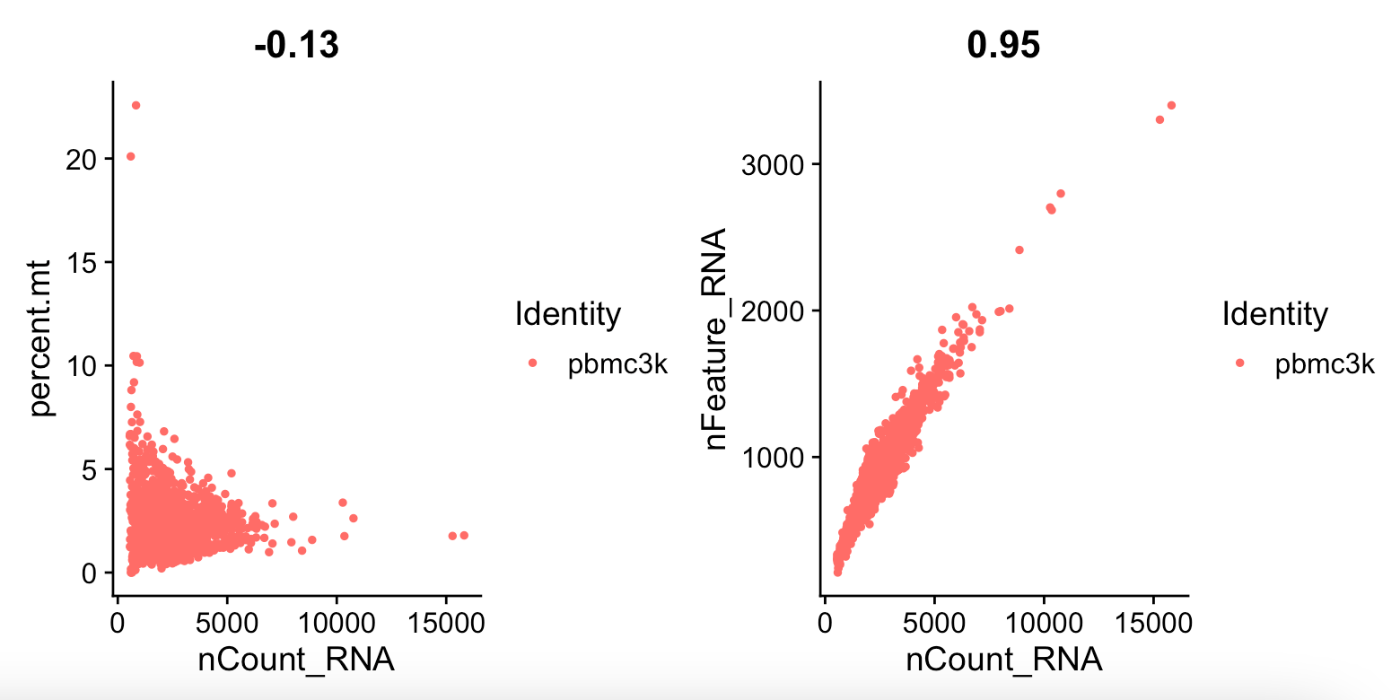
为了查看各变量之间的相关性,可以使用散点图。请执行以下代码:
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
运行上述代码后,你会看到两个散点图:
percent.mt与nCount_RNA之间没有显著的相关性(相关系数为 -0.13)。nFeature_RNA与nCount_RNA之间存在显著的相关性(相关系数为 0.95)。
重新筛选符合 QC 标准的细胞¶
在进行质量控制后,我们需要将筛选出的细胞重新存储到 pbmc 对象中:
pbmc <- subset(
pbmc,
subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5
)
数据的规范化和缩放¶
在通过 QC 从数据集中移除不需要的细胞后,需要对数据进行规范化处理。
默认情况下,Seurat 采用全局缩放规范化方法“LogNormalize”。这种方法通过对每个细胞的特征表达量进行规范化,除以总表达量,然后乘以缩放因子(默认是 10,000),并对结果进行对数转换。
# 数据规范化
pbmc <- NormalizeData(
pbmc,
normalization.method = "LogNormalize",
scale.factor = 10000
)
# 将规范化后的数据重新存储到pbmc对象中
pbmc <- NormalizeData(pbmc)
到此为止,scRNA-seq 数据的预处理已完成。接下来的步骤将是对规范化数据的可视化方法进行解释。