5.2 为每个簇自动添加细胞注释标签
实践结束后,进行了 UMAP 绘制,可能会得到以下输出结果。
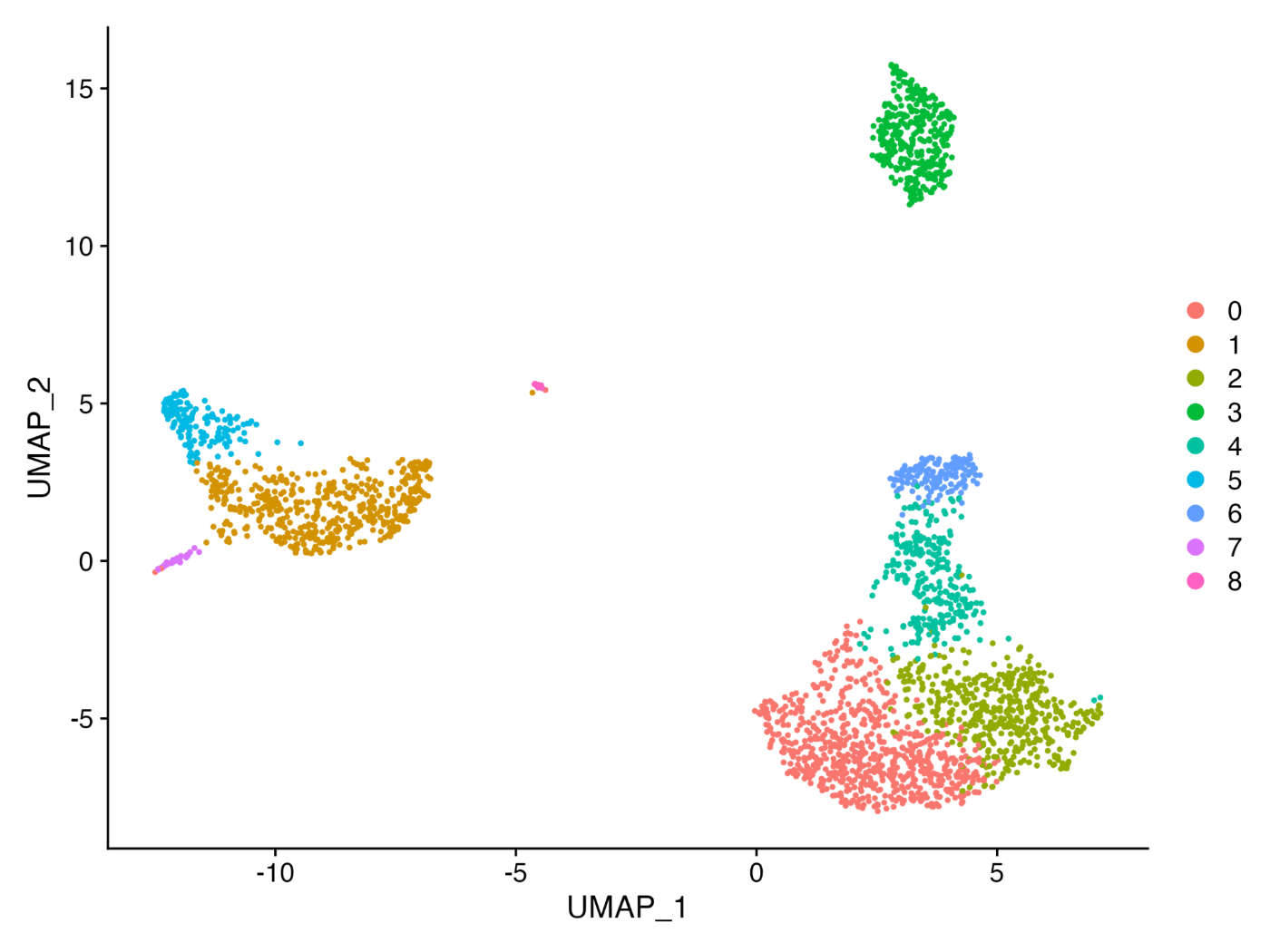
上述步骤中,我们已经将数据分为 8 个簇。但是,以下问题可能会出现:
- 各个簇是什么细胞?
- 是否存在特异性表达的生物标志物?
我们将使用 Seurat 来解析这些问题。
确认所用的数据集¶
现在我们开始进行分析。此次使用的数据集如下:
- 论文标题: Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19
- 数据集: GSE149689
在这篇论文中,通过 Single Cell RNA-seq 分析,研究了 I 型干扰素响应在重症 COVID-19 炎症进展中的作用。论文中使用的数据集 GSE149689 在另一篇论文中被重新分析:
- 论文标题: Early peripheral blood MCEMP1 and HLA-DRA expression predicts COVID-19 prognosis
这篇论文通过重新分析包括 GSE149689 在内的 7 个数据集,研究了重症 COVID-19 的预后生物标志物:CD14+ 细胞的 MCEMP1 和 HLA-DRA 基因表达。
由此可见,公共数据库中的 scRNA-seq 数据通过重新分析,可以用于发表新的论文。
实际操作¶
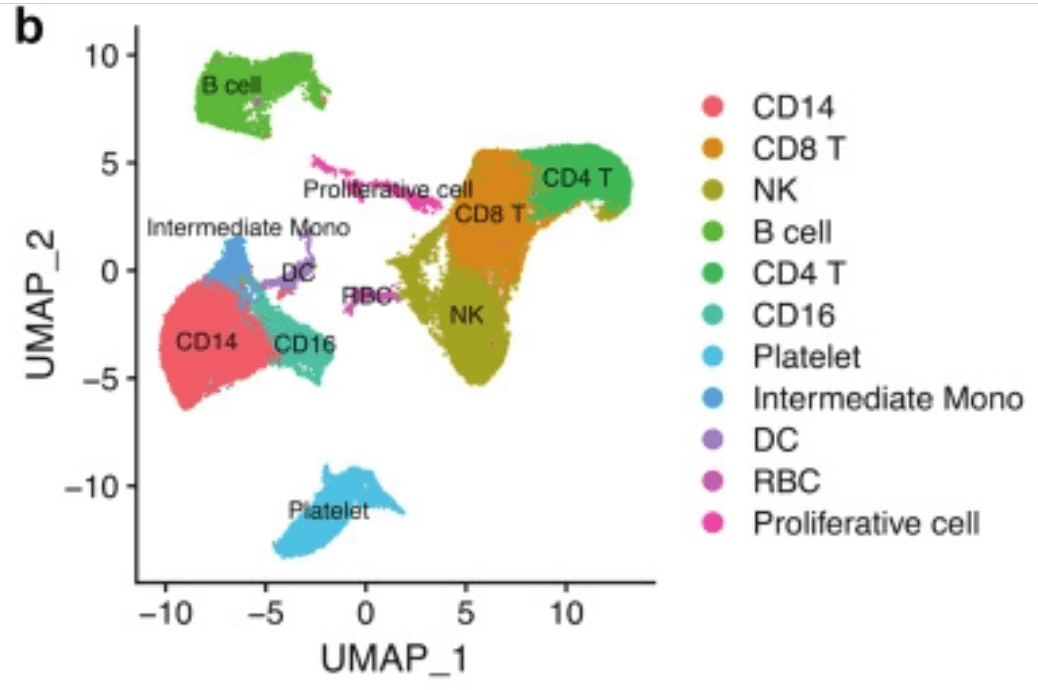
我们将尝试通过 Seurat 对 GSE149689 数据集进行分析,重现论文 Early peripheral blood MCEMP1 and HLA-DRA expression predicts COVID-19 prognosis 中的图 4b。虽然完全相同可能比较困难,但可以生成相似的图。
scRNA-seq 数据的预处理¶
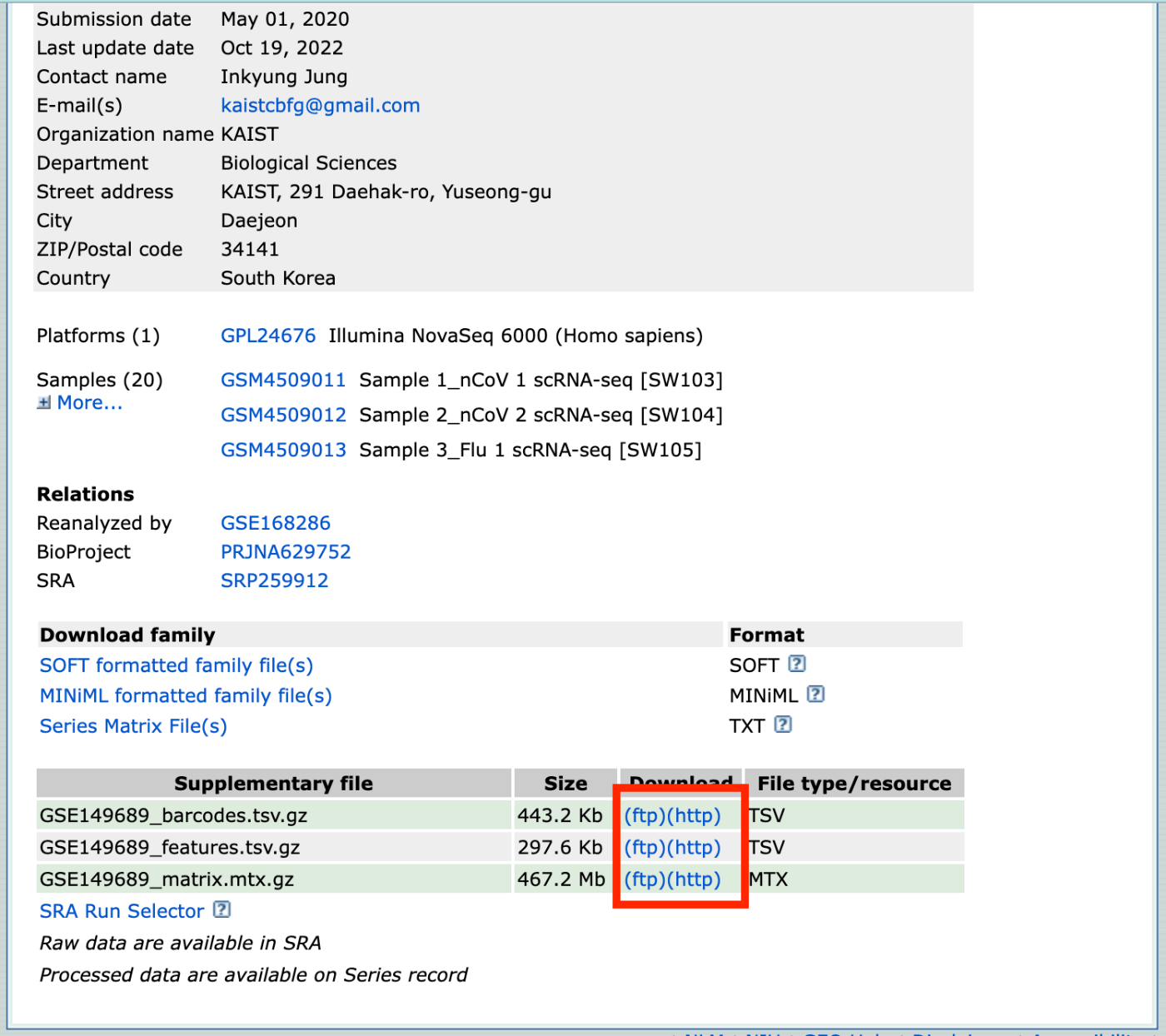
下载 GSE149689。[此处](https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE149689)。
转到页面底部,您将看到补充文件,点击 ftp 或 http 下载所有三个文件。
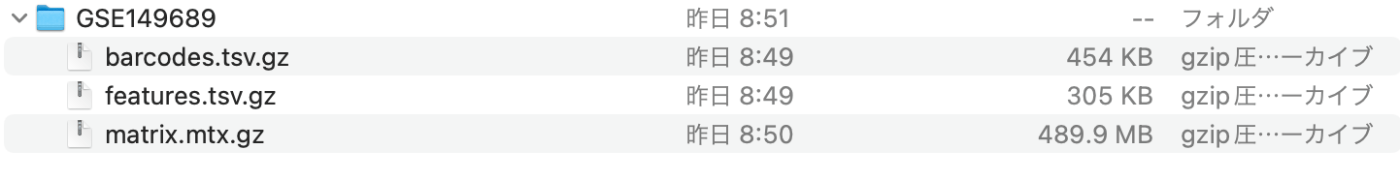
下载后,重新命名,只保留条形码、特征和矩阵的名称,如下图所示。方法与实践课程中的相同。
启动 Rstudio 并设置工作目录。
这次,我们在桌面上创建了一个名为 "GSE149689 " 的文件夹,将之前下载的文件存储在其中,并将桌面文件夹设置为工作目录。如果稍后加载数据时能正确指定路径,就没有问题了。
接下来,创建一个 SeuratObject。在编辑器中输入以下代码。
接下来,我们将创建 Seurat 对象并导入数据。请在编辑器中编写以下代码。
install.packages("dplyr")
install.packages("Seurat") # 使用Docker镜像的用户建议跳过此步骤
install.packages("patchwork")
library(dplyr)
library(Seurat)
library(patchwork)
# 加载数据集
pbmc.data <- Read10X(data.dir = "./GSE149689/")
# 使用原始(非规范化)数据初始化Seurat对象
pbmc <- CreateSeuratObject(
counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200
)
CreateSeuratObject 函数中的各参数说明如下,几乎使用默认值:
counts: 包含每个细胞基因表达数据的矩阵。在此例中输入的是pbmc.data。project: 分配给创建的 Seurat 对象的项目名称。在此例中指定为 "PBMC"。min.cells: 只包含在此数量以上的细胞中检测到的基因的阈值。在此例中,包含至少在 3 个细胞中检测到的基因。min.features: 只包含检测到此数量以上基因的细胞的阈值。在此例中,包含至少检测到 200 个基因的细胞。
接下来进行质量控制(QC)和规范化¶
条件在论文中有描述,如下:
单细胞数据处理和分析
使用 Seurat R 包(版本 3)进行单细胞数据的综合分析,使用 UMAP 进行单细胞可视化。在质量控制期间,去除线粒体基因比率超过 15% 的细胞,这些细胞可能是潜在的死亡细胞。仅保留基因数量在 200-5000 范围内或检测到的 RNA 数量在 1000 到 30000 个细胞之间的细胞。质量控制后,包含 48583 个 PBMC 和 78666 个 BALF 单细胞用于后续分析。数据使用 Seurat 包进行规范化,并对变异系数最大的前 2000 个基因进行主成分分析。
总结 QC 和规范化的要点如下:
- 去除线粒体基因比率超过 15% 的细胞
- 只保留基因数量在 200-5000 范围内或检测到的 RNA 数量在 1000 到 30000 的细胞
- 使用 Seurat 包进行数据规范化
几乎所有论文都会描述 QC 和规范化方法,请参考论文内容继续操作。
编写代码进行质量控制和规范化¶
首先,去除线粒体基因比率超过 15% 的细胞,只保留基因数量在 200-5000 范围内或检测到的 RNA 数量在 1000 到 30000 的细胞,最后进行规范化。
# 计算线粒体基因比例
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
# 质量控制
pbmc <- subset(
pbmc, subset = nFeature_RNA >= 200 & nFeature_RNA <= 5000 & percent.mt < 15
)
前处理 scRNA-seq 数据¶
使用第四章介绍的 Seurat 前处理步骤,并通过管道操作符 %>% 连接各步骤。如果不理解各步骤的含义,请回顾第四章内容。
pbmc <- pbmc %>%
NormalizeData() %>%
FindVariableFeatures(selection.method = "vst", nfeatures = 2000) %>%
ScaleData(features = VariableFeatures(object = pbmc)) %>%
RunPCA() %>%
RunUMAP(dims = 1:20) %>%
FindNeighbors(dims = 1:10) %>%
FindClusters(resolution = 0.5)
使用 DimPlot 可视化 UMAP¶
# 可视化
DimPlot(pbmc, reduction = "umap", label = TRUE)
如果成功,你将得到如下的 UMAP 图。到此为止,聚类的可视化已经完成。
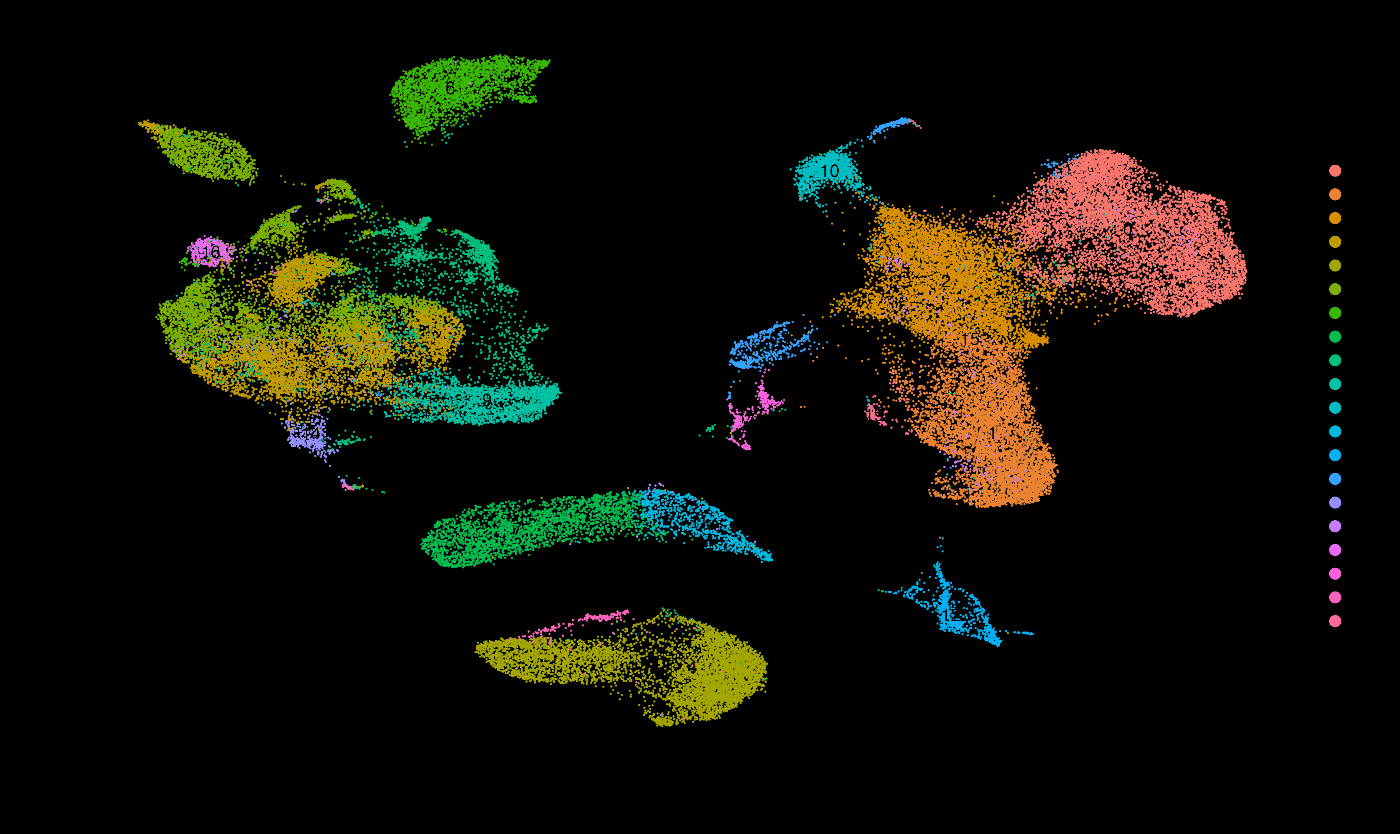
使用 SingleR 对细胞簇进行自动注释
到目前为止,我们已经从公共数据下载了实际数据,并在 UMAP 中显示了它们。然而,尽管我们可以观察到一些簇,但仍需要确定每个簇代表什么。
我们将介绍如何使用 celldex、SingleR 和 SingleCellExperiment 库为每个细胞自动添加细胞类型标签。以下是各库的说明:
- celldex: 一系列单细胞 RNA 测序(scRNA-seq)数据集的集合。
- SingleR: 一个库,通过将带有细胞类型注释的参考数据集与未知注释的样本数据进行比较,推断细胞类型。
- SingleCellExperiment: 为 R 编程语言和 Bioconductor 项目开发的包,提供了单细胞 RNA 测序(scRNA-seq)数据的存储、操作和分析的数据结构。
实际代码¶
请注意,当前由于 dbplyr 包的升级,可能会导致无法正常获取参考数据。您可以通过将 dbplyr 降级到版本 2.3.4 来暂时解决这个问题。
devtools::install_version("dbplyr", version = "2.3.4")
# 检查是否安装了BiocManager,如果没有则安装
if (!require("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
# 使用BiocManager安装所需的包
BiocManager::install("SingleR")
BiocManager::install("celldex")
BiocManager::install("SingleCellExperiment")
# 加载所需的库
library(SingleR)
library(celldex)
library(SingleCellExperiment)
# 从celldex加载参考数据
ref <- celldex::HumanPrimaryCellAtlasData()
# 使用参考数据运行SingleR推断pbmc数据集的细胞类型
results <- SingleR(
test = as.SingleCellExperiment(pbmc), ref = ref, labels = ref$label.main
)
# 将推断的细胞类型标签添加到pbmc对象
pbmc$singlr_labels <- results$labels
# 在UMAP图中可视化带有标签的细胞类型
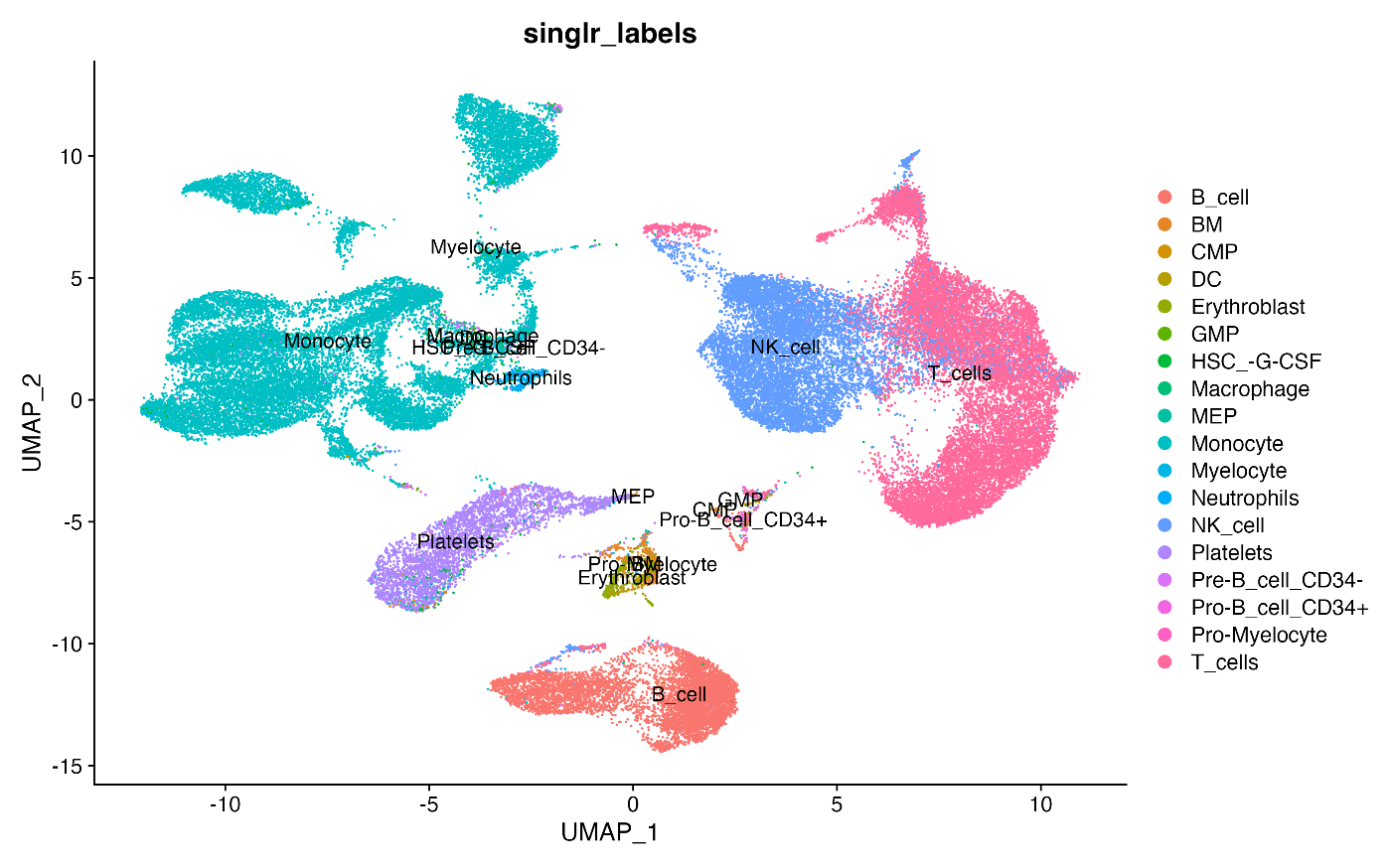
DimPlot(pbmc, reduction = "umap", group.by = "singlr_labels", label = TRUE)
运行上述代码后,如果得到如下图所示的数据,则表示成功!
代码解读
# Check if BiocManager is installed, if not, install it
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
# Install required packages using BiocManager
BiocManager::install("SingleR")
BiocManager::install("celldex")
BiocManager::install("SingleCellExperiment")
# Load required libraries
library(SingleR)
library(celldex)
library(SingleCellExperiment)
以上代码用于安装并加载所需的库。
# Load reference data from celldex
ref <- celldex::HumanPrimaryCellAtlasData()
使用 HumanPrimaryCellAtlasData 函数从 celldex 包中获取 Human Primary Cell Atlas 的参考数据,并存储在变量 ref 中。
# Run SingleR to infer cell types of pbmc dataset using reference data
results <- SingleR(
test = as.SingleCellExperiment(pbmc), ref = ref, labels = ref$label.main
)
使用 SingleR 函数推断 pbmc 数据集的细胞类型。该推断使用第一行获取的 ref 作为参考数据,并指定 ref$label.main 作为细胞类型标签。推断结果存储在变量 results 中。
# Add inferred cell type labels to pbmc object
pbmc$singlr_labels <- results$labels
将 SingleR 推断的细胞类型标签添加到 pbmc 对象的 singlr_labels 元数据中,使得细胞类型信息可以在可视化和后续分析中使用。
# Visualize cell types in a UMAP plot with labels
DimPlot(pbmc, reduction = "umap", group.by = "singlr_labels", label = TRUE)
使用 DimPlot 函数创建一个使用 UMAP(Uniform Manifold Approximation and Projection)降维技术的图。图中的细胞根据 SingleR 推断的细胞类型标签(singlr_labels)进行分组,并为每个组添加标签。这使得可以直观地查看数据集中细胞类型的分布。