5.4 将单细胞 RNA seq 数据集相互结合
关于 merge 函数
scRNA-seq 数据集的合并是指将两个或多个独立的 scRNA-seq 数据集合并成一个综合数据集的过程。您可能会想,为什么需要将分别获取的数据合并呢?主要有两个原因。
第一,是为了汇总不同时间点或条件下的样本。研究人员为了捕捉时间变化或不同实验条件下细胞的变化,可能会对多个样本进行测序。通过合并这些样本,可以捕捉整体的动态变化和条件之间的差异。
第二,是由于技术原因,一次能够测序的细胞数量有限,为了研究大量细胞,必须将样本分成多个数据集进行测序。
在 Seurat 包的上下文中,merge 函数是用于将多个 Seurat 对象合并为一个对象的函数。使用此函数,可以将多个数据集作为一个整体进行处理。
需要注意的是,合并并不包括批次效应的校正,因此仅仅将 Seurat 对象合并并不能解决批次效应问题。如果批次效应较小或者其影响在可接受范围内,可以使用 merge 进行简单的数据合并。
数据集准备¶
现在我们来尝试合并数据集。
这部分内容基于以下教程进行解说。
首先,访问以下数据集:
向下滚动页面,找到红色框标记的 "Gene / cell matrix(filtered)",并分别下载每个数据集。
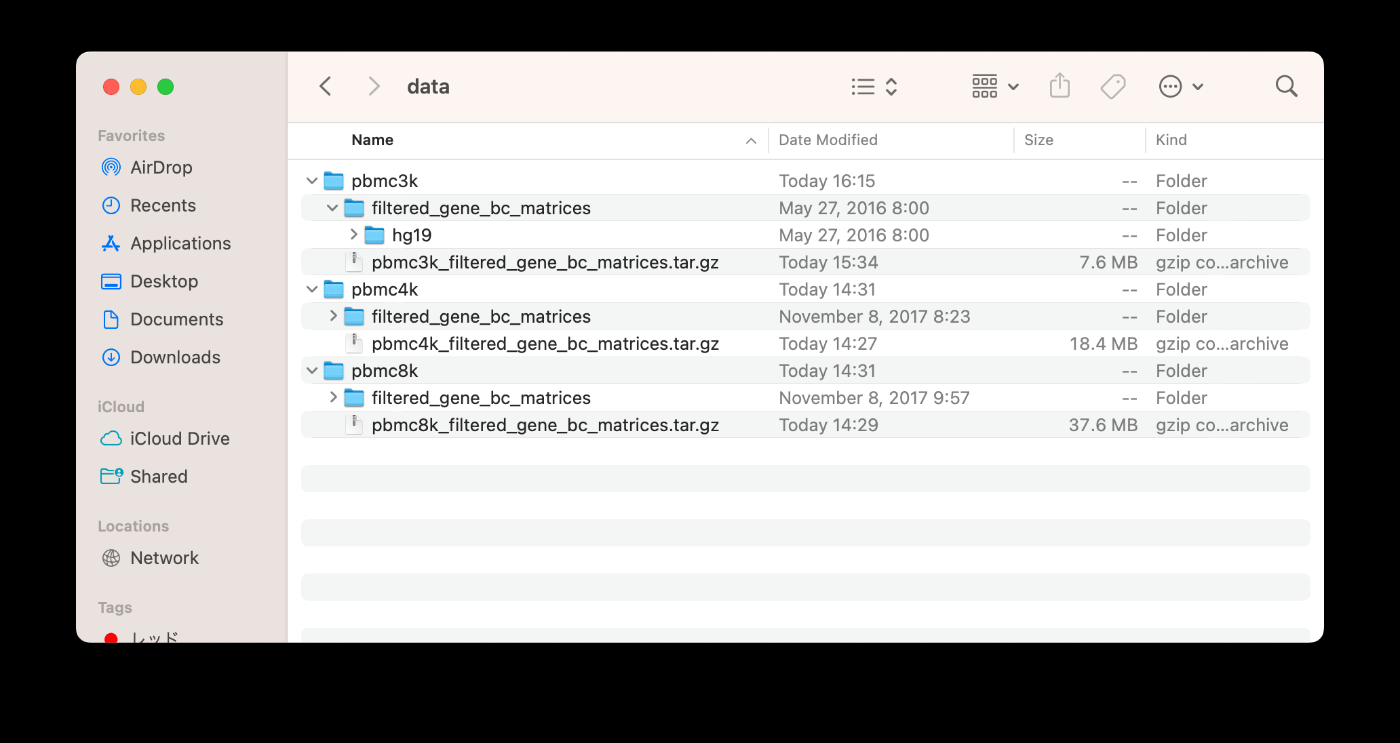
在桌面上创建一个名为 data 的文件夹,并按照以下图片中的目录结构进行组织。每个文件夹中的 filtered_gene_bc_matrices 是通过解压 gzip 文件生成的。目录路径如下:
# pbmc4k
"./data/pbmc4k/filtered_gene_bc_matrices/GRCh38/"
# pbmc8k
"./data/pbmc8k/filtered_gene_bc_matrices/GRCh38/"
# pbmc3k
"./data/pbmc3k/filtered_gene_bc_matrices/hg19/"
这里完成了准备工作。
结合两个 scRNA-seq 数据集¶
现在让我们实际将 scRNA-seq 数据结合在一起。在结合前后,我们将不断检查输出以确保每一步都正确。
首先,我们来查看 pbmc4k 数据。
pbmc4k.data <- Read10X(data.dir = "./data/pbmc4k/filtered_gene_bc_matrices/GRCh38/")
pbmc4k <- CreateSeuratObject(counts = pbmc4k.data, project = "PBMC4K")
pbmc4k
pbmc4k 的输出如下。它包含 4340 个样本。
> pbmc4k
An object of class Seurat
33694 features across 4340 samples within 1 assay
Active assay: RNA (33694 features, 0 variable features)
接下来,我们查看 pbmc8k 数据。
pbmc8k.data <- Read10X(data.dir = "./data/pbmc8k/filtered_gene_bc_matrices/GRCh38/")
pbmc8k <- CreateSeuratObject(counts = pbmc8k.data, project = "PBMC8K")
pbmc8k
pbmc8k 的输出如下。它包含 8381 个样本。
> pbmc8k
An object of class Seurat
33694 features across 8381 samples within 1 assay
Active assay: RNA (33694 features, 0 variable features)
现在我们将 pbmc4k 和 pbmc8k 结合在一起。
代码如下:
pbmc.combined <- merge(
pbmc4k,
y = pbmc8k,
add.cell.ids = c("4K", "8K"),
project = "PBMC12K"
)
pbmc.combined
此代码使用 Seurat 包的 merge 函数将两个 scRNA-seq 数据集(pbmc4k 和 pbmc8k)结合起来,并创建一个新的数据集 pbmc.combined。
> pbmc.combined
An object of class Seurat
33694 features across 12721 samples within 1 assay
Active assay: RNA (33694 features, 0 variable features)
我们看到 pbmc.combined 包含 12721 个样本。4340 (pbmc4k) + 8381 (pbmc8k) = 12721 个样本,这表明结合成功。
接下来,我们确认细胞的识别符是否正确添加。
head(colnames(pbmc.combined))
tail(colnames(pbmc.combined))
输出显示每个细胞名已添加前缀 4K_ 和 8K_。
> head(colnames(pbmc.combined))
[1] "4K_AAACCTGAGAAGGCCT-1" "4K_AAACCTGAGACAGACC-1" "4K_AAACCTGAGATAGTCA-1"
[4] "4K_AAACCTGAGCGCCTCA-1" "4K_AAACCTGAGGCATGGT-1" "4K_AAACCTGCAAGGTTCT-1"
> tail(colnames(pbmc.combined))
[1] "8K_TTTGTCAGTTACCGAT-1" "8K_TTTGTCATCATGTCCC-1" "8K_TTTGTCATCCGATATG-1"
[4] "8K_TTTGTCATCGTCTGAA-1" "8K_TTTGTCATCTCGAGTA-1" "8K_TTTGTCATCTGCTTGC-1"
接着我们统计 pbmc4k 和 pbmc8k 的数量。
table(pbmc.combined$orig.ident)
输出如下,确认了我们之前的结果。
> table(pbmc.combined$orig.ident)
PBMC4K PBMC8K
4340 8381
结合两个以上的 scRNA-seq 数据集¶
我们已经成功结合了两个 scRNA-seq 数据集。现在,我们尝试结合三个数据集。
首先读取第三个数据集 pbmc3k。
pbmc3k.data <- Read10X(data.dir = "./data/pbmc3k/filtered_gene_bc_matrices/hg19/")
pbmc3k <- CreateSeuratObject(counts = pbmc3k.data, project = "PBMC3K")
pbmc3k
pbmc3k 的输出如下。它包含 2700 个样本。
> pbmc3k
An object of class Seurat
32738 features across 2700 samples within 1 assay
Active assay: RNA (32738 features, 0 variable features)
现在使用 merge 函数结合三个数据集。
pbmc.big <- merge(
pbmc3k,
y = c(pbmc4k, pbmc8k),
add.cell.ids = c("3K", "4K", "8K"),
project = "PBMC15K"
)
pbmc.big
pbmc.big 的输出如下。它包含 15421 个样本。
> pbmc.big
An object of class Seurat
36116 features across 15421 samples within 1 assay
Active assay: RNA (36116 features, 0 variable features)
检查细胞识别符是否正确添加:
unique(sapply(X = strsplit(colnames(pbmc.big), split = "_"), FUN = "[", 1))
输出如下,显示 "3K" "4K" "8K" 已正确添加。
> unique(sapply(X = strsplit(colnames(pbmc.big), split = "_"), FUN = "[", 1))
[1] "3K" "4K" "8K"
统计 pbmc3k、pbmc4k 和 pbmc8k 的数量。
table(pbmc.big$orig.ident)
输出如下,确认了之前的结果。
> table(pbmc.big$orig.ident)
PBMC3K PBMC4K PBMC8K
2700 4340 8381
基于规范化数据进行结合¶
merge 函数可以结合已规范化的数据集。规范化可以保持数据的一致性,避免后续分析步骤中的问题。
首先规范化 pbmc4k 和 pbmc8k 数据。
pbmc4k <- NormalizeData(pbmc4k)
pbmc8k <- NormalizeData(pbmc8k)
pbmc.normalized <- merge(
pbmc4k,
y = pbmc8k,
add.cell.ids = c("4K", "8K"),
project = "PBMC12K",
merge.data = TRUE
)
使用 GetAssayData 函数查看规范化数据。
GetAssayData(pbmc.normalized)[1:10, 1:15]
输出如下。为了更清晰,我们也查看规范化前的输出。
> GetAssayData(pbmc.normalized)[1:10, 1:15]
10 x 15 sparse Matrix of class "dgCMatrix"
[[ suppressing 15 column names ‘4K_AAACCTGAGAAGGCCT-1’, ‘4K_AAACCTGAGACAGACC-1’, ‘4K_AAACCTGAGATAGTCA-1’ ... ]]
RP11-34P13.3 . . . . . . . . . . . . . .
FAM138A . . . . . . . . . . . . . .
OR4F5 . . . . . . . . . . . . . .
RP11-34P13.7 . . . . . . . . . . . . . .
RP11-34P13.8 . . . . . . . . . . . . . .
RP11-34P13.14 . . . . . . . . . . . . . .
RP11-34P13.9 . . . . . . . . . . . . . .
FO538757.3 . . . . . . . . . . . . . .
FO538757.2 . . . . . . . . . 0.7721503 . . . .
AP006222.2 . . . . . . . . . . . 1.087928 . .
查看规范化前的输出。
GetAssayData(pbmc.combined)[1:10, 1:15]
生的计数数据通常只包含非负整数,而规范化后的数据通常会转换为浮点数值,并且一般包含大于 0 的浮点值。
> GetAssayData(pbmc.combined)[1:10, 1:15]
10 x 15 sparse Matrix of class "dgCMatrix"
[[ suppressing 15 column names ‘4K_AAACCTGAGAAGGCCT-1’, ‘4K_AAACCTGAGACAGACC-1’, ‘4K_AAACCTGAGATAGTCA-1’ ... ]]
RP11-
34P13.3 . . . . . . . . . . . . . . .
FAM138A . . . . . . . . . . . . . . .
OR4F5 . . . . . . . . . . . . . . .
RP11-34P13.7 . . . . . . . . . . . . . . .
RP11-34P13.8 . . . . . . . . . . . . . . .
RP11-34P13.14 . . . . . . . . . . . . . . .
RP11-34P13.9 . . . . . . . . . . . . . . .
FO538757.3 . . . . . . . . . . . . . . .
FO538757.2 . . . . . . . . . 1 . . . . .
AP006222.2 . . . . . . . . . . . 1 . . .