5.5 纠正批次效应,比较对照组和刺激组
本节中,我们将基于 Seurat 手册中的这篇文章编写代码。
单个样本的 scRNA-seq 数据分析可以在一定范围内进行可视化,但要比较多个样本,尤其是像对照组和刺激组之间的条件比较,就需要将这些样本进行整合后再进行比较。掌握数据整合的方法,可以清晰地了解不同条件下的细胞响应变化和复杂细胞类型之间的差异。
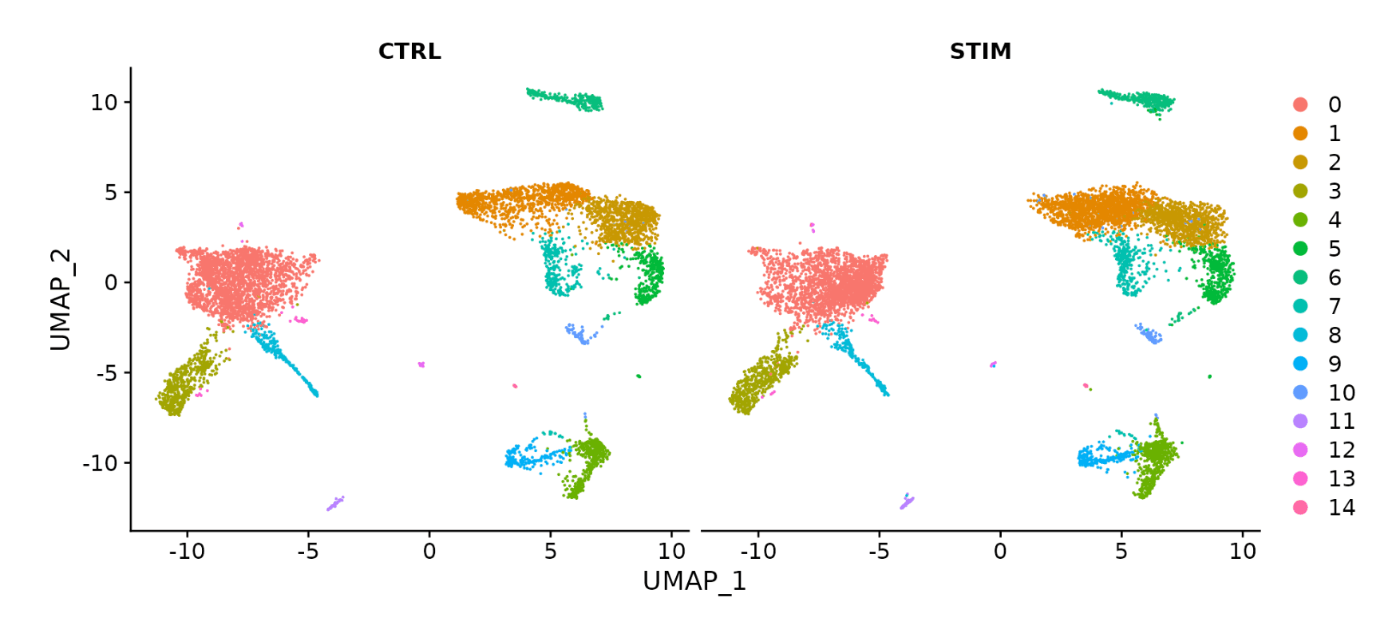
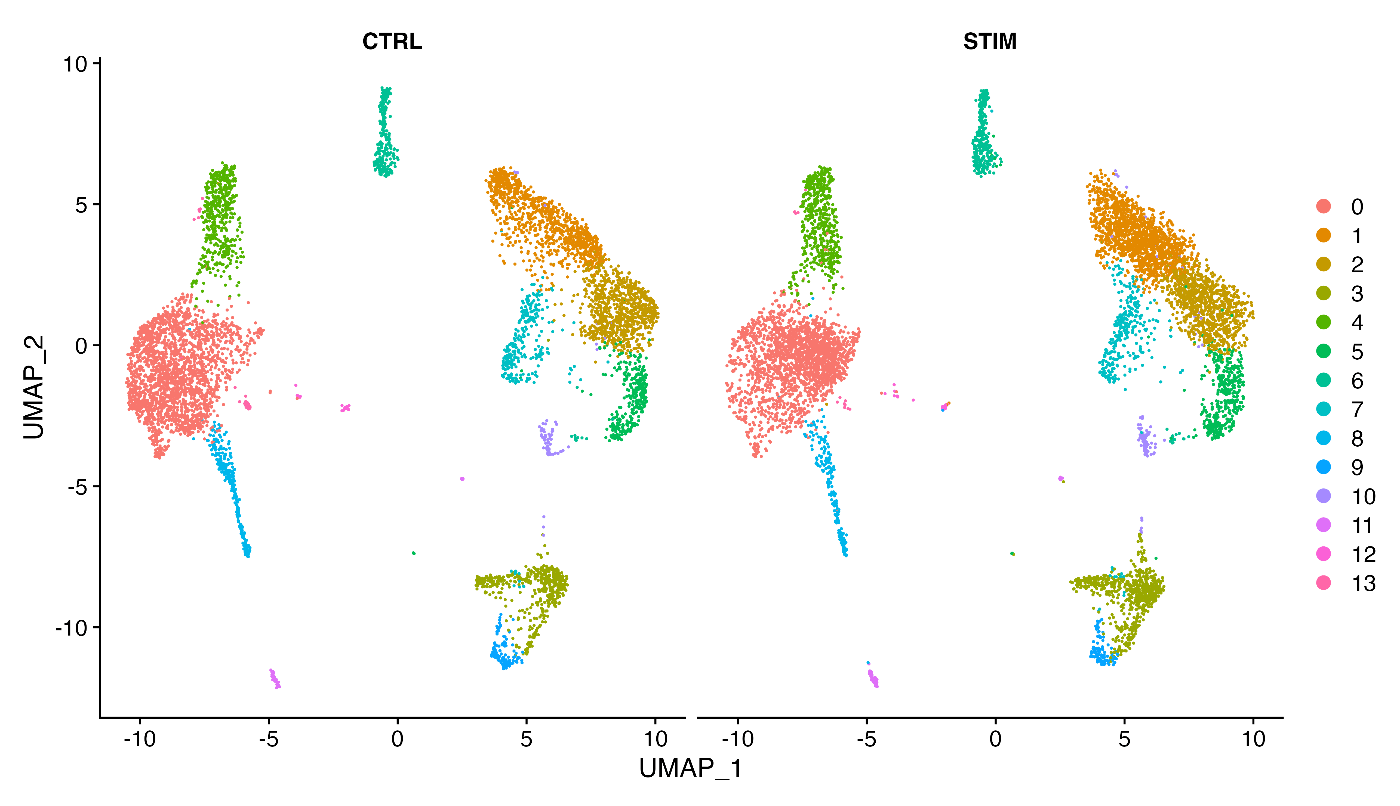
Seurat 提供了将两个以上的单细胞数据集整合并进行分析的方法。 例如,通过如下图所示的数据整合,我们可以看到对照组(CTRL)和刺激组(STIM)的 UMAP 几乎完全一致。
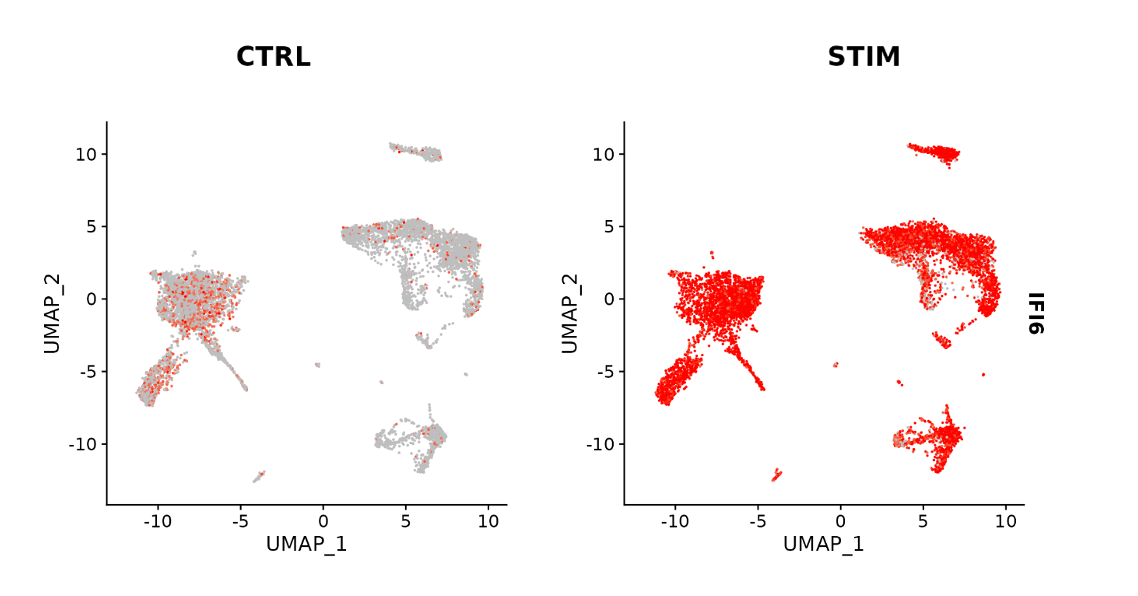
有了这个整合样本,就可以确定与对照组相比,每个基因中表达上调的基因(下图以 IFI6 为例)。
引用自 https://satijalab.org/seurat/articles/integration_introduction 并进行部分改编
关于批次效应校正¶
当整合不同条件或实验的数据集时,技术差异会导致批次效应。使用锚定点(anchors),可以识别并校正这些批次效应。锚定点是一种框架的一部分,用于在不同的单细胞 RNA 测序(scRNA-seq)数据集中找到生物学上一致(或非常相似)的细胞对。锚定点有助于评估不同数据集或条件之间基因表达的相对变化(例如,刺激前后的变化),了解细胞群如何转变,以及哪些细胞相互关联等生物学见解。锚定点使用在数据集中一致变化的基因(特征)。
数据集之间的批次效应校正¶
接下来介绍如何实现 scRNA-seq 数据整合。流程如下:创建 Seurat 对象→数据集之间的批次效应校正→解析。我们一步一步来看。
首先,创建 Seurat 对象。我们将使用 SeuratData 包来安装名为“ifnb”的数据集。
library(Seurat)
library(SeuratData)
library(patchwork)
# 安装数据集
InstallData("ifnb")
# 加载数据集
LoadData("ifnb")
在 ifnb 数据集中,包含了 IMMUNE_CTRL 和 IMMUNE_STIM 两个实验运行(Run)。orig.ident 列用于指示每个细胞的原始样本或运行。

这段代码通过对每个刺激状态的数据进行标准化和识别变异基因,进行前处理,以便在后续分析(例如聚类、基因表达比较等)中获得更准确的结果。此外,这段代码还为多个数据集的有效整合做准备。
# 将数据集拆分为两个Seurat对象的列表(stim和CTRL)
ifnb.list <- SplitObject(ifnb, split.by = "stim")
# 独立地对每个数据集进行标准化和识别变异基因
ifnb.list <- lapply(X = ifnb.list, FUN = function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000)
})
# 选择跨数据集重复可变的特征以进行整合
features <- SelectIntegrationFeatures(object.list = ifnb.list)
详细的代码解释如下:
SplitObject(ifnb, split.by = "stim"):根据 Seurat 对象 ifnb 中 meta.data 的 "stim" 列(表示刺激状态的元数据),将 ifnb 分割为两个不同的 Seurat 对象。这样可以根据刺激状态单独分析数据。
ifnb.list <- SplitObject(ifnb, split.by = "stim")

使用 lapply 函数对每个 Seurat 对象(每种刺激状态的数据)执行以下操作:
- NormalizeData(x): 对数据进行归一化。这是通过根据每个细胞的 RNA 总量对计数进行缩放来实现的。
- FindVariableFeatures(...): 查找变量基因(在每个细胞群中表现出不同表达的基因)。此步骤对于后续的聚类和其他分析中识别重要基因至关重要。
- SelectIntegrationFeatures(object.list = ifnb.list): 在整合两个或更多数据集之前,选择那些在数据集中表现出一致变动的基因(特征)。此步骤在使用
FindIntegrationAnchors()和IntegrateData()等函数整合数据集时非常重要。
顺便说一下,有些人可能会好奇,为什么一开始要将 seurat 对象拆分。对象拆分有以下几个原因:
-
理解每个数据集的特性: 对每种状态(此情况下为刺激状态或非刺激状态)进行细胞反应可能不同。为了理解这些差异,需要分别解析每个状态。通过这种独立解析,可以识别每种状态特有的变量基因或细胞簇。
-
校正批次效应: 通过对每种状态进行独立解析,可以在后续整合阶段更有效地校正批次效应(技术变动)。特别是当每种状态是在不同实验条件下获得时,这一点尤为重要。
-
高级比较分析: 将每种状态作为独立的对象进行分析,可以实现更高级的比较分析,例如基因表达的差异和细胞簇的差异。
-
提高整合精度: Seurat 的整合方法使用每个数据集(此情况下为每种状态)中选择的变量基因来找到“锚点”(表示相应细胞状态的点),然后基于这些锚点整合数据集。这个过程需要每个数据集独立预处理的信息。
以上这些原因使得在很多情况下,先将数据分成独立的对象分别进行预处理和基本分析,然后再进行整合是一个合理的方法。
接下来,使用锚点对两个数据集进行批次效应校正并进行整合。
# Find integration anchors
immune.anchors <- FindIntegrationAnchors(
object.list = ifnb.list, anchor.features = features
)
# This command creates an 'integrated' data assay
immune.combined <- IntegrateData(anchorset = immune.anchors)
- FindIntegrationAnchors 函数: 这个函数根据指定的特征量(变量基因)在多个 scRNA-seq 数据集之间找到称为“锚点”的匹配细胞对。
object.list = ifnb.list: 这个参数传递要整合的 Seurat 对象列表(这里是 ifnb.list)。-
anchor.features = features: 用于整合的特征量(基因)列表。通常是那些在多个数据集中变动较大的基因。 -
IntegrateData 函数: 这个函数使用前一步找到的锚点将多个数据集整合为一个“整合的”数据集。
anchorset = immune.anchors: 这个参数包含前一步找到的锚点。

对于 ifnb 数据集,两个条件存在于同一个 Seurat 对象 (IMMUNE_CTRL 和 IMMUNE_STIM) 中,但这并未进行数据整合。进行整合后的 immune.combined 对象中,新生成了一个 "integrated" 的分析层,包含了整合后的数据。
在 ifnb 对象阶段,仅存在 "RNA" 分析层,这可以解释为两个数据集的条件未整合,只是作为一个 Seurat 对象保存。
接下来,将整合后的数据集带入下游分析。
首先,通过以下代码将 DefaultAssay 设置为 integrated。这段代码将 immune.combined 对象的默认分析层设置为 "integrated",这样接下来的分析将以整合数据为目标:
DefaultAssay(immune.combined) <- "integrated"
这意味着在这个 Seurat 对象 (immune.combined) 中,默认的分析层已经被指定为 "integrated",因此后续的所有分析都将针对这个整合后的数据集进行。
这样设置 DefaultAssay 为 integrated 后,可以确保后续的分析步骤(如聚类、降维等)都基于整合后的数据进行。这一步非常重要,因为它明确了接下来分析的基准数据。
接下来进行下游处理。由于这是在 Seurat 中进行的常规预处理,因此省略详细步骤,但使用 magrittr 库和 %>% 运算符进行流水线处理。
library(magrittr) # 加载 magrittr 包以使用 %>% 运算符
# 现在可以使用管道运算符
immune.combined <- immune.combined %>%
ScaleData(verbose = FALSE) %>%
RunPCA(npcs = 30, verbose = FALSE) %>%
RunUMAP(reduction = "pca", dims = 1:30) %>%
FindNeighbors(reduction = "pca", dims = 1:30) %>%
FindClusters(resolution = 0.5)
接下来,让我们可视化 UMAP。为了比较有无整合处理的效果,分别输出 immune.combined(整合后的数据集)和 ifnb(未整合的数据集)。
# 可视化
p1 <- DimPlot(immune.combined, reduction = "umap", group.by = "stim")
p2 <- DimPlot(immune.combined, reduction = "umap", label = TRUE, repel = TRUE)
p1 + p2
这段代码将可视化使用 immune.combined(整合后的数据集)的 UMAP。
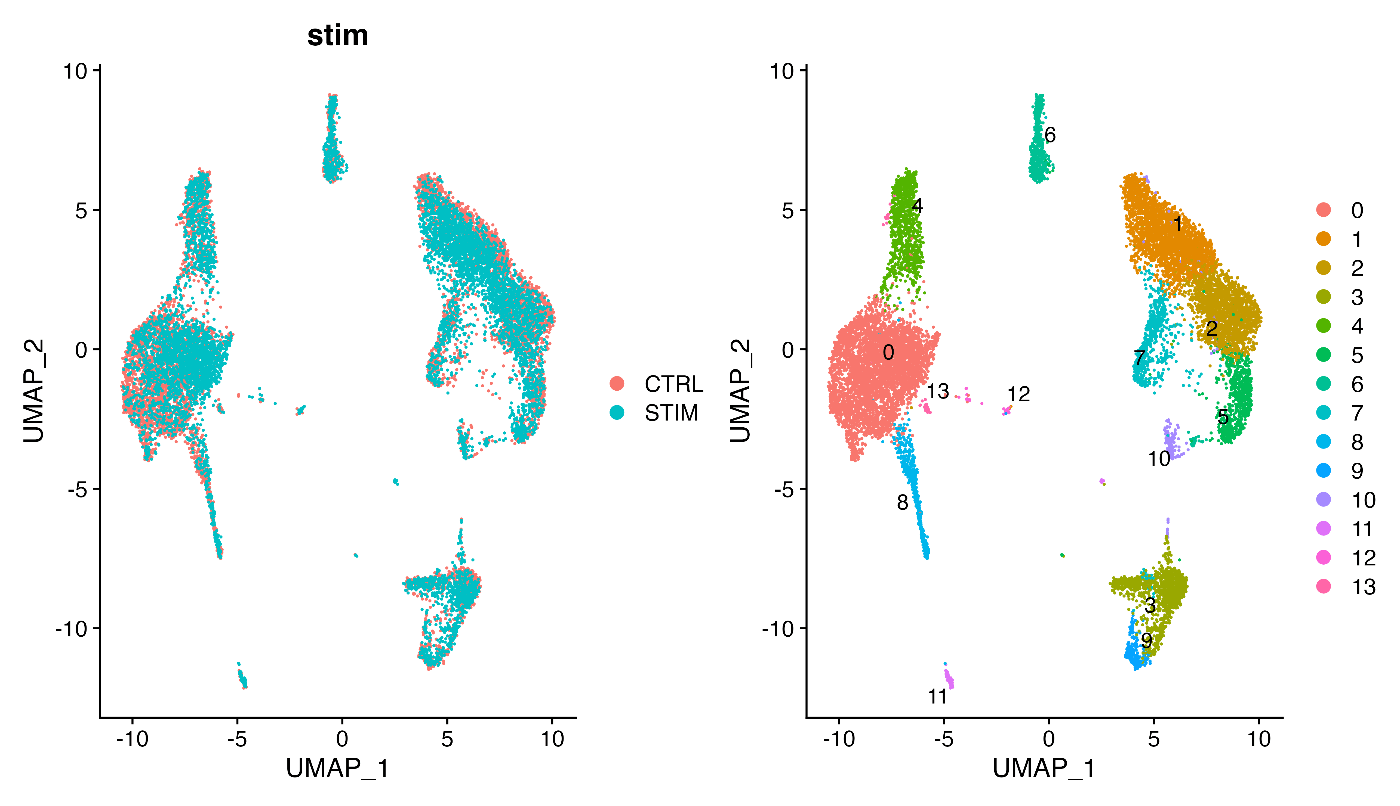
如果使用 ifnb 进行可视化(未整合的数据集),可以使用以下代码:
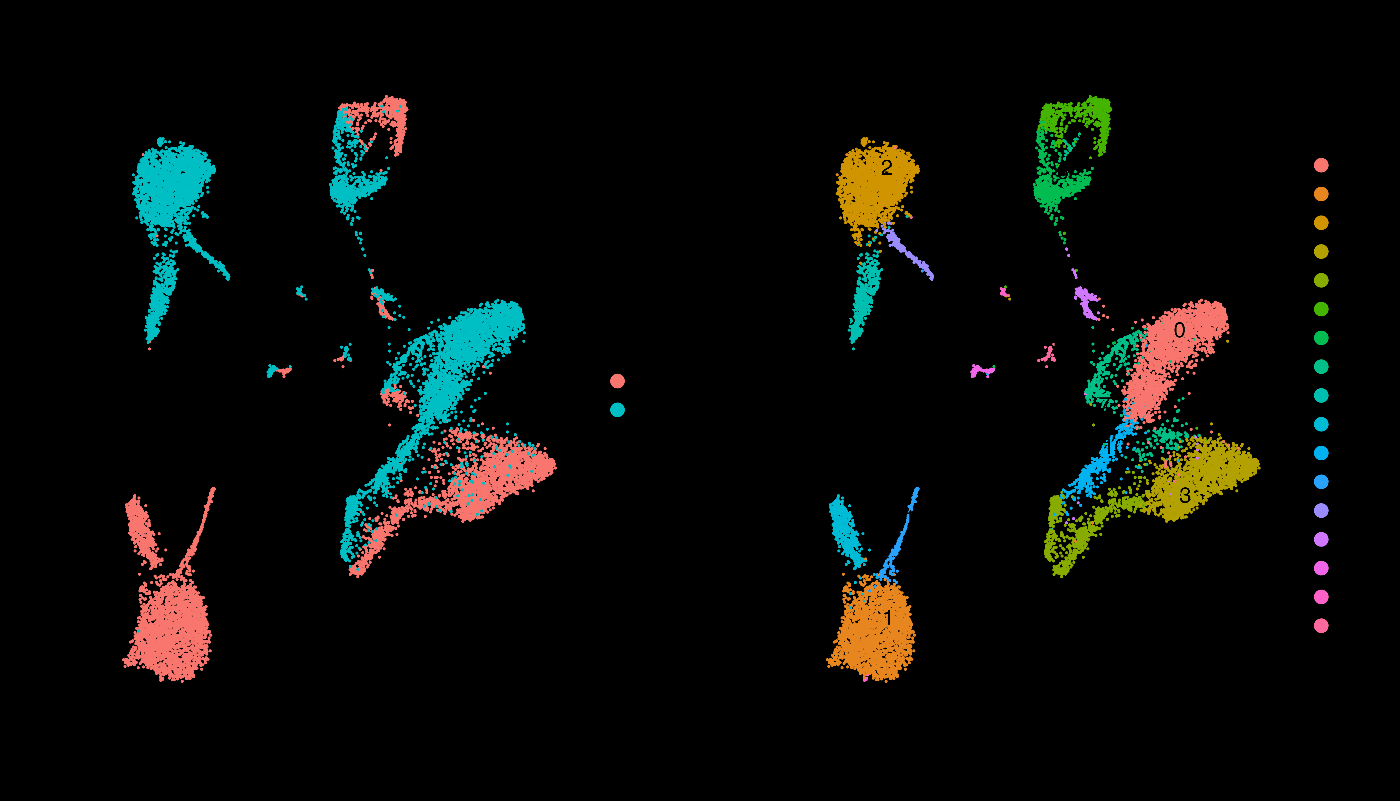
如你所见,在 ifnb 数据集中,左侧的 UMAP 图中可以确认 CTRL 和 STIM 完全没有重叠。因此,在左侧的聚类结果中,可以看到同样的细胞群(例如 0 和 3 可能属于同一细胞群)被检测为不同的聚类,这使得分析结果不准确。这说明了整合处理的重要性。
顺便说一下,指定 split.by = "stim" 可以将 CTRL 和 STIM 的图表合并。
DimPlot(immune.combined, reduction = "umap", split.by = "stim")
这样,您就可以在一张图中比较 CTRL 和 STIM 的 UMAP 分布,更清楚地看到它们的差异和相似之处。
在进入下一部分之前,我们先为每个聚类分配细胞类型。(这次将省略具体的细胞类型和标志基因的确定过程。)
immune.combined <- RenameIdents(immune.combined,
`0` = "CD14 Mono", `1` = "CD4 Naive T", `2` = "CD4 Memory T",
`3` = "CD16 Mono", `4` = "B", `5` = "CD8 T", `6` = "NK", `7` = "T activated",
`8` = "DC", `9` = "B Activated", `10` = "Mk", `11` = "pDC", `12` = "Eryth",
`13` = "Mono/Mk Doublets", `14` = "HSPC"
)
p1 <- DimPlot(immune.combined, reduction = "umap", group.by = "stim")
p2 <- DimPlot(
immune.combined,
reduction = "umap",
group.by = "seurat_annotations",
label = TRUE,
repel = TRUE
)
p1 + p2
以上代码将为每个聚类分配对应的细胞类型,并可视化结果。第一个图 (p1) 显示了根据刺激状态分组的 UMAP 图,第二个图 (p2) 显示了根据细胞类型标注的 UMAP 图。通过这种方式,可以直观地查看不同细胞类型在 UMAP 中的分布情况。
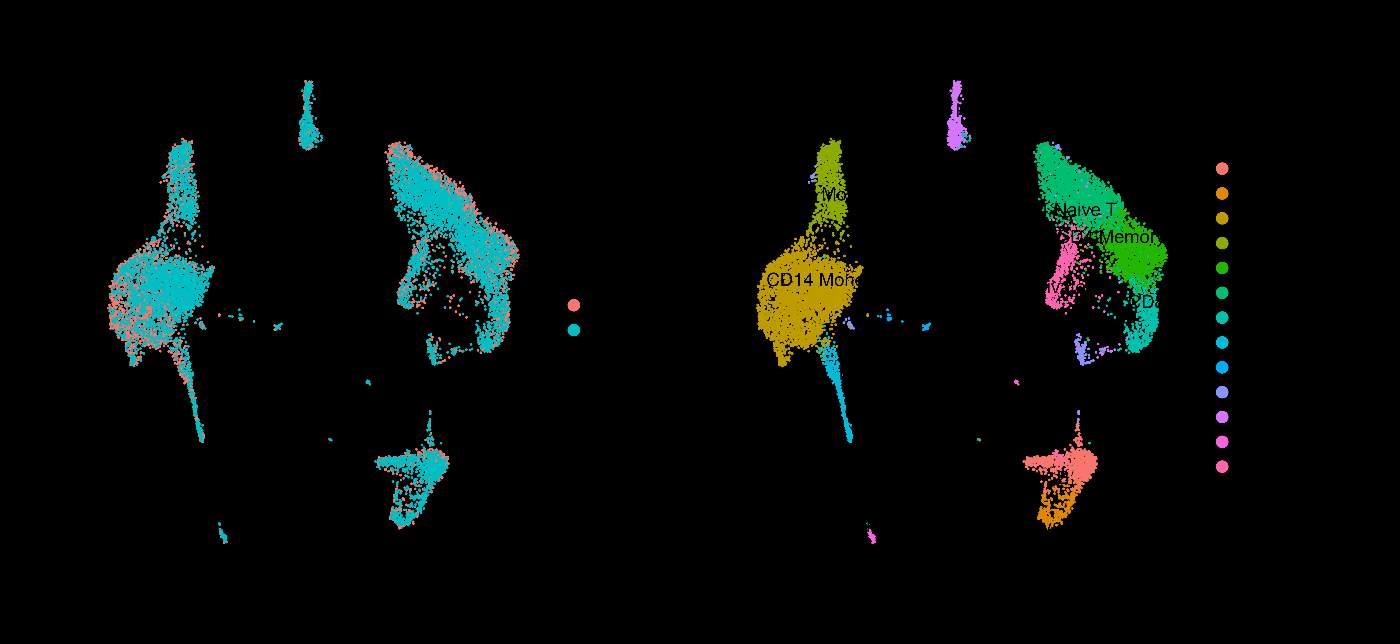
确定条件间的表达变化基因¶
那么,让我们实际比较一下对照组和刺激组。
以下代码用于可视化特定标记基因的表达水平,基于不同的聚类或类别。
# 设置细胞类型的顺序
Idents(immune.combined) <- factor(
Idents(immune.combined),
levels = c(
"HSPC", "Mono/Mk Doublets", "pDC", "Eryth", "Mk", "DC", "CD14 Mono",
"CD16 Mono", "B Activated", "B", "CD8 T", "NK", "T activated",
"CD4 Naive T", "CD4 Memory T"
)
)
# 定义要可视化的标记基因列表
markers.to.plot <- c(
"CD3D", "CREM", "HSPH1", "SELL", "GIMAP5", "CACYBP", "GNLY", "NKG7",
"CCL5", "CD8A", "MS4A1", "CD79A", "MIR155HG", "NME1", "FCGR3A", "VMO1",
"CCL2", "S100A9", "HLA-DQA1", "GPR183", "PPBP", "GNG11", "HBA2", "HBB",
"TSPAN13", "IL3RA", "IGJ", "PRSS57"
)
# 使用 DotPlot 可视化标记基因的表达
DotPlot(
immune.combined,
features = markers.to.plot,
cols = c("blue", "red"),
dot.scale = 8,
split.by = "stim"
) + RotatedAxis()
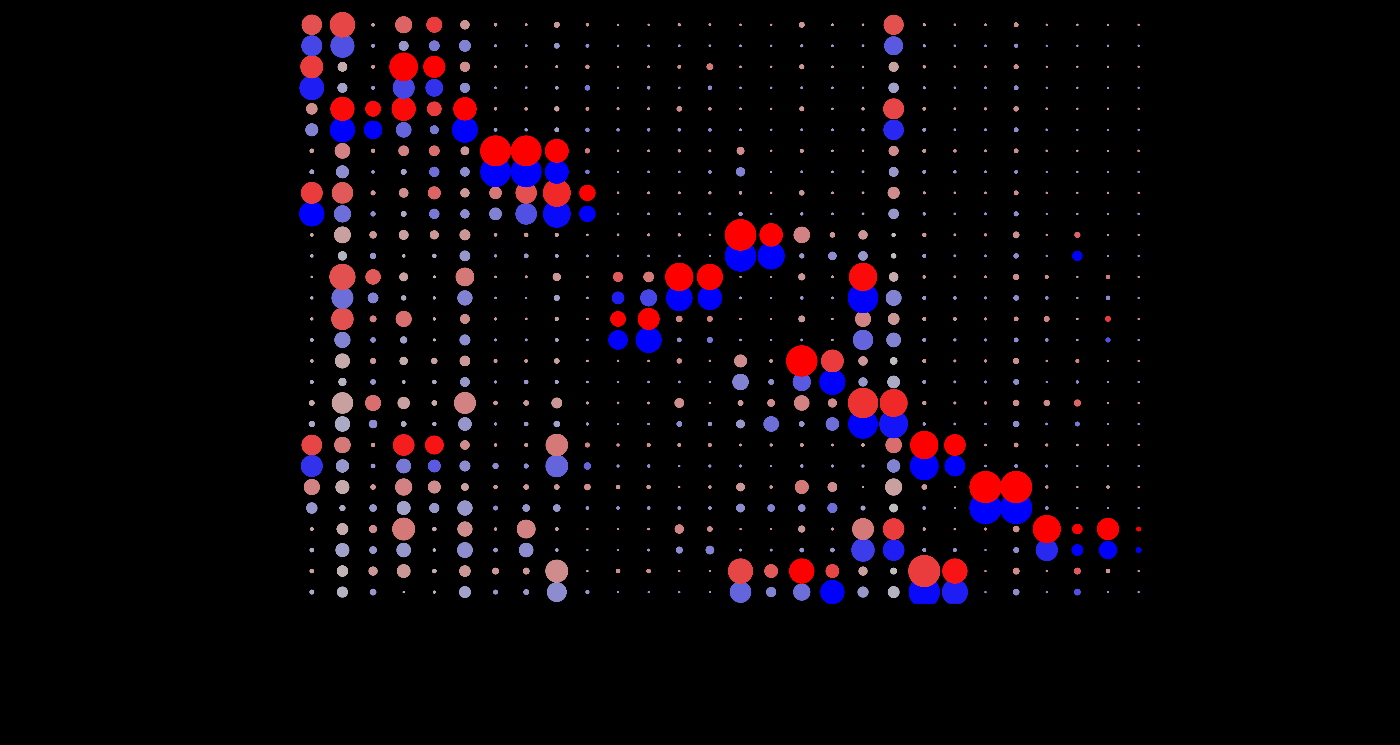
Idents函数用于从 Seurat 对象中获取当前的聚类 ID 或其他身份信息。通过重新排序immune.combined的身份,可以控制后续绘图中的显示顺序。markers.to.plot <- c("CD3D", "CREM", ...定义了要绘制的标记基因列表。DotPlot函数用于绘制点图,显示标记基因在不同条件下的表达水平。split.by = "stim"参数将对照组(CTRL)和刺激组(STIM)的比较显示在同一图中。
成功执行上述代码后,输出应为一个点图,其中行代表每个细胞类型在 CTRL 和 STIM 条件下的比较,列代表基因特征(markers)。这样,您可以直观地查看各基因在不同条件下的表达变化。
接下来,我们将确定条件间表达不同的基因。
这段代码旨在视觉比较刺激细胞和对照细胞的基因表达差异。具体来说,它比较了 Naive T 细胞和 CD14 单核细胞群体的表达,并在散点图中强调显示因干扰素刺激而反应的特定基因。
# 加载可视化库
library(ggplot2)
library(cowplot)
theme_set(theme_cowplot())
# 提取和处理 "CD4 Naive T" 细胞的数据
t.cells <- subset(immune.combined, idents = "CD4 Naive T")
Idents(t.cells) <- "stim"
avg.t.cells <- as.data.frame(
log1p(AverageExpression(t.cells, verbose = FALSE)$RNA)
)
avg.t.cells$gene <- rownames(avg.t.cells)
# 提取和处理 "CD14 Mono" 细胞的数据
cd14.mono <- subset(immune.combined, idents = "CD14 Mono")
Idents(cd14.mono) <- "stim"
avg.cd14.mono <- as.data.frame(
log1p(AverageExpression(cd14.mono, verbose = FALSE)$RNA)
)
avg.cd14.mono$gene <- rownames(avg.cd14.mono)
# 指定在图中要突出的基因
genes.to.label <- c(
"ISG15", "LY6E", "IFI6", "ISG20", "MX1", "IFIT2", "IFIT1", "CXCL10", "CCL8"
)
# 创建散点图并标注指定的基因
p1 <- ggplot(avg.t.cells, aes(CTRL, STIM)) +
geom_point() +
ggtitle("CD4 Naive T Cells")
p1 <- LabelPoints(plot = p1, points = genes.to.label, repel = TRUE)
p2 <- ggplot(avg.cd14.mono, aes(CTRL, STIM)) +
geom_point() +
ggtitle("CD14 Monocytes")
p2 <- LabelPoints(plot = p2, points = genes.to.label, repel = TRUE)
# 并排显示图表
p1 + p2
代码的处理流程如下:
- 获取刺激的 Naive T 细胞和对照的 CD14 单核细胞群体的平均基因表达值。
- 使用 ggplot2 和 cowplot 包绘制这两种细胞类型的基因表达。
- 突出显示并标注特定基因(如 ISG15、LY6E 等)。
- 最后,分别显示两个图表(Naive T 细胞和 CD14 单核细胞)并排显示。
成功执行上述代码后,您将会看到以下的散点图,其中显示了 Naive T 细胞和 CD14 单核细胞在 CTRL 和 STIM 条件下的基因表达比较。

以下的代码针对单细胞 RNA-seq 数据集 immune.combined,根据刺激状态进行细胞类型识别,确定差异表达基因,并在 Feature plot 中可视化特定基因的表达水平。
# 创建一个结合细胞类型和刺激状态的新标识符
immune.combined$celltype.stim <- paste(
Idents(immune.combined),
immune.combined$stim,
sep = "_"
)
# 备份当前的细胞类型标识符,并更新为新的组合标识符
immune.combined$celltype <- Idents(immune.combined)
Idents(immune.combined) <- "celltype.stim"
# 识别刺激B细胞和对照B细胞之间差异表达的基因
b.interferon.response <- FindMarkers(
immune.combined,
ident.1 = "B_STIM",
ident.2 = "B_CTRL",
verbose = FALSE
)
# 可视化选定基因的表达,按刺激状态分割
FeaturePlot(
immune.combined,
features = c("CD3D", "GNLY", "IFI6"),
split.by = "stim",
max.cutoff = 3,
cols = c("grey", "red")
)
解释:
immune.combined$celltype.stim:创建一个结合细胞类型和刺激状态的新标识符。- 备份当前的细胞类型标识符,并使用新的组合标识符进行更新。
- 使用
FindMarkers函数识别刺激 B 细胞和对照 B 细胞之间差异表达的基因。 - 使用
FeaturePlot可视化选定基因(CD3D、GNLY、IFI6)的表达,并按刺激状态分割显示。
成功执行上述代码后,您将看到以下的 Feature plot。图中可以清晰地看到在 CTRL 和 STIM 条件下,哪些聚类的基因表达发生了变化(例如,IFI6 基因)。

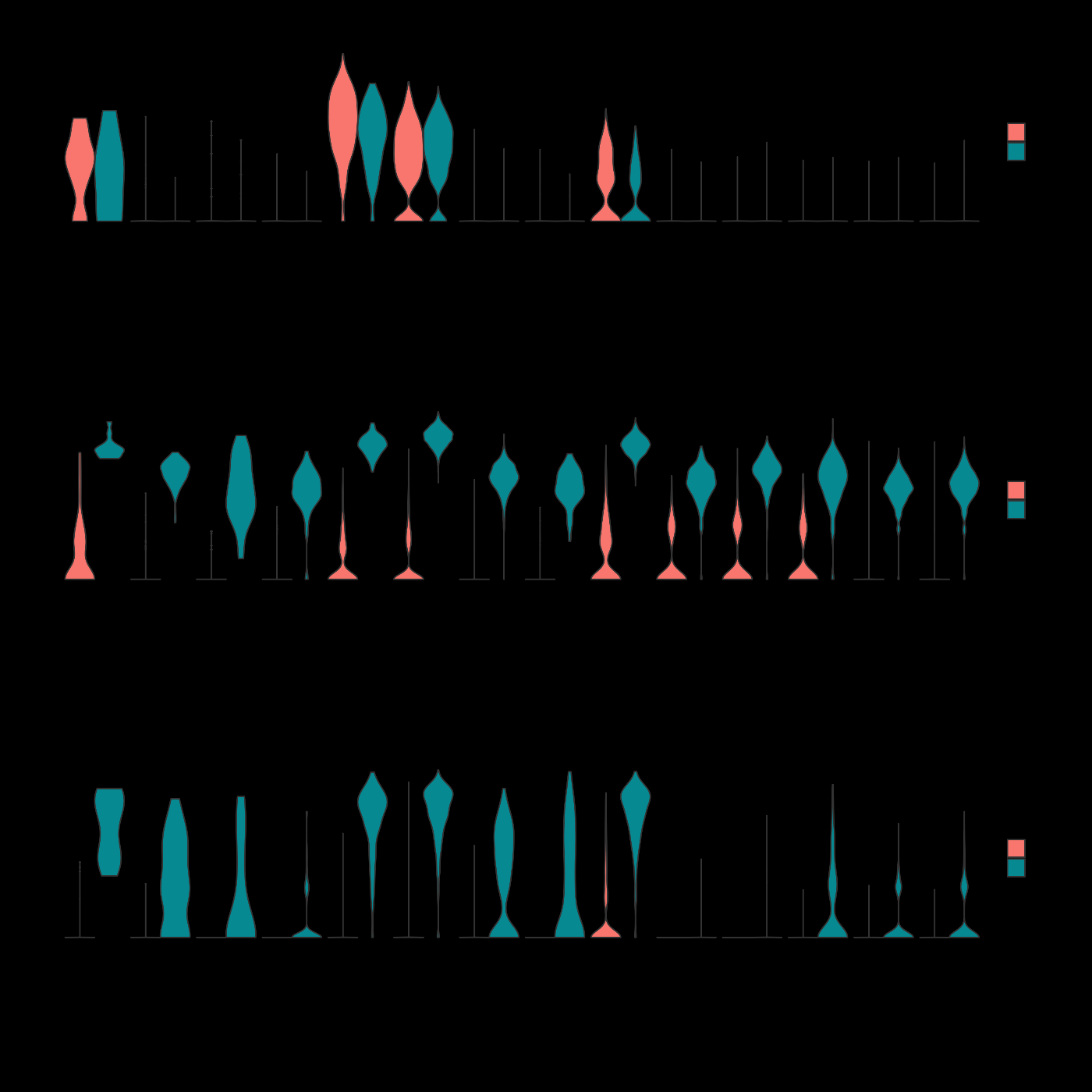
最后,让我们通过小提琴图可视化各细胞类型的基因表达。
# 为指定基因生成小提琴图,根据刺激状态分组并按细胞类型进行分组
plots <- VlnPlot(
immune.combined,
features = c("LYZ", "ISG15", "CXCL10"),
split.by = "stim",
group.by = "celltype",
pt.size = 0,
combine = FALSE
)
# 以单列布局显示生成的小提琴图
wrap_plots(plots = plots, ncol = 1)
解释:
VlnPlot函数生成指定基因(LYZ、ISG15、CXCL10)的小提琴图,这些图根据刺激状态分割,并按细胞类型分组。wrap_plots函数将生成的小提琴图在单列布局中显示。
成功执行上述代码后,您将看到如图所示的小提琴图。您可以清楚地看到在刺激后的样本中,ISG15和CXCL10的表达水平在各细胞类型中显著上升。