5.6 确定每个群组的标记基因
scRNA-seq 数据分析可视化到一定程度后,您可能会好奇各个聚类中特征性的基因表达。Seurat 提供了查看每个聚类的标记基因表达的功能。当您能够确认标记基因的表达时,便可以研究各个聚类的特征。
用于标记基因识别的函数¶
在 Seurat 中,有三种用于标记基因识别的函数:FindMarkers、FindAllMarkers 和 FindConservedMarkers。这些函数分别适用于不同的目的和情况。
FindMarkers:
- 这个函数用于检测特定聚类与其他所有聚类或两个特定聚类之间的标记基因。
- 使用 ident.1 和 ident.2 参数指定要比较的聚类。
- 例如:如果想找到聚类 0 和聚类 1 之间的标记基因,可以使用 FindMarkers(object, ident.1 = 0, ident.2 = 1)。
FindAllMarkers:
- 这个函数一次性检测数据集中所有聚类的标记基因。
- 相当于重复应用 FindMarkers 函数,但更高效。
- 例如:FindAllMarkers(object) 返回数据集中所有聚类的标记基因。
FindConservedMarkers:
- 这个函数用于寻找在多个条件、时间点或实验组间保持一致的标记基因。
- 例如,在多个样本或条件中一致表达的标记基因。
- 使用 grouping.var 参数指定基于哪些条件或组寻找保持一致的标记基因。
- 例如:FindConservedMarkers(object, grouping.var = "condition") 返回在不同 condition 值中保持一致的标记基因。
总结: - 想知道两个聚类间的标记基因时使用 FindMarkers。 - 想知道所有聚类的标记基因时使用 FindAllMarkers。 - 想知道在多个条件或时间点间保持一致的标记基因时使用 FindConservedMarkers。
这次我们将使用这些函数来解释实际数据的应用。
scRNA-seq 数据准备¶
我们将使用“比较 scRNA-seq 数据集中的对照组和刺激组”一节中介绍的数据集。详细信息可以参阅那一部分,此处省略详细解释。
library(Seurat)
library(SeuratData)
library(patchwork)
# 安装数据集
InstallData("ifnb")
# 加载数据集
LoadData("ifnb")
# 将数据集拆分为两个 Seurat 对象(stim 和 CTRL)
ifnb.list <- SplitObject(ifnb, split.by = "stim")
# 独立地标准化并识别每个数据集的可变特征
ifnb.list <- lapply(X = ifnb.list, FUN = function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000)
})
# 选择跨数据集反复变化的特征用于整合
features <- SelectIntegrationFeatures(object.list = ifnb.list)
# 找到整合锚点
immune.anchors <- FindIntegrationAnchors(
object.list = ifnb.list, anchor.features = features
)
# 创建一个“integrated”数据分析层
immune.combined <- IntegrateData(anchorset = immune.anchors)
DefaultAssay(immune.combined) <- "integrated"
FindMarkers 函数¶
用于特定细胞聚类或群体与其他细胞之间的差异表达基因(DEGs)的识别。这个函数的主要目的是揭示与特定细胞状态、类型或任何其他分类相关的基因表达变化。
FindMarkers 函数有两个主要用途: 1. 探索特定两个聚类(或聚类集)间的 DEG。 2. 探索特定聚类与其他所有聚类间的 DEG。
让我们分别来看。
探索特定两个聚类(或聚类集)间的 DEG¶
这个用例适合希望明确了解两个特定聚类间差异表达基因的情况。可以详细调查特定聚类间的差异。
例如,如果对 T 细胞和 B 细胞,或健康细胞和疾病细胞等两种明显不同的细胞群体间的基因表达差异特别感兴趣,这个选项非常有用。
示例:CD14 Mono vs CD16 Mono
运行以下代码,详细调查特定聚类对之间的比较:
# 查找区分聚类5和聚类0及3的所有标记基因
cluster5.markers <- FindMarkers(
immune.combined,
ident.1 = 5,
ident.2 = c(0, 3),
min.pct = 0.25
)
head(cluster5.markers, n = 5)
执行后,您会得到如下输出:
p_val avg_log2FC pct.1 pct.2 p_val_adj
CXCL10 0 -6.169698 0.555 0.951 0
CCL5 0 4.553574 0.897 0.474 0
HLA-DRA 0 -3.732949 0.459 0.986 0
FCER1G 0 -4.505242 0.213 0.869 0
IFITM3 0 -4.493810 0.577 0.926 0
解释各列的含义:
- p_val: p 值,用于判断特定基因在两个聚类间的差异表达是否显著。p 值越小,表示基因表达差异越显著。
- avg_log2FC (平均对数 2 倍数变化): 显示两个聚类间的基因表达相对变化。正值表示在 ident.1 指定的聚类中表达高于 ident.2,负值则相反。
- pct.1 和 pct.2: 显示特定基因在 ident.1 和 ident.2 各自聚类中表达的细胞百分比。例如,pct.1 为 0.555 表示 ident.1 聚类中有 55.5% 的细胞表达该基因。
- p_val_adj: 调整后的 p 值,考虑多重检验的假阳性率。调整后 p 值越小,基因的差异表达越显著。
注意:p_val 和 p_val_adj 为 0 是因为它们的值接近 R 内设置的浮点数极限。
可视化顶级基因表达¶
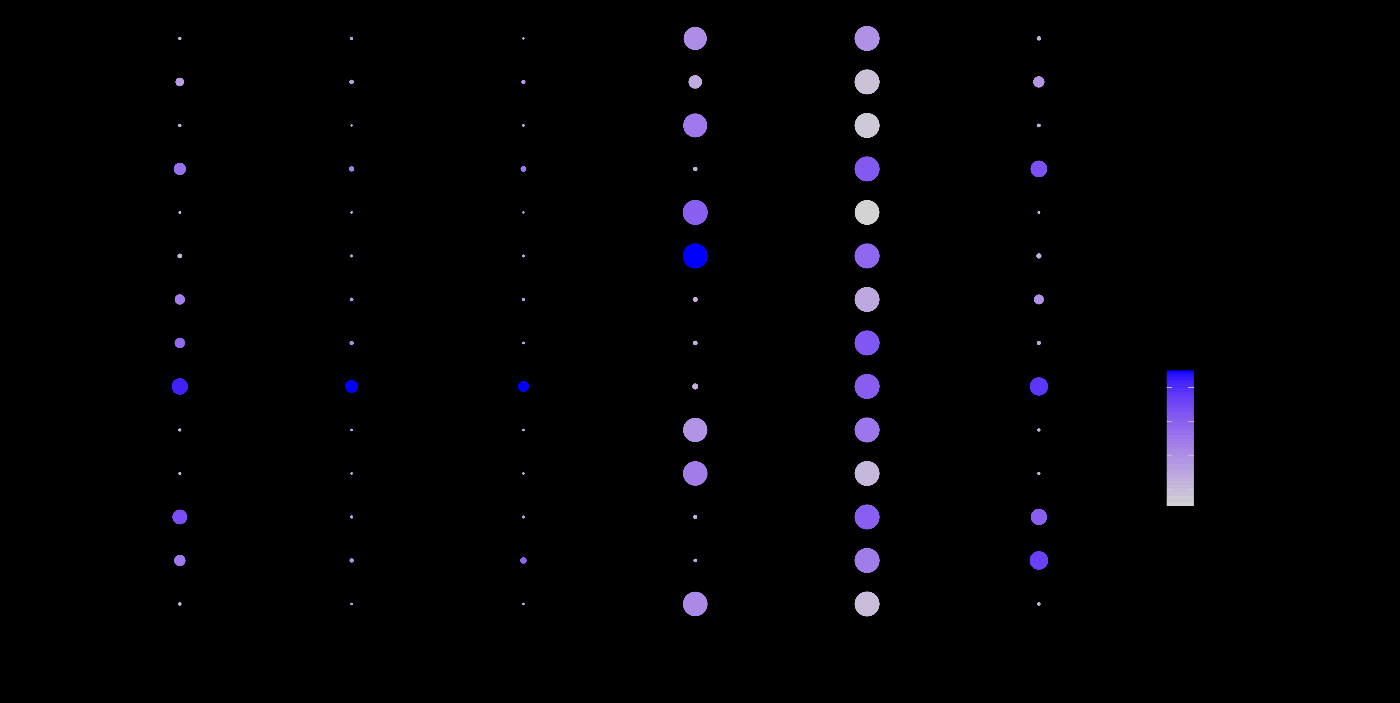
让我们用小提琴图和点图来查看前六个基因的表达。
# 选择顶级基因
top_genes <- head(row.names(cluster5.markers))
# 创建小提琴图
VlnPlot(immune.combined, features = top_genes, group.by = "seurat_clusters")
# 创建点图
top_genes <- head(row.names(cluster5.markers), n = 5)
DotPlot(immune.combined, features = top_genes, group.by = "seurat_clusters")
探索特定聚类与其他所有聚类之间的 DEG¶
例如:CD14 Mono 与所有其他细胞(CD16 Mono、CD4 Memory T、CD4 Naive T 等)之间的差异表达基因。
以下代码识别聚类 0 与其他所有细胞之间的差异表达基因。
cluster0.markers <- FindMarkers(
immune.combined,
ident.1 = 0,
logfc.threshold = 0.25,
test.use = "roc",
only.pos = TRUE
)
head(cluster0.markers, n = 5)
执行后,您会得到如下输出:
myAUC avg_diff power avg_log2FC pct.1 pct.2
FTL 0.975 2.294724 0.950 3.310586 1.000 0.877
CD63 0.954 1.834068 0.908 2.646001 0.994 0.324
ANXA5 0.941 1.759063 0.882 2.537791 0.987 0.322
C15orf48 0.936 1.999672 0.872 2.884916 0.986 0.193
LGALS3 0.923 2.018229 0.846 2.911689 0.953 0.239
解释各列的含义:
myAUC: ROC 曲线下面积(AUC)值,表示基因在两个聚类间的分类能力。值越接近 1,分类能力越强。avg_diff: 平均表达差异。power: 检测能力。avg_log2FC: 平均对数 2 倍数变化,显示基因在两个聚类间的相对表达变化。pct.1和pct.2: 显示特定基因在ident.1和其他聚类中表达的细胞百分比。
可视化基因表达¶
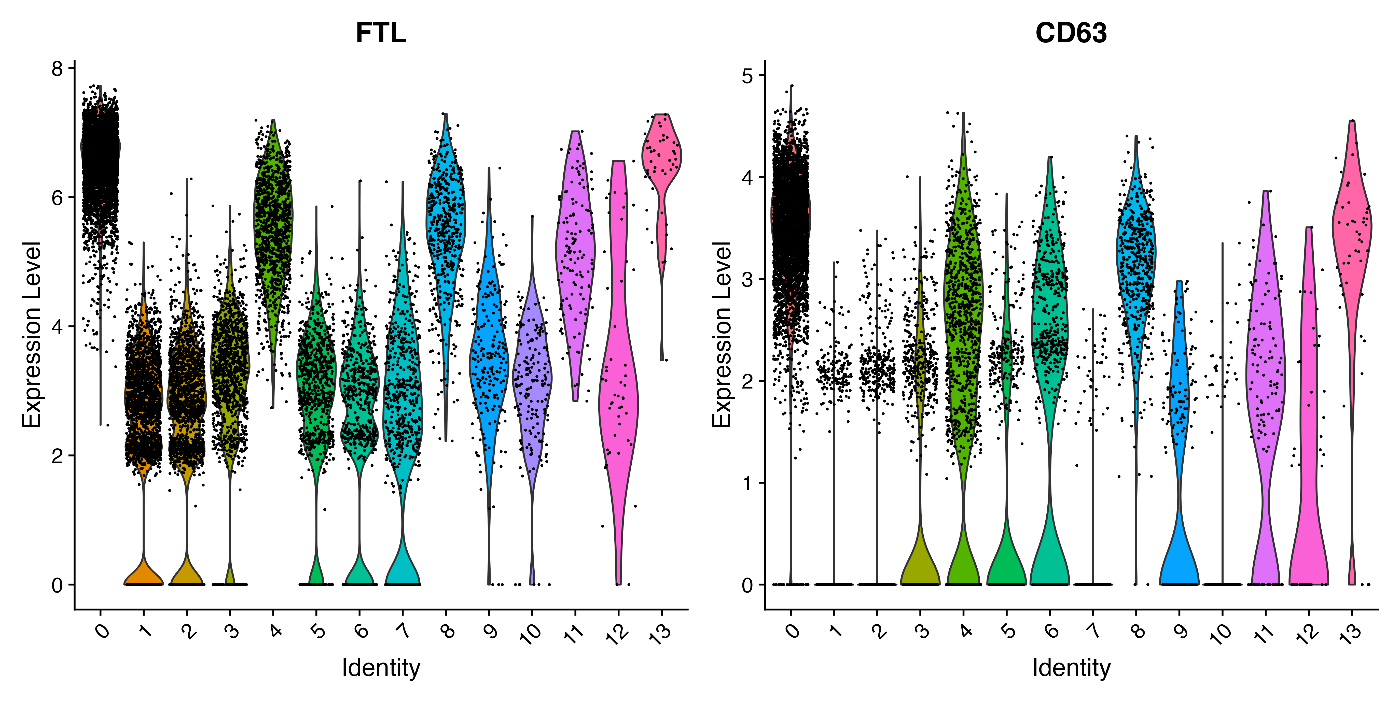
使用小提琴图查看 "FTL" 和 "CD63" 的表达。
VlnPlot(
immune.combined,
features = c("FTL", "CD63"),
pt.size = 0.1,
group.by = "seurat_clusters"
)
成功执行上述代码后,您将看到以下的小提琴图,其中显示了 "FTL" 和 "CD63" 基因在不同聚类中的表达情况。这有助于直观地比较这些基因在特定聚类与其他所有聚类中的表达差异。
探索特定聚类与其他所有聚类之间的 DEG¶
例如:CD14 Mono 与所有其他细胞(CD16 Mono、CD4 Memory T、CD4 Naive T 等)之间的差异表达基因。
以下代码识别聚类 0 与其他所有细胞之间的差异表达基因。
cluster0.markers <- FindMarkers(
immune.combined,
ident.1 = 0,
logfc.threshold = 0.25,
test.use = "roc",
only.pos = TRUE
)
head(cluster0.markers, n = 5)
执行后,您会得到如下输出:
myAUC avg_diff power avg_log2FC pct.1 pct.2
FTL 0.975 2.294724 0.950 3.310586 1.000 0.877
CD63 0.954 1.834068 0.908 2.646001 0.994 0.324
ANXA5 0.941 1.759063 0.882 2.537791 0.987 0.322
C15orf48 0.936 1.999672 0.872 2.884916 0.986 0.193
LGALS3 0.923 2.018229 0.846 2.911689 0.953 0.239
在聚类 0 中,FTL 和 CD63 的表达高于其他聚类,并且在许多细胞中(黑色点)高表达。FTL 和 CD63 可能作为聚类 0 的标记基因显著发挥作用。
FindAllMarkers 函数¶
FindMarkers 函数适用于特定聚类对之间的详细比较,或者特定聚类与其他所有聚类之间的比较。虽然能够指定特定聚类进行详细分析,但对每个聚类都应用该函数会非常耗时。
执行 FindAllMarkers 可以快速获取全局差异。也就是说,它同时探索 Seurat 对象中所有聚类的细胞与其他所有细胞之间的差异表达基因(DEG)。
# Identify markers for every cluster compared to all remaining cells, report only the positive ones
cluster.markers <- FindAllMarkers(
immune.combined,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.25
)
cluster.markers %>%
group_by(cluster) %>%
slice_max(n = 2, order_by = avg_log2FC) %>%
print(n = 28)
执行后,您会得到如下输出:
# A tibble: 28 × 7
# Groups: cluster [14]
p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
<dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
1 0 5.39 0.96 0.537 0 0 CCL8
2 0 5.36 0.987 0.512 0 0 CCL2
3 0 2.34 0.929 0.659 0 1 SELL
4 0 1.93 0.98 0.685 0 1 GIMAP7
5 0 1.82 0.785 0.31 0 2 ALOX5AP
6 0 1.70 0.743 0.379 0 2 TRAT1
7 4.30e- 8 3.61 0.285 0.085 8.59e- 5 3 IGLL5
8 0 3.41 0.734 0.128 0 3 CD79A
9 0 4.31 0.774 0.187 0 4 VMO1
10 0 3.94 0.992 0.314 0 4 FCGR3A
11 0 2.97 0.897 0.418 0 5 CCL5
12 1.16e-241 2.86 0.77 0.432 2.32e-238 5 LAG3
13 0 6.08 0.975 0.35 0 6 GNLY
14 0 4.56 0.944 0.344 0 6 GZMB
15 2.03e- 95 2.60 0.773 0.563 4.07e- 92 7 GADD45B
16 1.20e- 96 2.46 0.797 0.609 2.39e- 93 7 CD69
17 8.79e-181 3.11 0.998 0.67 1.76e-177 8 TXN
18 1.49e-277 3.10 0.998 0.541 2.99e-274 8 HLA-DPB1
19 1.73e-133 4.46 0.941 0.451 3.46e-130 9 MIR155HG
20 5.76e- 95 3.71 0.882 0.313 1.15e- 91 9 MYC
21 1.38e-105 5.15 0.92 0.282 2.75e-102 10 PPBP
22 1.18e- 97 3.78 0.823 0.253 2.36e- 94 10 PF4
23 2.57e-163 4.54 0.917 0.149 5.15e-160 11 TSPAN13
24 5.62e- 47 3.73 0.977 0.678 1.12e- 43 11 TXN
25 6.05e- 39 10.4 1 0.386 1.21e- 35 12 HBB
26 2.98e- 45 9.63 1 0.276 5.96e- 42 12 HBA2
27 3.93e- 32 3.74 0.979 0.287 7.87e- 29 13 PPBP
28 5.05e- 25 2.78 0.851 0.233 1.01e- 21 13 GNG11
可视化标记基因表达¶
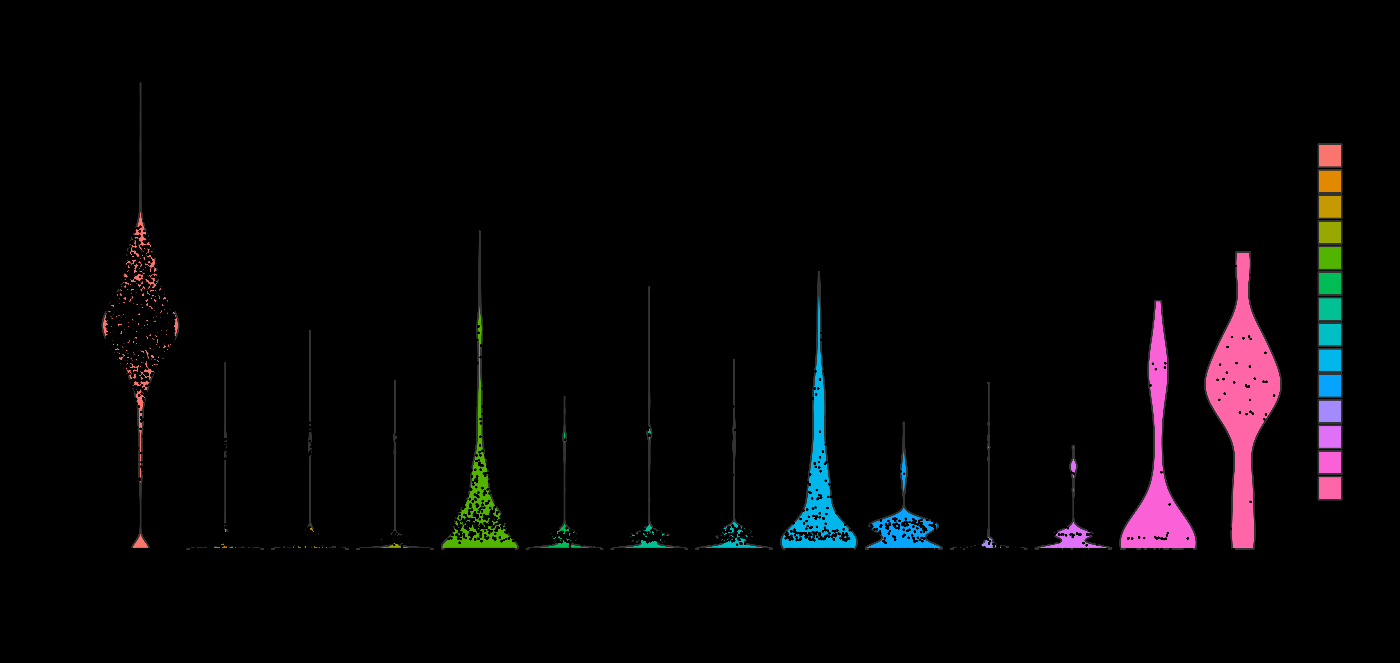
让我们用小提琴图查看 CCL8 的表达。
plots <- VlnPlot(
immune.combined,
features = c("CCL8"),
group.by = "seurat_clusters",
pt.size = 0.1,
combine = FALSE
)
wrap_plots(plots = plots, ncol = 1)
成功执行上述代码后,您将看到如下的小提琴图。可以确认聚类 0 的表达水平高于其他聚类。
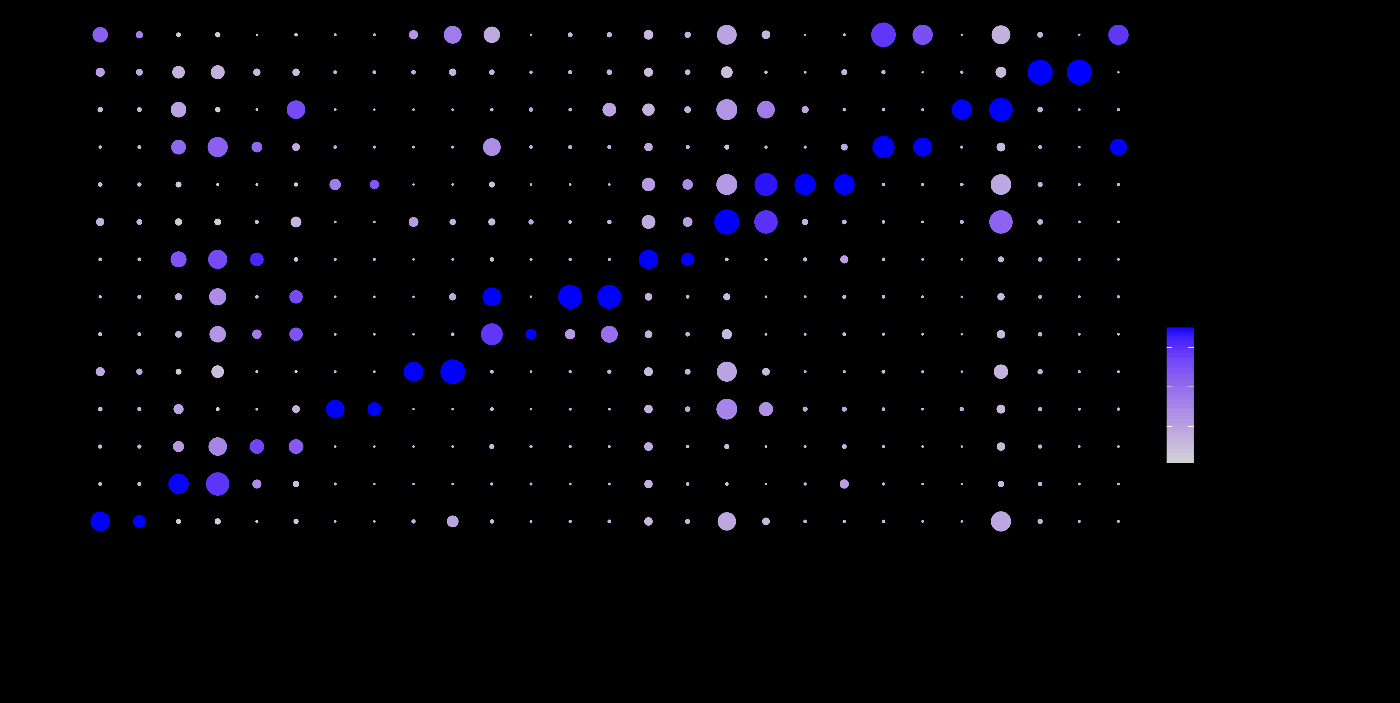
使用 DotPlot 可视化大量标记基因¶
当像 FindAllMarkers 这样的函数检测到大量标记基因时,DotPlot 更适合查看所有标记基因的列表。
# 提取每个聚类的顶级标记基因
marker_genes <- cluster.markers %>%
group_by(cluster) %>%
slice_max(order_by = avg_log2FC, n = 2) %>%
pull(gene)
# 获取唯一的标记基因列表
unique_marker_genes <- unique(marker_genes)
# 使用DotPlot可视化标记基因
DotPlot(
immune.combined,
features = unique_marker_genes,
group.by = "seurat_clusters"
) + theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1))
成功执行上述代码后,您将看到一个点图,显示各个聚类中标记基因的高表达情况。每个点的大小和颜色表示基因在相应聚类中的表达水平,横轴表示基因,纵轴表示聚类。通过这种方式,可以直观地查看不同聚类中哪些基因具有高表达,帮助识别和理解每个聚类的特征。
使用 FindConservedMarkers 函数¶
FindConservedMarkers 函数用于识别在控制组和刺激组等不同条件下保持一致表达的标记基因。这可以帮助我们在刺激条件变化时,确定特定细胞聚类中一致表达的标记基因。这些信息有助于细胞类型识别和功能分析。
FindMarkers 函数用于在单一条件或单一实验数据集内的两个聚类间寻找差异表达基因,而 FindConservedMarkers 函数考虑多个条件或多个实验数据集,寻找一致差异表达的基因。例如,在健康个体和患病个体的两个不同数据集中,找到在特定聚类中一致上升或下降的基因。
以下是具体代码:
# 为进行整合后的差异表达分析,切换回原始数据
DefaultAssay(immune.combined) <- "RNA"
# 使用 FindConservedMarkers 识别在不同刺激条件下的一致标记基因
cluster6.markers <- FindConservedMarkers(
immune.combined,
ident.1 = 6,
grouping.var = "stim"
)
head(cluster6.markers)
解释:
DefaultAssay(immune.combined) <- "RNA":将整合数据集 immune.combined 的默认分析层切换回 "RNA"。这样可以基于原始 RNA-seq 数据进行差异表达分析。FindConservedMarkers:该函数在特定细胞身份(这里是 ident.1 = 6)和特定分组变量(这里是 "stim")下寻找一致表达的标记基因。head(cluster6.markers):显示找到的标记基因列表的前几行。
执行上述代码后,您可能会得到如下输出:
p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
1 0 -3.169698 0.555 0.951 0 6 GeneA
2 0 4.553574 0.897 0.474 0 6 GeneB
3 0 -3.732949 0.459 0.986 0 6 GeneC
4 0 -4.505242 0.213 0.869 0 6 GeneD
5 0 -4.493810 0.577 0.926 0 6 GeneE
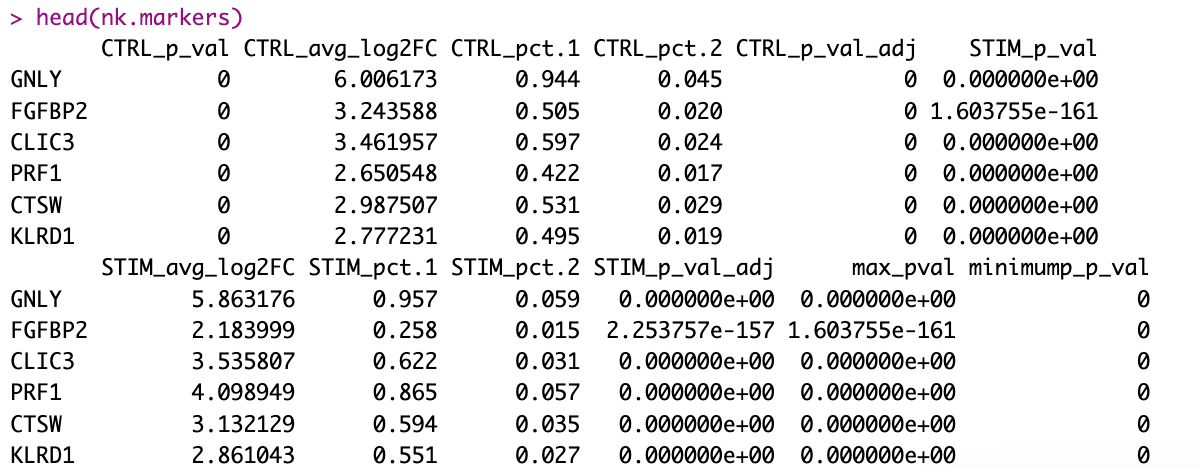
FindConservedMarkers 函数输出¶
FindConservedMarkers() 函数的输出是一个矩阵,包含指定聚类的基因 ID 的潜在标记基因的排序列表,以及相关的统计信息。需要注意的是,对于每个组(此处为 Ctrl 和 Stim),计算相同的统计集,最后两列是两个组整体的合并 p 值。以下是一些列的说明:
- gene: 基因符号
- 〇〇 _p_val: 针对条件未调整的 p 值
- 〇〇 _avg_logFC: 条件下的平均对数倍数变化。正值表示该基因在聚类中高表达
- 〇〇 _pct.1: 在条件下,聚类中检测到该基因的细胞比例
- 〇〇 _pct.2: 在条件下,其他聚类中检测到该基因的细胞比例
- 〇〇 _p_val_adj: 基于数据集中所有基因的 Bonferroni 校正的调整 p 值,用于确定显著性
- max_pval: 每个组/条件下计算的最大 p 值
- minimump_p_val: 合并后的 p 值
在查看输出时,建议寻找 pct.1 和 pct.2 之间表达差异大、fold change 大的标记基因。例如,pct.1 = 0.90 和 pct.2 = 0.80 可能不是有效标记,但如果 pct.2 = 0.1,则差异较大且有说服力。如果 pct.1 = 0.3 也可能不是很有趣的标记。
实例分析:Granulysin (GNLY)¶
Granulysin (GNLY) 是已知在 NK 细胞中特异性表达的细胞毒性蛋白质。由于 GNLY 在对照组和刺激组中均被保留为标记基因,聚类 6 可能被分配为 NK 细胞。我们可以通过 FeaturePlot 来比较 GNLY 和其他免疫细胞的标记基因。
# 使用 FeaturePlot 比较 GNLY 和其他免疫细胞的标记基因
FeaturePlot(
immune.combined,
features = c(
"CD3D", "SELL", "CREM", "CD8A", "GNLY", "CD79A", "FCGR3A", "CCL2", "PPBP"
),
min.cutoff = "q9"
)
执行上述代码后,您将看到一个 FeaturePlot,其中显示了所选标记基因(包括 GNLY)在不同细胞中的表达水平。通过这种方式,可以直观地比较不同免疫细胞标记基因的表达情况,进一步确认聚类 6 为 NK 细胞的可能性。
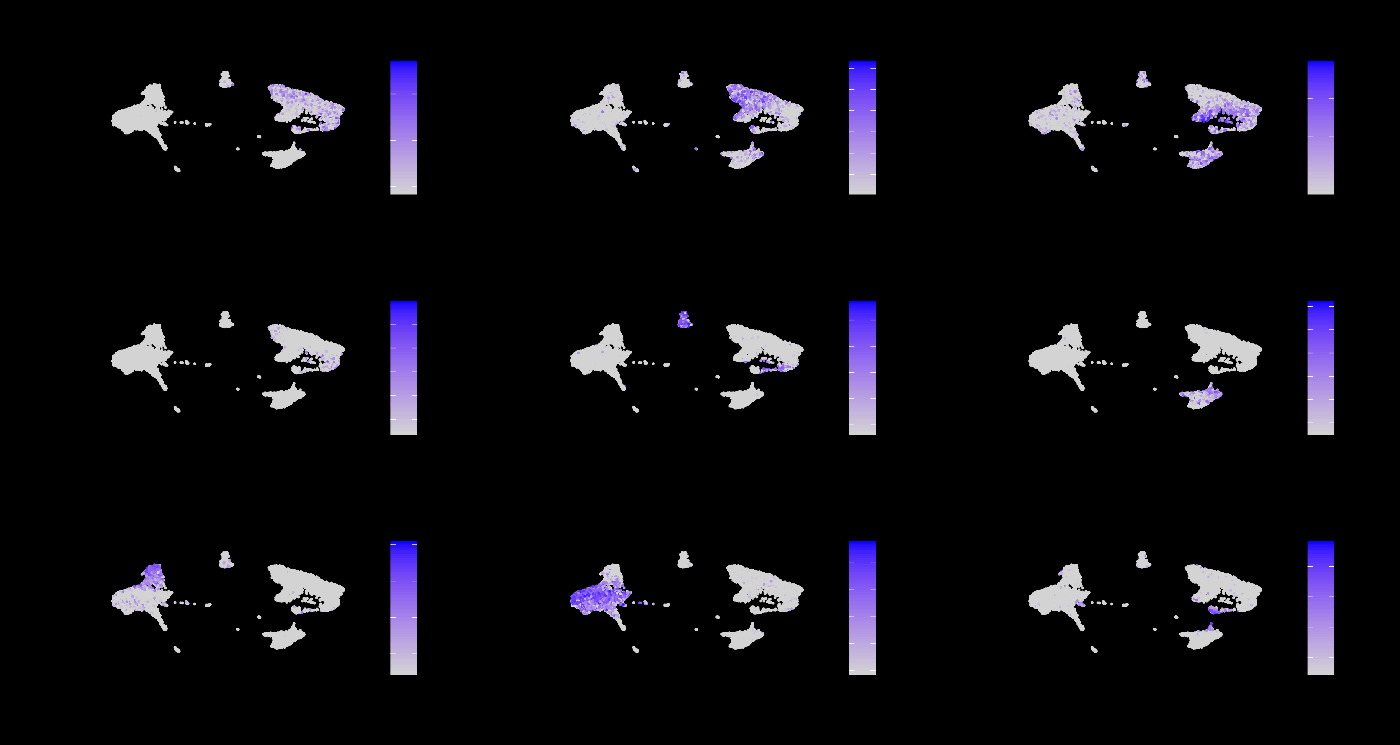
使用 FeaturePlot 确认标记基因表达¶
通过 FeaturePlot 确认各聚类的标记基因特异性表达后,我们可以使用 RenameIdents 函数为每个聚类分配细胞类型。
# 使用 FeaturePlot 确认标记基因表达
FeaturePlot(
immune.combined,
features = c(
"CD3D", "SELL", "CREM", "CD8A", "GNLY", "CD79A", "FCGR3A", "CCL2", "PPBP"
),
min.cutoff = "q9"
)
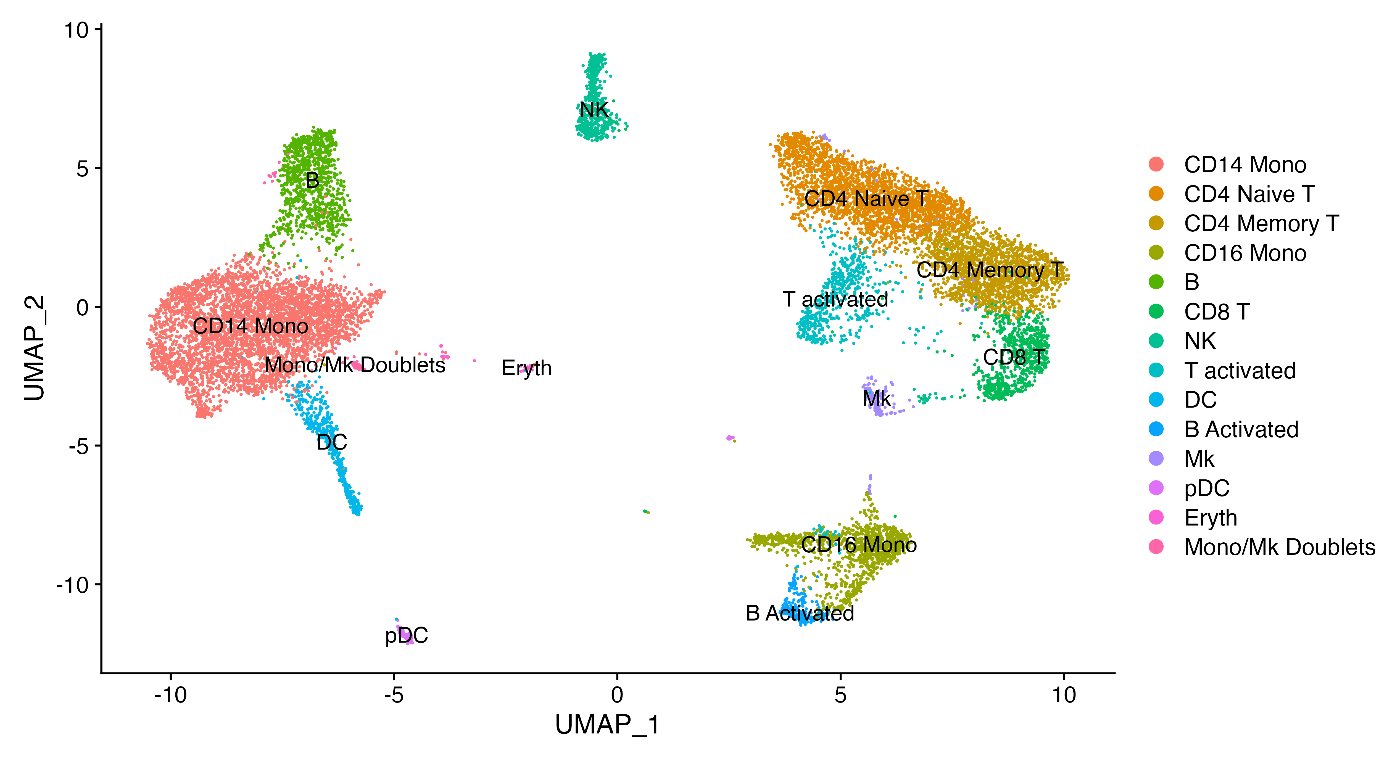
# 为每个聚类分配细胞类型
immune.combined <- RenameIdents(
immune.combined,
`0` = "CD14 Mono",
`1` = "CD4 Naive T",
`2` = "CD4 Memory T",
`3` = "CD16 Mono",
`4` = "B",
`5` = "CD8 T",
`6` = "NK",
`7` = "T activated",
`8` = "DC",
`9` = "B Activated",
`10` = "Mk",
`11` = "pDC",
`12` = "Eryth",
`13` = "Mono/Mk Doublets"
)
# 可视化分配后的细胞类型
DimPlot(immune.combined, label = TRUE)
通过上述代码,您可以在 FeaturePlot 中查看特定标记基因的表达情况,确认每个聚类的特异性表达。然后使用 RenameIdents 函数为每个聚类分配适当的细胞类型,最后通过 DimPlot 可视化分配后的细胞类型。
这样可以直观地查看各个聚类的细胞类型分布,确保每个聚类被正确标识为相应的细胞类型。这对于进一步的分析和解释结果非常重要。
补充说明¶
在使用 FindConservedMarkers 函数之前,补正批次效应是非常重要的。比较多个条件或实验组时,如果未补正批次效应,可能会将批次来源的变动误认为是实际的生物学差异。Seurat 包提供了如 IntegrateData 等工具来整合多个数据集并补正批次效应,因此应预先进行这些处理。