6.1 如何使用 Cell Ranger 统计 scRNA seq 数据中的基因表达水平
什么是 Cell Ranger?¶
Cell Ranger 是由 10x Genomics 公司开发的软件包,用于单细胞 RNA 测序 (scRNA-seq) 数据的预处理和分析。它专门用于分析使用 10x Genomics 的 Chromium 系统进行单细胞 RNA-seq 实验所得的数据。下图展示了在测序之后的数据处理部分由 Cell Ranger 负责。
Cell Ranger 的输出结果可以用于后续的分析,如数据降维(t-SNE 或 PCA)、聚类分析和差异表达分析等。
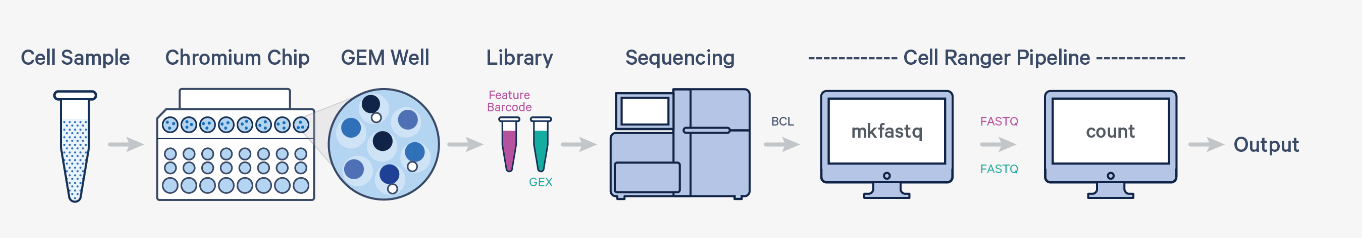
Cell Ranger 的主要管道¶
Cell Ranger 包含以下主要管道:
- cellranger mkfastq: 将原始 BCL 文件(直接从测序仪输出的文件格式)转换为 FASTQ 格式(生物信息分析中常用的格式)。
- cellranger count: 以 FASTQ 文件为输入,进行读段比对、过滤和 UMI 计数(表示每个细胞/基因的表达量),生成细胞基因表达矩阵。
- cellranger aggr: 结合多个 cellranger count 的结果,使这些数据在同一分析空间中可以比较。
- cellranger reanalyze: 基于已获得的 cellranger count 或 cellranger aggr 结果,使用新参数进行聚类和降维等分析。
- cellranger multi: 同时处理不同类型的单细胞数据(如 RNA-seq 和 ATAC-seq),进行整合分析。
通过组合这些管道,可以管理从原始测序数据到获取分析结果的一系列过程。
主要管道介绍¶
接下来,我们将参考 10X Genomics 提供的实操教程,介绍主要使用的 cellranger mkfastq、cellranger count 和 cellranger aggr 管道的具体实现方法。
环境构建¶
首先,需要构建运行 Cell Ranger 的环境。
重要提示:Cell Ranger 只能在 Linux 操作系统上运行。这意味着我们通常使用的 Windows 或 Mac 系统不能直接运行它。因此,Windows 用户需要安装 Ubuntu,Mac 用户则需要使用 Docker 来运行(当然,Windows 用户也可以使用 Docker)。
这次,我们将使用 Docker image 在 Mac 环境中运行 Cell Ranger。以下步骤假设 Docker Desktop 已经安装。Windows 用户可以参考本文中的“Ubuntu 安装”部分来安装 Ubuntu。对于不需要准备 Docker 环境的 Windows 用户,可以直接跳到“运行 cellranger mkfastq”部分。
安装 Cell Ranger 的 Docker image¶
docker pull litd/docker-cellranger
在 Docker UI 中按下终端按钮以启动终端。
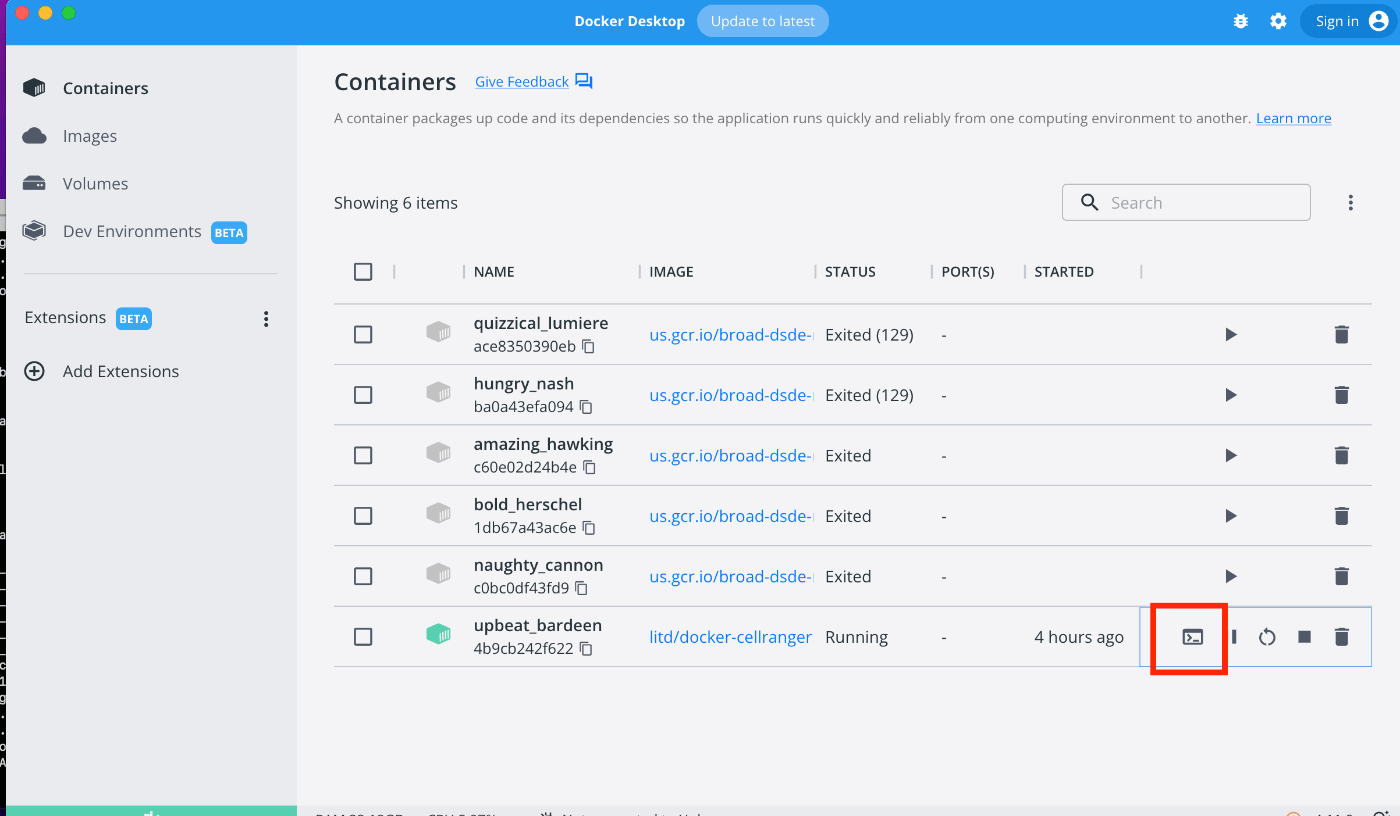
如果无法从 UI 启动,可以使用 docker run -it 命令来启动。可以从 UI 复制容器 ID。¶
docker run -it [ee0a61935c4f49f03a18ee3a9c1f8c17e4437dda43ca1b5e9d07577565632 (容器ID示例)]
启动后,请创建 yard 目录。基本操作将在这个 yard 目录中进行。
mkdir yard
运行 cellranger mkfastq¶
如果公共数据库中已经有 FASTQ 文件,则无需执行此步骤。如果没有,需要在分析前使用 mkfastq 创建 FASTQ 文件。
mkfastq 通过两个步骤运行:
- 去重多重标签:去重多重标签是将一个测序运行中包含的多个样本分离的过程。由于每个样本使用唯一的条形码,mkfastq 读取这些条形码并准确识别每个样本的读段。
- 生成 Fastq 文件:去重多重标签后,mkfastq 使用这些信息生成 Fastq 文件。Fastq 文件包含测序读段及其质量评分,是后续测序分析(如读段比对、基因计数等)的输入。
以下是运行 mkfastq 的实际步骤:
- 创建
run_cellranger_mkfastq目录并进入:
mkdir /yard/run_cellranger_mkfastq
cd /yard/run_cellranger_mkfastq
- 下载并解压示例文件:
wget https://cf.10xgenomics.com/supp/cell-exp/cellranger-tiny-bcl-1.2.0.tar.gz
wget https://cf.10xgenomics.com/supp/cell-exp/cellranger-tiny-bcl-simple-1.2.0.csv
tar -zxvf cellranger-tiny-bcl-1.2.0.tar.gz
解压成功后,将看到以下文件:
cellranger-tiny-bcl-1.2.0/
cellranger-tiny-bcl-1.2.0/Data/
cellranger-tiny-bcl-1.2.0/Data/Intensities/
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/L001/
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/L001/C25.1/
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/L001/C25.1/s_1_1101.bcl.gz
...
- 运行 mkfastq:
cellranger mkfastq --id=tutorial_walk_through.\ \$
--run=/yard/run_cellranger_mkfastq/cellranger-tiny-bcl-1.2.0.\ \$
--csv=/yard/run_cellranger_mkfastq/cellranger-tiny-bcl-simple-1.2.0.csv
成功运行后,将看到以下输出:
sh-4.4# cellranger mkfastq --id=tutorial_walk_through.\ \$
> --run=/yard/run_cellranger_mkfastq/cellranger-tiny-bcl-1.2.0.\ \$
> --csv=/yard/run_cellranger_mkfastq/cellranger-tiny-bcl-simple-1.2.0.csv
/opt/cellranger-7.1.0/bin
cellranger mkfastq (cellranger-7.1.0)
Copyright (c) 2021 10x Genomics, Inc. All rights reserved.
-------------------------------------------------------------------------------
Martian Runtime - v4.0.10
2023-06-11 11:57:52 [runtime] Reattaching in local mode.
Serving UI at http://4b9cb242f622:39525?auth=TKb2F_7AUK6OGai2jN2JBiMtSEysYDXQV3OIbXZURdo
Outputs:
- FASTQ output folder: /yard/run_cellranger_mkfastq/tutorial_walk_through/outs/fastq_path
- Interop output folder: /yard/run_cellranger_mkfastq/tutorial_walk_through/outs/interop_path
- Input samplesheet: /yard/run_cellranger_mkfastq/tutorial_walk_through/outs/input_samplesheet.csv
Waiting 6 seconds for UI to do final refresh.
Pipestance completed successfully!
2023-06-11 11:57:58 Shutting down.
Saving pipestance info to "tutorial_walk_through/tutorial_walk_through.mri.tgz"
生成的 FASTQ 文件位于 /yard/run_cellranger_mkfastq/tutorial_walk_through/outs/fastq_path/H35KCBCXY/test_sample 目录中。可以使用以下命令查看:
cd tutorial_walk_through/outs/fastq_path/H35KCBCXY/test_sample
ls
您将看到类似如下的 FASTQ 文件:
sh-4.4# ls
test_sample_S1_L001_I1_001.fastq.gz test_sample_S1_L001_R1_001.fastq.gz test_sample_S1_L001_R2_001.fastq.gz
运行 cellranger count¶
为了从 FASTQ 文件中获得基因表达计数,运行 cellranger count。这是 Cell Ranger 管道的主要步骤,进行读段比对、过滤、条形码计数和 UMI(唯一分子标识符)计数等一系列分析。
- 创建
run_cellranger_count目录并进入:
mkdir /yard/run_cellranger_count
cd /yard/run_cellranger_count
- 下载并解压示例文件:
wget https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_v3/pbmc_1k_v3_fastqs.tar
tar -xvf pbmc_1k_v3_fastqs.tar
- 下载参考基因组:
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz
tar -zxvf refdata-gex-GRCh38-2020-A.tar.gz
- 运行 cellranger count:
cellranger count --id=run_count_1kpbmcs.\ \$
--fastqs=/yard/run_cellranger_count/pbmc_1k_v3_fastqs.\ \$
--sample=pbmc_1k_v3.\ \$
--transcriptome=/yard/run_cellranger_count/refdata-gex-GRCh38-2020-A
参数说明:
--id=run_count_1kpbmcs:指定输出目录的名称。--fastqs=/yard/run_cellranger_count/pbmc_1k_v3_fastqs:指定输入 FASTQ 文件所在目录。--sample=pbmc_1k_v3:指定样本名称,FASTQ 文件名中应包含此样本名。--transcriptome=/yard/run_cellranger_count/refdata-gex-GRCh38-2020-A:指定使用的转录组参考数据的路径。
完成后,结果将存储在 /yard/run_cellranger_count/tutorial_count/outs 目录中。可以使用以下命令查看输出文件:
cd /yard/run_cellranger_count/tutorial_count/outs
ls
输出文件可能包括:
analysis filtered_feature_bc_matrix.h5 metrics_summary.csv
cloupe.cloupe filtered_feature_bc_matrix molecule_info.h5
filtered_feature_bc_matrix possorted_genome_bam.bam web_summary.html
运行 cellranger aggr¶
cellranger aggr 是用于组合多个 cellranger count 输出(即来自多个样本或实验的单细胞 RNA-seq 数据)的管道。该管道规范化每个数据集并将它们合并为一个表达矩阵,使得可以跨多个样本或条件进行分析。
- 创建
run_cellranger_aggr目录并进入:
mkdir run_cellranger_aggr
cd run_cellranger_aggr
- 下载示例文件:
wget https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_v3/pbmc_1k_v3_molecule_info.h5
wget https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_10k_v3/pbmc_10k\_v3_molecule_info.h5
- 创建一个包含文件路径的 CSV 文件:
使用 vi 创建 pbmc_aggr.csv:
vi pbmc_aggr.csv
添加以下内容:
sample_id,molecule_h5
1k_pbmcs,/path/to/run_cellranger_aggr/pbmc_1k_v3_molecule_info.h5
10k_pbmcs,/path/to/run_cellranger_aggr/pbmc_10k_v3_molecule_info.h5
- 运行 cellranger aggr:
cellranger aggr --id=1k_10k_pbmc_aggr --csv=pbmc_aggr.csv
输入的每个样本的数据将被整合为一个计数矩阵,输出文件存储在 /yard/run_cellranger_aggr/tutorial_aggr/outs 目录中。可以使用以下命令查看输出文件:
cd /yard/run_cellranger_aggr/tutorial_aggr/outs
ls
输出文件可能包括:
analysis filtered_matrices_mex raw_feature_bc_matrix
cloupe.cloupe raw_feature_bc_matrix.h5 web_summary.html
通过这些步骤,您可以成功运行 cellranger 的主要管道,从原始测序数据到综合分析结果。