6.2 使用 Cell Ranger 处理 NCBI Sequence Read Archive (SRA) 数据
从 NCBI SRA 下载的测序数据进行 Cell Ranger 数据处理¶
接下来,我们将介绍如何使用 Cell Ranger 处理从 NCBI(国家生物技术信息中心)提供的 Sequence Read Archive(SRA)中获取的测序数据。
从 NCBI SRA 下载 FASTQ 文件¶
我们将使用 fastq-dump 命令,因此需要先安装 sra-toolkit。以下是安装步骤的相关链接:
- Mac 安装步骤
- Windows 安装步骤(如果未安装 Ubuntu,请从安装 Ubuntu 开始)
下载 FASTQ 文件¶
首先,使用 --split-files 参数通过 NCBI 的 fastq-dump 实用工具获取 FASTQ 文件。命令如下:
fastq-dump --split-files --gzip SRR6334436
此命令将下载以下两个 FASTQ 文件:
SRR6334436_1.fastq.gzSRR6334436_2.fastq.gz
要确定这是什么原因,让我们先看看 SRR6334436 的元数据。值得注意的是 L=26 和 L=98 这两个数字,它们分别代表读取 1 和读取 2 的读取长度。
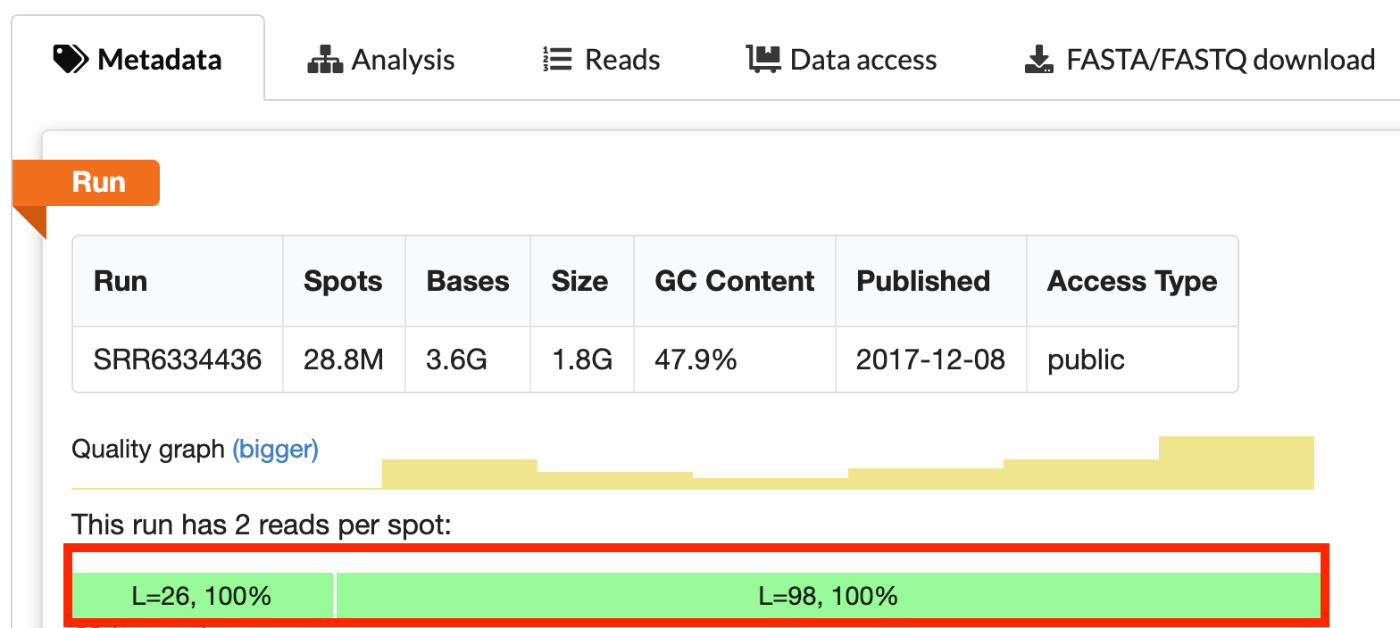
Read1 和 Read2 的含义¶
Read1 是细胞条形码和 UMI 的数据,Read2 是插入片段(Insert)。它们的长度分别是 28 和 90。由此可以推测,SRR6334436_1.fastq 是 Read1,SRR6334436_2.fastq 是 Read2。
引用自 10x Genomics 的文档:
通常,测序数据的样本有两个读段(Read 1 和 Read 2),但有时会有三个或四个读段,这通常是因为包含了索引读段(Index Read)。
示例:从 SRR9291388 获取 FASTQ 文件¶
例如,从 SRR9291388 获取 FASTQ 文件的命令如下:
fastq-dump --split-files --gzip SRR9291388
执行此命令后,输出将是三个 FASTQ 文件:
SRR9291388_1.fastq.gzSRR9291388_2.fastq.gzSRR9291388_3.fastq.gz
这与 SRR9291388 的元数据报告的信息一致,即该测序运行每个点(spot)有三个读段(read)。
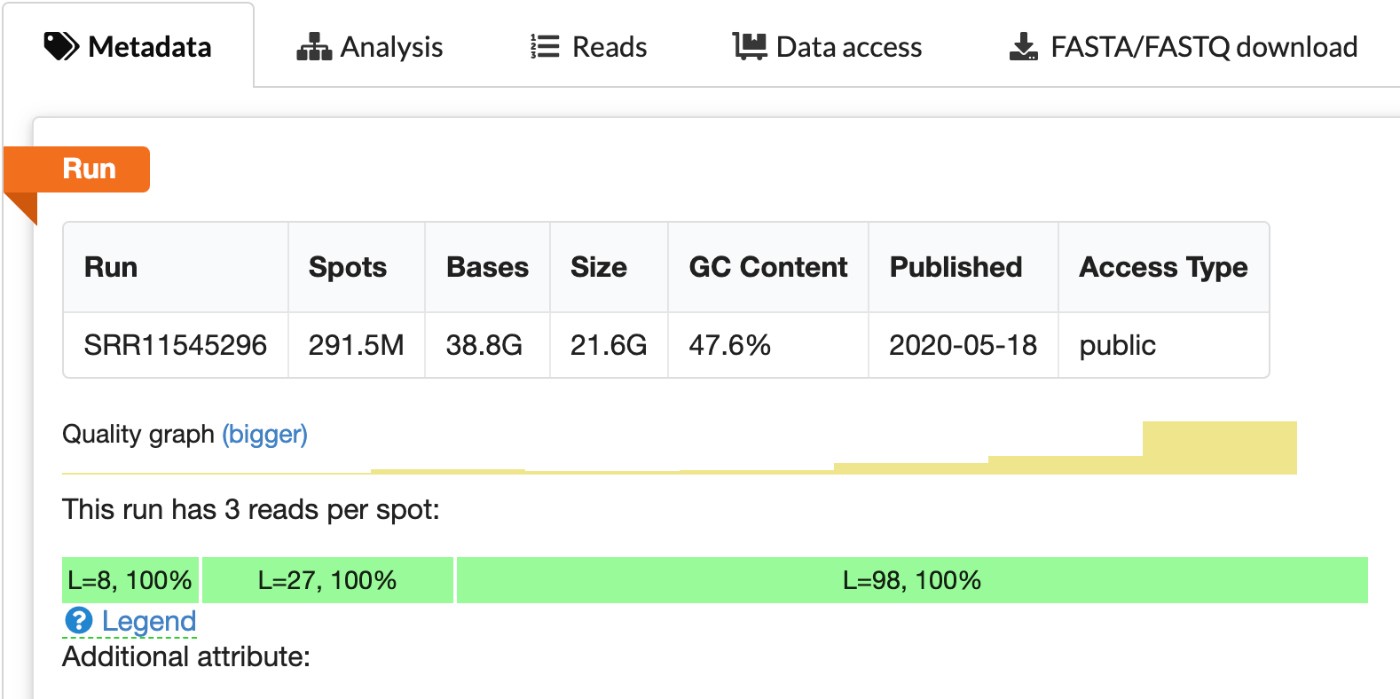
从元数据分配文件名¶
根据元数据,我们可以如下分配文件:
SRR9291388_1.fastq: Read 1SRR9291388_2.fastq: Read 2SRR9291388_3.fastq: Index
将 SRA 数据重命名为适合 Cell Ranger 的文件名¶
Cell Ranger 需要遵循 bcl2fastq 的文件命名规则。bcl2fastq 的文件命名规则如下:
[Sample Name]_S1_L00[Lane Number]_[Read Type]_001.fastq.gz
各部分说明:
- [Sample Name]: 代表样本名称或 ID,用于识别数据所属的样本。
- S1: 代表样本的序列号,多个样本时依次递增(如 S2、S3 等)。
- L00[Lane Number]: 代表流动单元中的通道号,多个通道时依次递增(如 L001、L002 等)。
- [Read Type]: 代表读段类型,R1 代表正向读段(Read 1),R2 代表反向读段(Read 2),I1 和 I2 代表索引读段。
- 001: 代表段号,通常为 001。
基于以上规则,将 SRR6334436_1.fastq.gz 和 SRR6334436_2.fastq.gz 重命名为:
- Read 1:
SRR6334436_S1_L001_R1_001.fastq.gz - Read 2:
SRR6334436_S1_L001_R2_001.fastq.gz
将 SRR9291388_1.fastq, SRR9291388_2.fastq, SRR9291388_3.fastq 重命名为:
- Read 1:
SRR9291388_S1_L001_R1_001.fastq.gz - Read 2:
SRR9291388_S1_L001_R2_001.fastq.gz - Index 1:
SRR9291388_S1_L001_I1_001.fastq.gz
在 SRA 数据上运行 Cell Ranger count¶
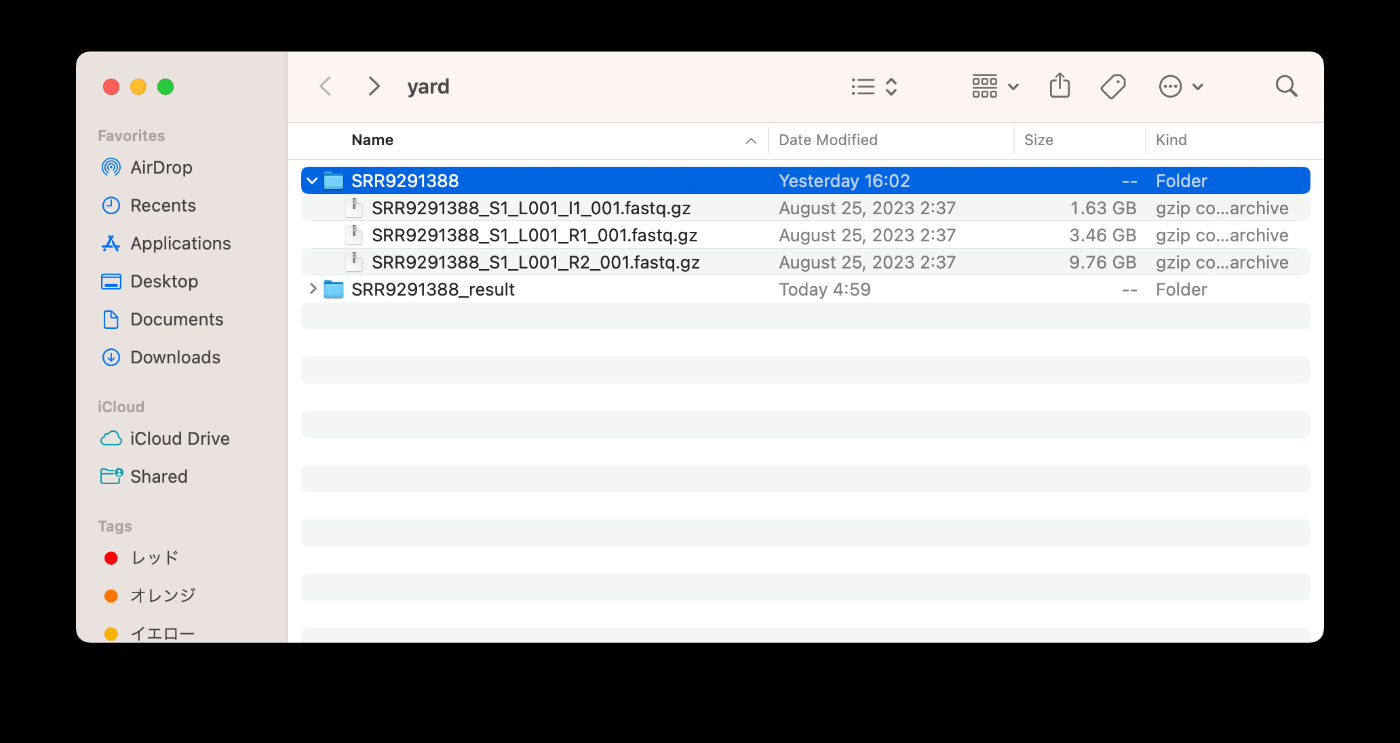
实际运行时,请记得创建 yard 文件夹。
通常,我会在 yard 文件夹中创建 SRR9291388 文件夹和 SRR9291388_result 文件夹,并将重命名后的文件放入 SRR9291388 文件夹中。
接下来,我们将使用 SRR9291388_1.fastq, SRR9291388_2.fastq, SRR9291388_3.fastq 来运行 Cell Ranger。
记得创建 yard 文件夹以运行 Cell Ranger。
示例步骤¶
通常,我会在 yard 文件夹中创建 SRR9291388 文件夹和 SRR9291388_result 文件夹,并将重命名后的文件放入 SRR9291388 文件夹中。
准备好文件夹后,运行以下命令:
cellranger count --id=SRR9291388_result --fastqs=/yard/SRR9291388 --sample=SRR9291388 --transcriptome=/yard/run_cellranger_count/refdata-gex-GRCh38-2020-A
各选项的说明如下:
--id=SRR9291388_result:此选项指定的 ID 将用作分析结果输出目录的名称。将创建名为SRR9291388_result的目录,分析结果将保存在该目录中。--fastqs=/yard/SRR9291388:此选项指定的路径指向保存输入 FASTQ 文件的目录。/yard/SRR9291388目录中的 FASTQ 文件将用于分析。--sample=SRR9291388:此选项指定的样本名用于识别 FASTQ 文件中要分析的样本。在本例中,将分析名为SRR9291388的样本。--transcriptome=/yard/run_cellranger_count/refdata-gex-GRCh38-2020-A:此选项指定的路径指向保存参考转录组数据的目录。在本例中,将使用目录/yard/run_cellranger_count/refdata-gex-GRCh38-2020-A内的参考转录组(GRCh38 版本 2020-A)。
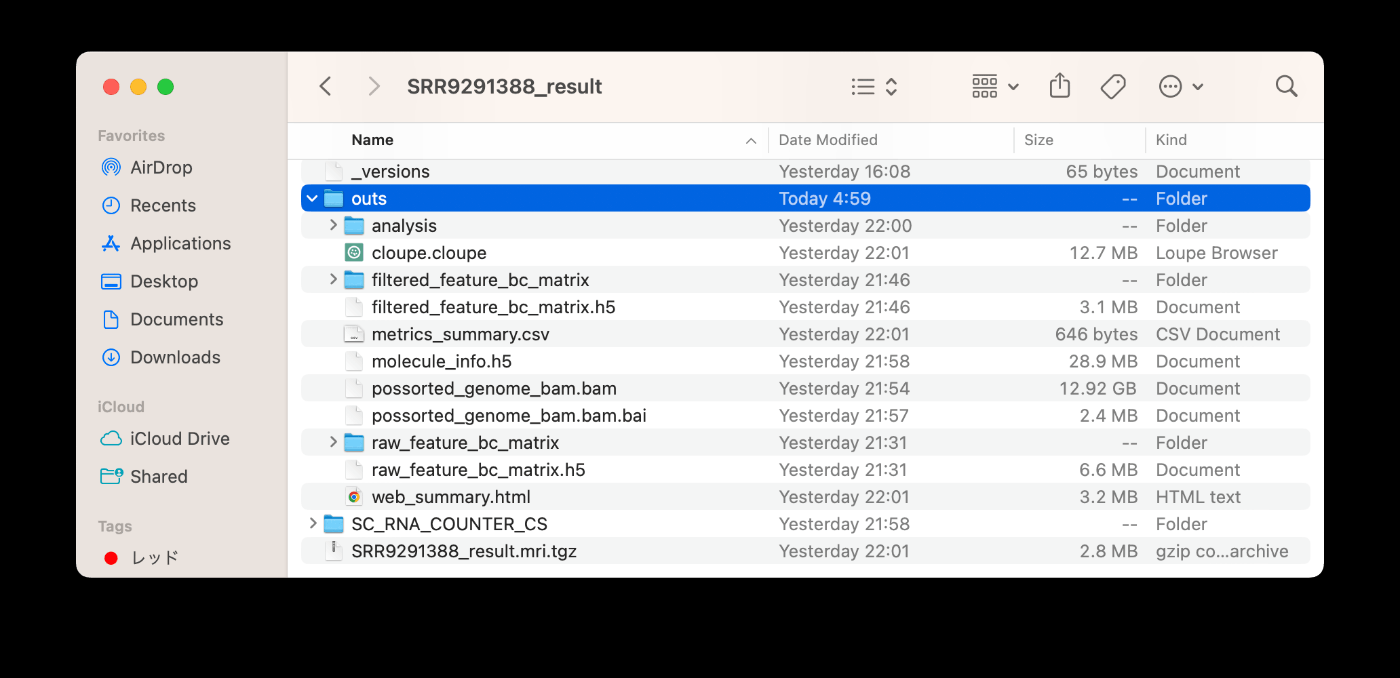
成功运行后,文件将输出到 SRR9291388_result 文件夹中。查看 outs 目录,您会发现所需的文件已生成。