7.1 关于 scRNA seq 数据的质量控制
QC 和过滤指标¶
以下是 QC 和过滤中常用的指标和方法。通过掌握这些 QC 要点,scRNA-seq 数据的质量将显著提升。
-
UMI 计数的细胞条形码过滤 由于 UMI 计数异常高或低的条形码可能不代表单个细胞,因此需要排除这些条形码。
-
特征数的细胞过滤 特征数异常高或低的条形码也可能不代表单个细胞,因此需要排除这些条形码。
-
根据线粒体(mt)读数百分比的细胞过滤 来自 mtDNA 的转录水平升高与细胞状态不健康相关,因此需要根据线粒体读数百分比进行过滤。
-
双细胞检测的细胞过滤 由于双细胞或多细胞会干扰分析,需要识别并移除这些细胞。
-
识别并移除空滴 10x Genomics 的 Chromium 技术设计了大多数滴中含有一个以下的细胞,因此存在许多不含完整细胞的空滴。需要识别并移除这些空滴。
-
去除背景 RNA 滴基方法中,滴内可能会封入周围环境中的 RNA,这些 RNA 可能来自于细胞释放或污染物。需要去除这些 RNA。
使用 Seurat 进行 UMI 计数、特征数和线粒体(mt)读数百分比的细胞过滤¶
下面是使用 Seurat 进行这些过滤的代码示例。这种处理方法在大多数论文中作为基本的 QC 步骤非常常见。(以下是样本代码,实际运行时需要将 raw.data 替换为实际使用的文件。)
# 加载Seurat包并创建Seurat对象
library(Seurat)
pbmc <- CreateSeuratObject(counts = raw.data)
# 计算线粒体基因的百分比
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
# 根据UMI计数、特征数和线粒体基因的百分比进行细胞过滤
pbmc <- subset(
pbmc,
subset = nFeature_RNA > 200 &
nFeature_RNA < 2500 &
nCount_RNA > 500 &
nCount_RNA < 25000 &
percent.mt < 5
)
# 再次计算线粒体基因的百分比
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
这里,pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-") 是计算每个细胞中线粒体基因表达的百分比,pattern = "^MT-" 是用于识别线粒体基因的正则表达式模式。
使用 subset 函数进行过滤,仅保留满足特定条件的细胞:
nFeature_RNA > 200 & nFeature_RNA < 2500:选择特征数在 200 到 2500 之间的细胞。特征数低于 200 可能是空滴,高于 2500 可能是双细胞或多细胞,因此需要除去。nCount_RNA > 500 & nCount_RNA < 25000:选择 UMI 计数在 500 到 25000 之间的细胞。percent.mt < 5:选择线粒体基因表达占比低于 5% 的细胞。
QC 前后可视化¶
通过小提琴图输出 QC 前后的图表差异,可以更直观地理解 QC 对样本的影响。QC 后,符合上述条件外的细胞将被剔除。
# 使用MT-开头的所有基因集合作为线粒体基因集合
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
# 绘制小提琴图
VlnPlot(
pbmc,
features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
ncol = 3
)
QC 前
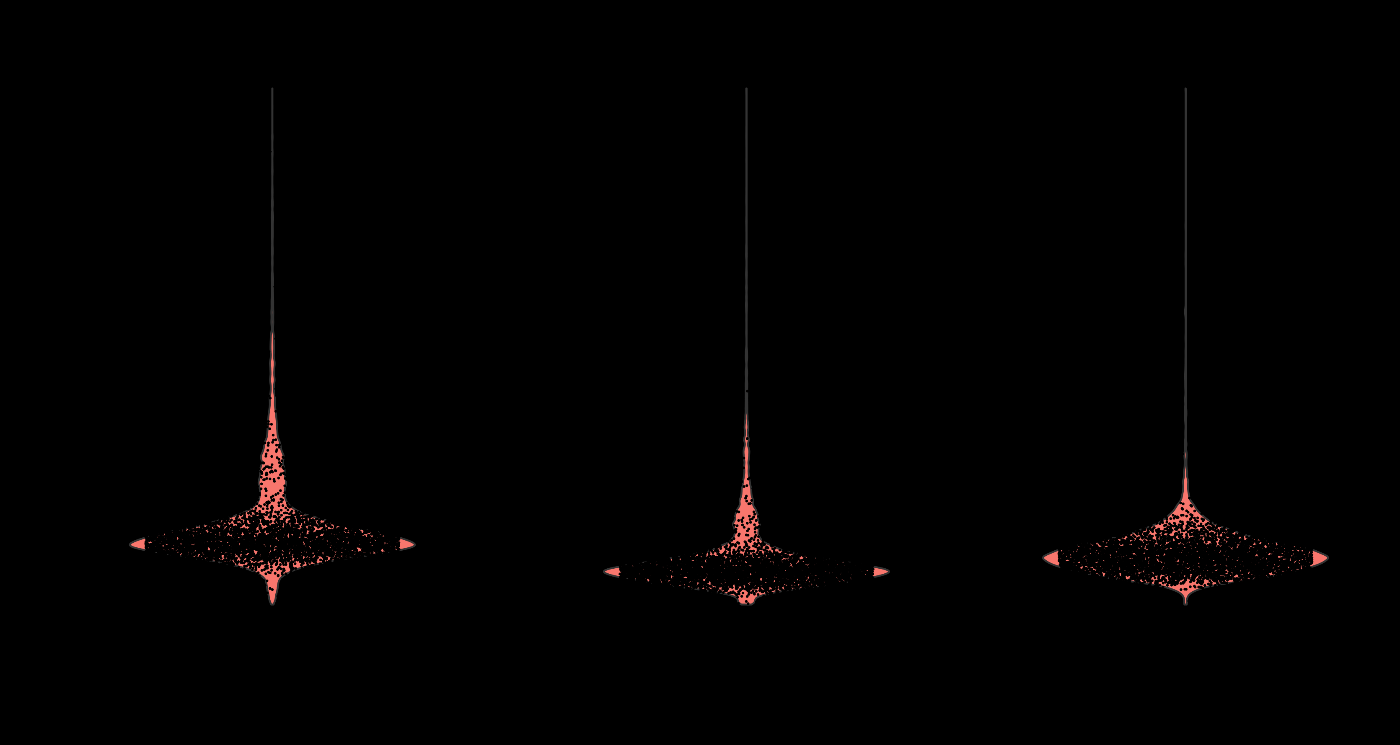
QC 后
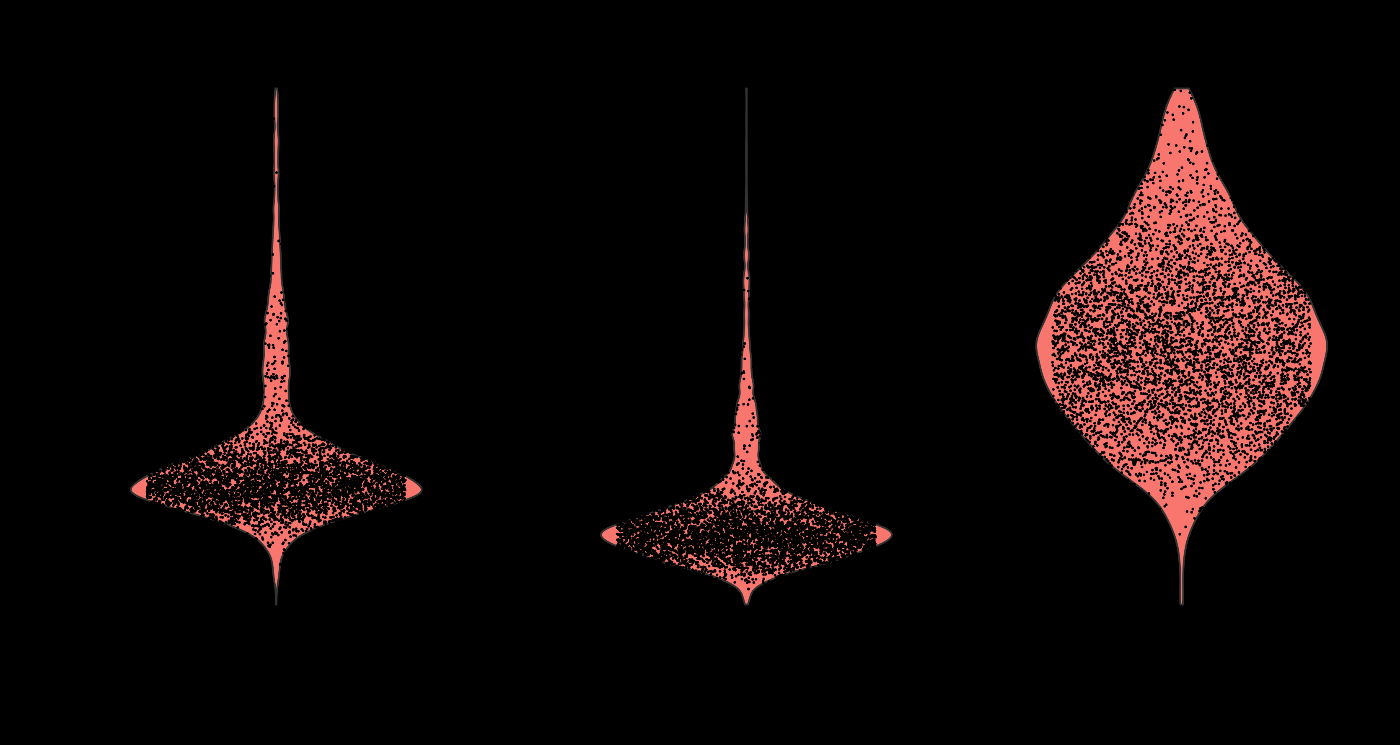
使用 Seurat 进行上述方法时,由于 QC 阈值是自行设定的,因此可能会包含一些主观因素。此外,被 QC 排除的细胞也可能是稀有细胞,不能一概而论它们是质量差的细胞。在介绍 scRNA-seq 库的网站 scRNA-tools 上,通过“Quality Control”进行筛选,可以找到近 100 个库。这表明 QC 领域非常深入,且备受关注。
因此,在本章中,我们将介绍使用 DoubletFinder 包进行“4.双细胞检测的细胞过滤”,以及使用 CellBender 包进行“5.识别并移除空滴”和“6.去除背景 RNA”的解析方法。