7.2 使用 DoubleFinder 检测双重细胞
什么是双细胞检测?¶
双细胞(Doublet)指的是两个或多个细胞偶然一起被捕获并作为单个细胞进行处理的现象。在单细胞数据分析中,这成为一个重大问题。因为双细胞会将来自两种不同细胞类型的基因表达模式混合,从而可能误导细胞类型的鉴定和基因表达的分析。
根据 10x Genomics 发布的质量控制方法,使用社区工具进行双细胞检测以过滤细胞时提到:“双细胞或多细胞可能会干扰分析。可以使用 DoubletFinder、Scrublet、Solo 等工具。”
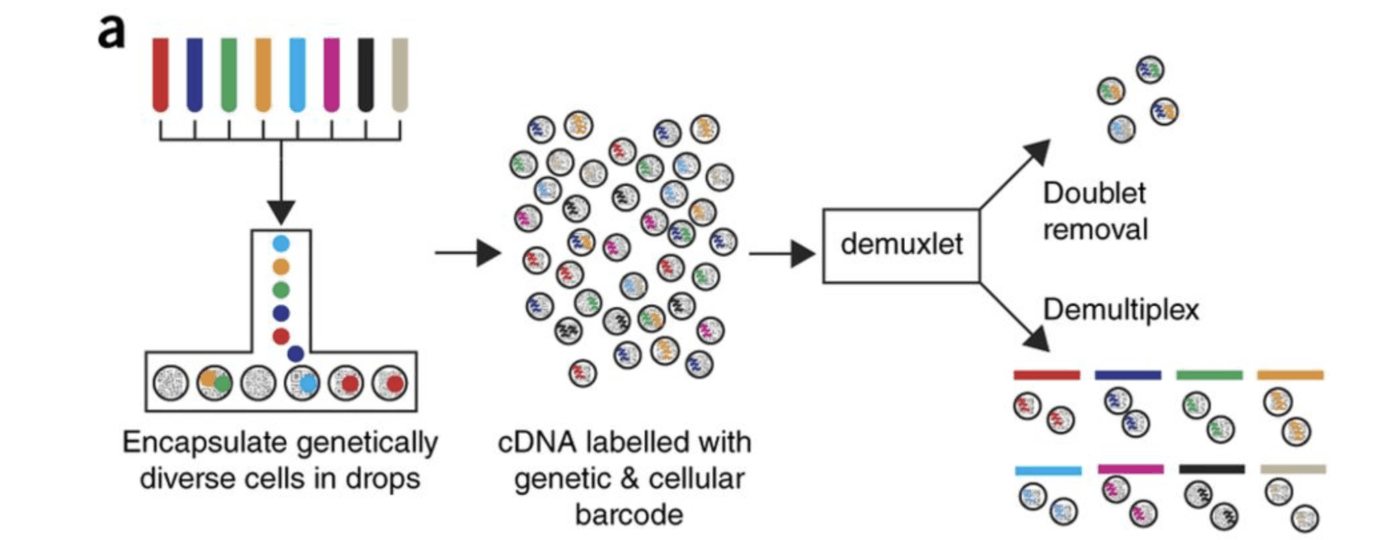
双细胞检测的综合基准测试¶
在本论文中,对九种工具进行了双细胞检测的综合基准测试,结果表明 DoubletFinder 具有最高的检测精度,并且在下游分析中具有最大的改进效果。
DoubletFinder 的双细胞检测步骤¶
DoubletFinder 通过以下步骤检测双细胞:
- 人工生成双细胞:通过将数据集内每个细胞的基因表达谱与从其他细胞中随机选择的基因表达谱相加,生成人工双细胞。
- 主成分分析(PCA):进行 PCA 分析,并在图中绘制双细胞和单细胞(仅由一个细胞组成的数据)。人工双细胞通常在 PCA 空间中比单细胞更远。
- 二项混合模型:使用二项混合模型分离 PCA 空间中的双细胞和单细胞簇。
- 计算概率:根据这些簇计算每个细胞是双细胞的概率。
通过以上步骤,DoubletFinder 能够有效地从单细胞 RNA-seq 数据中移除双细胞。
使用 DoubletFinder 进行双细胞检测¶
接下来,我们使用 DoubletFinder 进行双细胞检测。我们将使用 10k Human PBMCs, 3′ v3.1, Chromium Controller 数据集。请按照以下步骤下载和解压数据集:
安装必要的库¶
首先,我们需要安装和加载必要的库:
install.packages("Seurat")
install.packages("ggplot2")
install.packages("tidyverse")
install.packages("DoubletFinder")
library(Seurat)
library(ggplot2)
library(tidyverse)
library(DoubletFinder)
读取下载的文件¶
接下来,我们读取之前下载的文件。由于 UMAP 之前的步骤与往常相同,这里将省略详细说明。如果不熟悉这些操作,请复习第 4 章中的 Seurat 数据分析基本步骤。
# 创建计数矩阵
cts <- Read10X(data.dir = "./raw_feature_bc_matrix/")
# 创建Seurat对象
pbmc.seurat <- CreateSeuratObject(counts = cts)
# QC和过滤
# 探索QC
pbmc.seurat$mitoPercent <- PercentageFeatureSet(pbmc.seurat, pattern = "^MT-")
pbmc.seurat.filtered <- subset(pbmc.seurat, subset = nCount_RNA > 800 &
nFeature_RNA > 500 &
mitoPercent < 10)
# 标准预处理工作流程
pbmc.seurat.filtered <- NormalizeData(object = pbmc.seurat.filtered)
pbmc.seurat.filtered <- FindVariableFeatures(object = pbmc.seurat.filtered)
pbmc.seurat.filtered <- ScaleData(object = pbmc.seurat.filtered)
pbmc.seurat.filtered <- RunPCA(object = pbmc.seurat.filtered)
ElbowPlot(pbmc.seurat.filtered)
pbmc.seurat.filtered <- FindNeighbors(object = pbmc.seurat.filtered, dims = 1:20)
pbmc.seurat.filtered <- FindClusters(object = pbmc.seurat.filtered)
pbmc.seurat.filtered <- RunUMAP(object = pbmc.seurat.filtered, dims = 1:20)
使用 DoubletFinder 进行双细胞检测¶
DoubletFinder 通过创建模拟双细胞来计算双细胞分数,这些模拟双细胞模仿了数据集中不同细胞之间可能共享的表达模式。
在创建模拟双细胞时,使用的参数包括 pK 和 pN。
- pK指定了相对于整个细胞群体的簇比例,这个参数决定了从每个簇中选择的模拟双细胞数量。设置较高的 pK 值会创建更多的模拟双细胞。
- pN指定了用于创建模拟双细胞的预期双细胞率。例如,pN = 0.05 表示预计原始单细胞数据集中存在 5% 的双细胞。
以下代码用于计算 pK 值并生成相应的图表:
# pK识别(无地面实况)
sweep.res.list_pbmc <- paramSweep_v3(pbmc.seurat.filtered, PCs = 1:20, sct = FALSE)
sweep.stats_pbmc <- summarizeSweep(sweep.res.list_pbmc, GT = FALSE)
bcmvn_pbmc <- find.pK(sweep.stats_pbmc)
# 绘制pK值与BCmetric的关系图
ggplot(bcmvn_pbmc, aes(pK, BCmetric, group = 1)) +
geom_point() +
geom_line()
通过执行 find.pK,可以计算出最佳的 pK 值。在此示例中,当 pK 为 0.2 时,BCmetric 达到最大值。
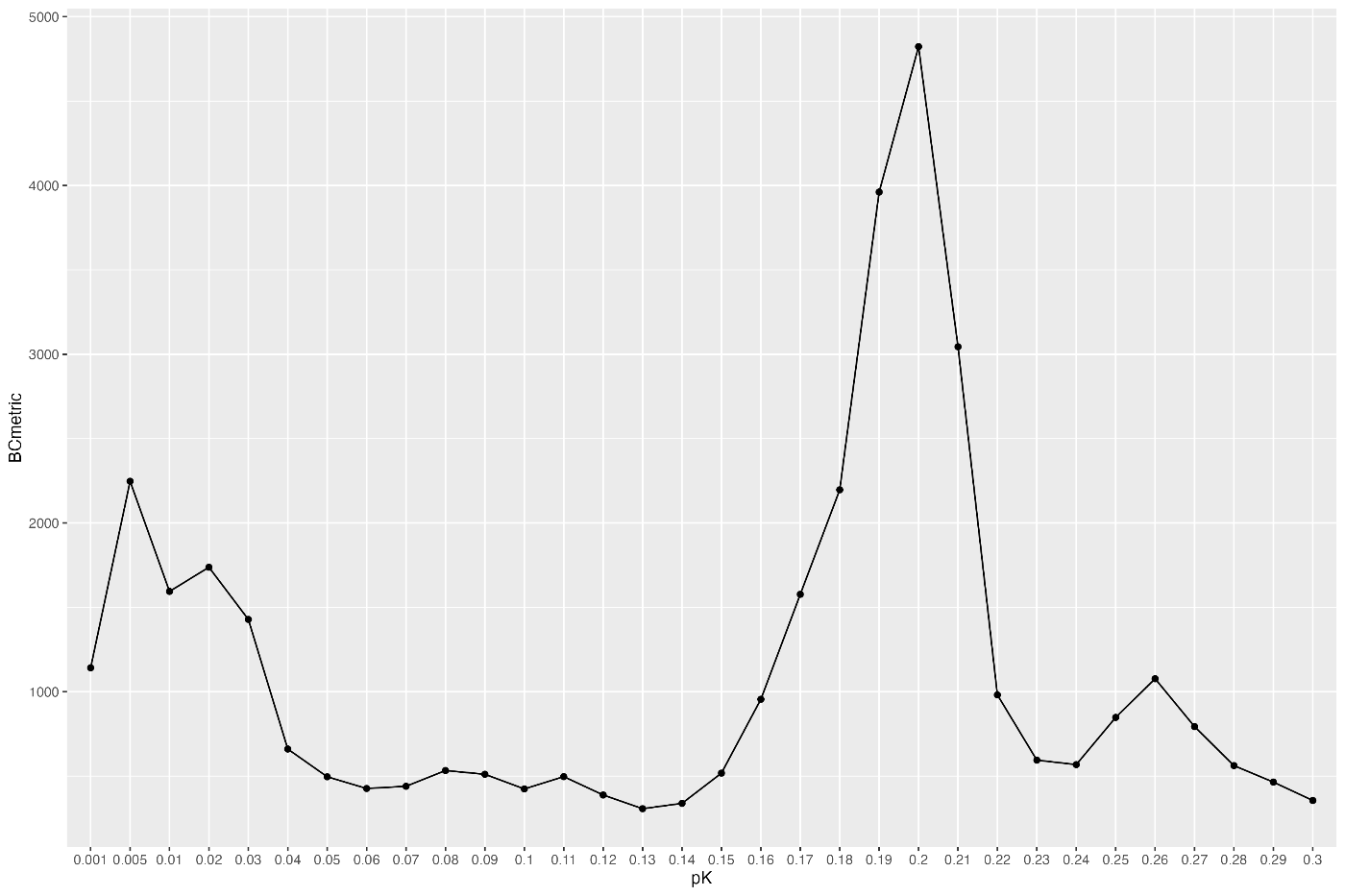
从生成的数据框 bcmvn_pbmc 中,我们确定了最优的 pK 值(最高的双细胞得分,BCmetric)。以下代码保存了前面计算的最优 pK 值:
pK <- bcmvn_pbmc %>%
filter(BCmetric == max(BCmetric)) %>%
select(pK)
pK <- as.numeric(as.character(pK[[1]]))
这段代码利用 doubletfinder 包的功能,为检测双细胞(同一孔中存在两个或更多细胞的状态)做准备。
## 同型双细胞比例估算 -------------------------------------------------------------------------------------
annotations <- pbmc.seurat.filtered@meta.data$seurat_clusters
homotypic.prop <- modelHomotypic(annotations) ## 例如:annotations <- seu_kidney@meta.data$ClusteringResults
nExp_poi <- round(0.076 * nrow(pbmc.seurat.filtered@meta.data)) ## 假设双细胞形成率为7.6% - 根据您的数据集调整
nExp_poi.adj <- round(nExp_poi * (1 - homotypic.prop))
解释:
- annotations <- pbmc.seurat.filtered@meta.data$seurat_clusters:从 Seurat 对象(这里是 pbmc.seurat.filtered)的元数据中获取聚类结果(seurat_clusters),并保存到 annotations 中。这样可以获得每个细胞所属的聚类信息。
- homotypic.prop <- modelHomotypic(annotations):使用 doubletfinder 包的 modelHomotypic 函数,估算同一聚类内(同型)双细胞的比例。
- nExp_poi <- round(0.076 * nrow(pbmc.seurat.filtered@meta.data)):假设双细胞的比例为 7.6%,计算预期的双细胞数量。
- nExp_poi.adj <- round(nExp_poi * (1 - homotypic.prop)):考虑同型双细胞的比例,调整预期的双细胞数量。这个值将在双细胞检测算法中使用。
运行 DoubletFinder¶
使用以下代码运行 DoubletFinder:
# 运行 DoubletFinder
pbmc.seurat.filtered <- doubletFinder_v3(pbmc.seurat.filtered,
PCs = 1:20,
pN = 0.25,
pK = pK,
nExp = nExp_poi.adj,
reuse.pANN = FALSE, sct = FALSE
)
可视化结果¶
使用 DimPlot 可视化双细胞检测的结果:
# 可视化双细胞
DimPlot(
pbmc.seurat.filtered,
reduction = "umap",
group.by = "DF.classifications_0.25_0.22_691"
)
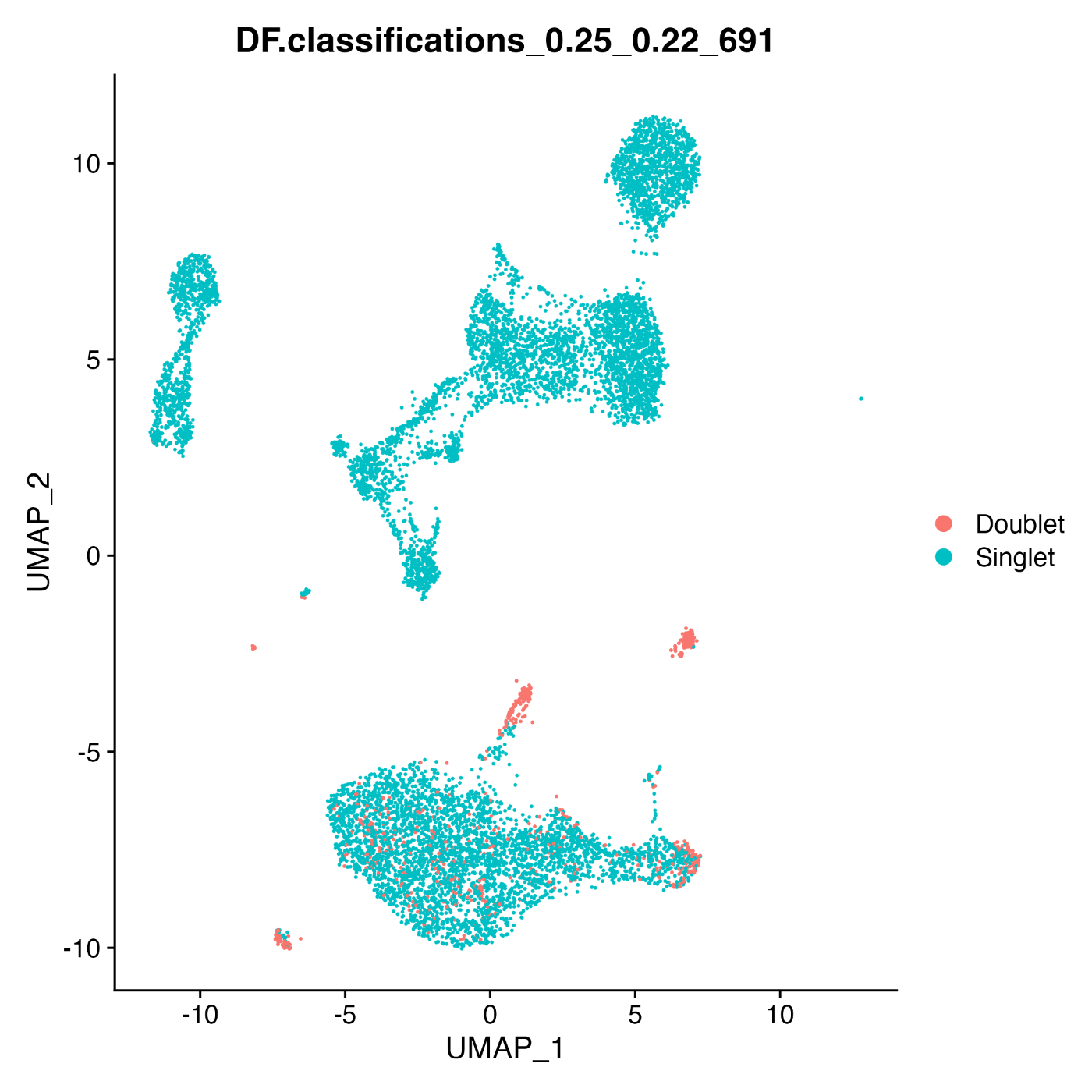
Doublet 检测结果¶
在上面的可视化中,检测到的双细胞用红色表示。
最后,让我们输出双细胞的数量:
# 输出单细胞和双细胞的数量
table(pbmc.seurat.filtered@meta.data$DF.classifications_0.25_0.22_691)
输出结果¶
Doublet Singlet
691 9326
从结果中可以看到,数据集中包含 691 个双细胞。