7.3 用 CellBender 去除背景 RNA
CellBender 简介¶
CellBender 是一个处理 scRNA-seq 数据中常见的实验噪音和伪影(如背景噪音、基因丢失、空滴等)的软件。它提供了详细的解说页面,以下是对其主要功能的介绍:
空滴的识别¶
在滴序列技术中,即使细胞未被捕获,滴中也可能检测到 RNA。这些称为“空滴”,通常被视为噪音。CellBender 能够有效地识别并从数据中删除这些空滴。
基因丢失的校正¶
在单细胞 RNA 测序数据中,某些基因可能未被检测到(即基因丢失)。CellBender 使用贝叶斯统计方法来校正这些基因丢失现象。
基于深度学习的模型¶
CellBender 采用基于深度学习的模型(特别是变分自编码器,VAE),能够高效地学习高维 scRNA-seq 数据中的复杂模式,从而去除噪音。
HDF5 文件格式¶
CellBender 使用 HDF5 文件进行分析,而不是使用条形码、特征和矩阵文件。以下是对这两种文件格式的简单介绍:
条形码、特征和矩阵格式¶
这种格式通常用于 10x Genomics 等 scRNA-seq 平台。每个文件包含以下信息:
- 条形码文件:包含唯一标识每个细胞的条形码列表
- 特征(基因)文件:包含测量到的所有基因列表
- 矩阵文件:包含每个细胞中每个基因的表达水平的稀疏矩阵
HDF5 文件¶
HDF5 将条形码、特征和矩阵信息整合到一个文件中。HDF5 是一种分层数据格式,能够高效存储大量数据。
这两种格式的主要区别在于读取和分析所需的步骤数量。使用条形码、特征和矩阵格式时,需要分别读取每个文件,并组装成一个矩阵进行分析。而 HDF5 格式将所有信息包含在一个文件中,因此只需一次操作即可读取文件。
CellBender 的背景 RNA 去除机制¶
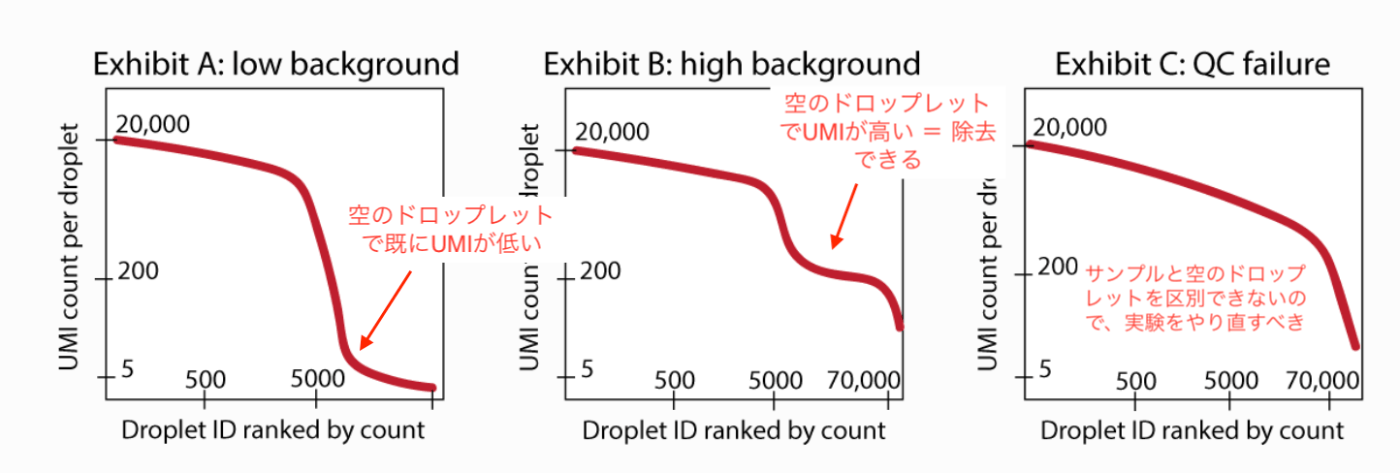
remove-background 功能用于去除空滴中包含的一定量的背景 RNA,这种现象称为“环境平台”。
- Exhibit A:对于 UMI 计数非常少的数据集,背景 RNA 几乎不存在,因此此功能可能效果不明显。
- Exhibit B:对于 UMI 计数在 50、100、几百的空滴数据集,
remove-background功能可以显著改善数据集。 - Exhibit C:对于背景 RNA 非常多,无法视觉上确认空滴平台的数据集,建议重新进行实验。
使用 CellBender 进行 QC¶
前面的准备工作已经介绍完毕,现在我们将开始使用 CellBender。以下步骤参考 CellBender 的官方文档。
安装 Docker 镜像¶
首先,我们需要安装 CellBender 的 Docker 镜像:
docker pull us.gcr.io/broad-dsde-methods/cellbender:latest
启动 Docker¶
接着,启动 Docker 容器:
docker run -it us.gcr.io/broad-dsde-methods/cellbender:latest /bin/bash
切换到 remove-background 目录¶
在 Docker 容器中,切换到 remove-background 目录:
cd /cellbender/remove-background
下载分析所需文件¶
接下来,下载用于分析的文件。访问以下链接并点击“下载全部”。如果没有看到“下载全部”按钮,可以点击上方标签中的“▼”并选择“下载”进行下载。
链接: 下载链接
运行 CellBender¶
完成以上步骤后,我们可以开始使用 CellBender 进行分析。下载并解压文件后,按照官方文档的指引运行 CellBender 即可。具体步骤如下:
- 将下载的文件解压到指定目录。
- 在 Docker 容器中运行 CellBender 的命令进行背景去除。
例如:
cellbender remove-background.\ \$
--input raw_feature_bc_matrix.h5.\ \$
--output filtered_feature_bc_matrix.h5.\ \$
--cuda
以上命令中:
- --input:指定输入文件
- --output:指定输出文件
- --cuda:如果有 GPU 支持,可以使用此选项加速计算
下载文件,只留下文件名 "tiny_raw_gene_bc"。
下载的文件应在本地解压缩。
将下载并解压的文件复制到 docker 容器中,从 Docker Desktop 复制容器 ID。
使用 CellBender 进行 QC¶
准备工作已经完成,现在我们将使用 CellBender 进行分析。以下是具体步骤:
启动一个新的终端并复制文件¶
打开一个新的终端(注意:不是在 Docker 终端中),使用先前复制的 Docker 容器 ID 将文件复制到容器内。
以下命令将 Desktop 目录中的 raw_gene_bc_matrices 文件夹复制到 Docker 容器的 remove-background 目录中:
docker container cp /Users/hogeuser/Desktop/raw_gene_bc_matrices [复制的ID]:/cellbender/remove-background
在 Docker 容器中运行 CellBender¶
在 Docker 容器中,运行以下命令以执行 CellBender。整个过程大约需要 6 到 12 小时。如果可以使用 GPU,可以加上 --cuda 标志以缩短执行时间。
cellbender remove-background.\ \$
--input ./tiny_raw_gene_bc_matrices/GRCh38.\ \$
--output ./tiny_10x_pbmc.h5.\ \$
--expected-cells 500.\ \$
--total-droplets-included 5000
参数解释¶
--input ./tiny_raw_gene_bc_matrices/GRCh38:指定输入数据的路径。这里指定了当前目录下的tiny_raw_gene_bc_matrices/GRCh38文件夹。--output ./tiny_10x_pbmc.h5:指定去除背景噪音后的数据保存路径。这里将在当前目录下创建一个名为tiny_10x_pbmc.h5的文件。--expected-cells 500:指定预期的实际细胞数量。这个参数对于算法解释数据非常重要,通常根据实验信息设置。这里预期有 500 个细胞。--total-droplets-included 5000:指定包含在分析中的总滴数量。这里预期包含 5000 个滴。
成功运行的标志¶
成功运行后,应该能够看到如下文件列表:
root@ba0a43efa094:/cellbender/examples/remove_background# ls
generate_tiny_10x_pbmc.py tiny_10x_pbmc.h5 tiny_10x_pbmc.log tiny_10x_pbmc.pdf tiny_10x_pbmc_cell_barcodes.csv tiny_10x_pbmc_filtered.h5
各输出文件的解释¶
tiny_10x_pbmc.h5:这是一个 HDF5 格式的文件,包含推断过程的详细输出,包括环境转录本的归一化丰度、每个滴的污染程度、背景校正后的基因表达低维嵌入和背景校正后的计数矩阵(CSC 稀疏矩阵格式)。tiny_10x_pbmc_filtered.h5:包含与上述相同的信息,但只包括后验细胞概率超过 0.5 的滴。tiny_10x_pbmc_cell_barcodes.csv:包含后验细胞概率超过 0.5 的条形码列表。tiny_10x_pbmc.pdf:一个 PDF 总结文件,显示训练过程中的损失函数演变、UMI 图和后验细胞概率的排名、包含细胞的滴的表达潜在嵌入的二维 PCA 散点图。请注意,UMI 排名超过大约 500 时,细胞概率急剧下降。
将生成的文件复制回本地¶
将生成的文件复制到本地,以便进行分析。再次从 Docker Desktop 复制容器 ID。
使用新的终端复制文件¶
打开一个新的终端(注意:不是在 Docker 终端中),使用之前复制的 Docker 容器 ID 将文件从容器复制到本地 Desktop。
以下命令将容器中的 examples 目录复制到本地的 Desktop 目录:
docker container cp [复制的ID]:/cellbender/examples /Users/hogeuser/Desktop/
确保在 Desktop 目录中下载了 examples 文件夹,并且确认生成了 tiny_10x_pbmc_filtered.h5 文件。这是非常重要的。
使用 Seurat 解析 tiny_10x_pbmc_filtered.h5 文件¶
为了使用 Read10X_h5 函数,我们需要安装并使用 hdf5r 库。以下是具体步骤:
安装并加载必要的库¶
# 加载Seurat(版本3)库
library(Seurat)
install.packages("hdf5r")
library(hdf5r)
读取过滤后的 H5 文件数据¶
# 从过滤后的h5文件加载数据
data.file <- 'tiny_10x_pbmc_filtered.h5'
data.data <- Read10X_h5(filename = data.file, use.names = TRUE)
初始化 Seurat 对象¶
# 使用原始(未标准化)数据初始化Seurat对象
obj <- CreateSeuratObject(counts = data.data)
数据标准化¶
# 数据标准化
obj <- NormalizeData(obj)
识别可变特征¶
# 识别可变特征
obj <- FindVariableFeatures(obj)
数据缩放和 PCA 分析¶
# 缩放数据并进行PCA分析
obj <- ScaleData(obj)
obj <- RunPCA(obj, features = VariableFeatures(object = obj))
细胞聚类¶
# 聚类细胞
obj <- FindNeighbors(obj)
obj <- FindClusters(obj)
运行非线性降维(UMAP/tSNE)¶
# 运行UMAP进行非线性降维
obj <- RunUMAP(obj, dims = 1:10)
绘制 UMAP¶
# 绘制UMAP
DimPlot(obj, group.by = "seurat_clusters")
成功运行以上代码后,应该能够生成如下的 UMAP 图:
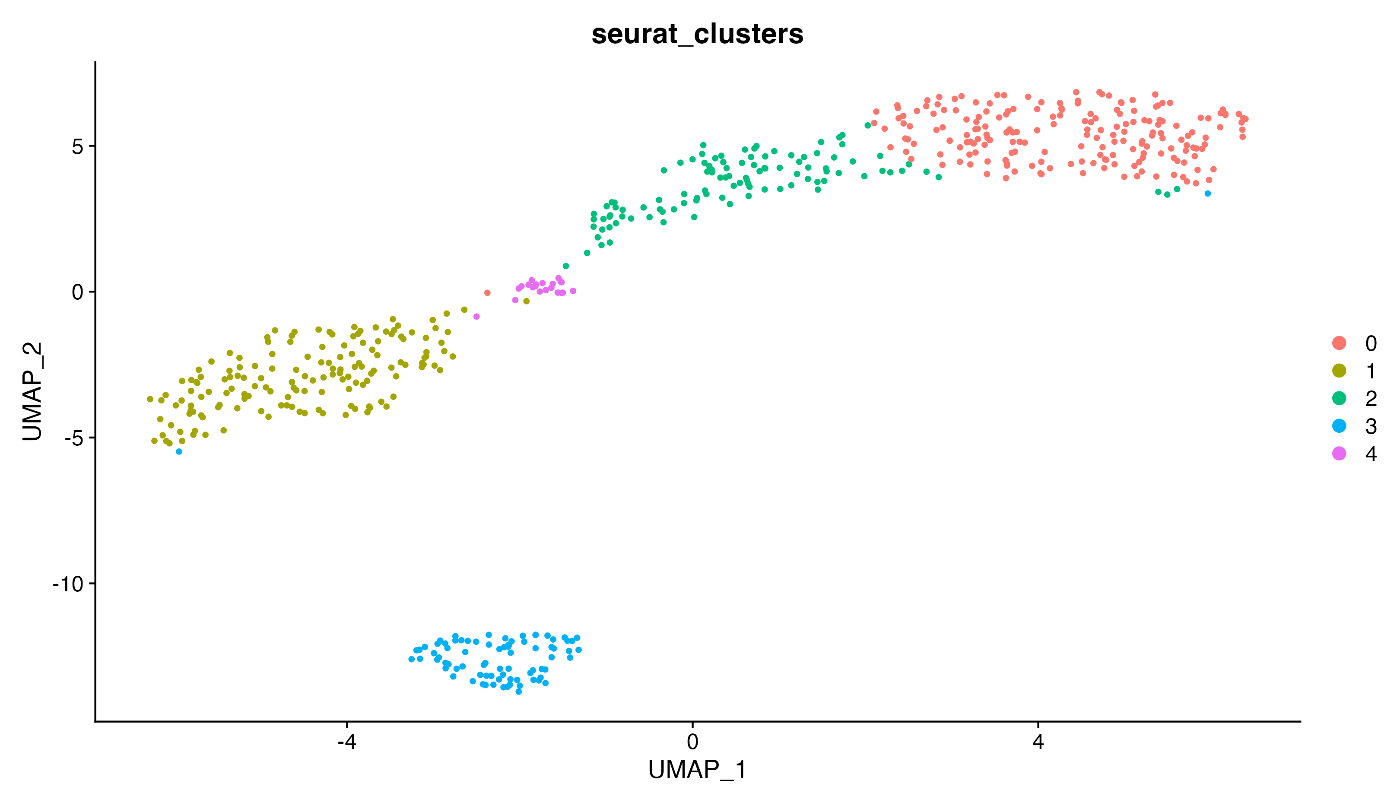
解说与补充¶
将 tiny_10x_pbmc.h5 和 tiny_10x_pbmc_filtered.h5 分别转换为 Seurat 对象,并输出样本数量如下:
# 从过滤后的h5文件加载数据
data.data_filtered <- Read10X_h5(
filename = "tiny_10x_pbmc_filtered.h5",
use.names = TRUE
)
data.data <- Read10X_h5(filename = "tiny_10x_pbmc.h5", use.names = TRUE)
# 使用原始(未标准化)数据初始化Seurat对象
obj_filtered <- CreateSeuratObject(counts = data.data_filtered)
obj <- CreateSeuratObject(counts = data.data)
# ---输出结果---
> obj_filtered
An object of class Seurat
100 features across 558 samples within 1 assay
Active assay: RNA (100 features, 0 variable features)
> obj
An object of class Seurat
100 features across 14650 samples within 1 assay
Active assay: RNA (100 features, 0 variable features)
结果表明,过滤前有 14650 个细胞,而过滤后只有 558 个细胞。大部分细胞都被过滤掉了。接下来,让我们查看 tiny_10x_pbmc.pdf 文件。
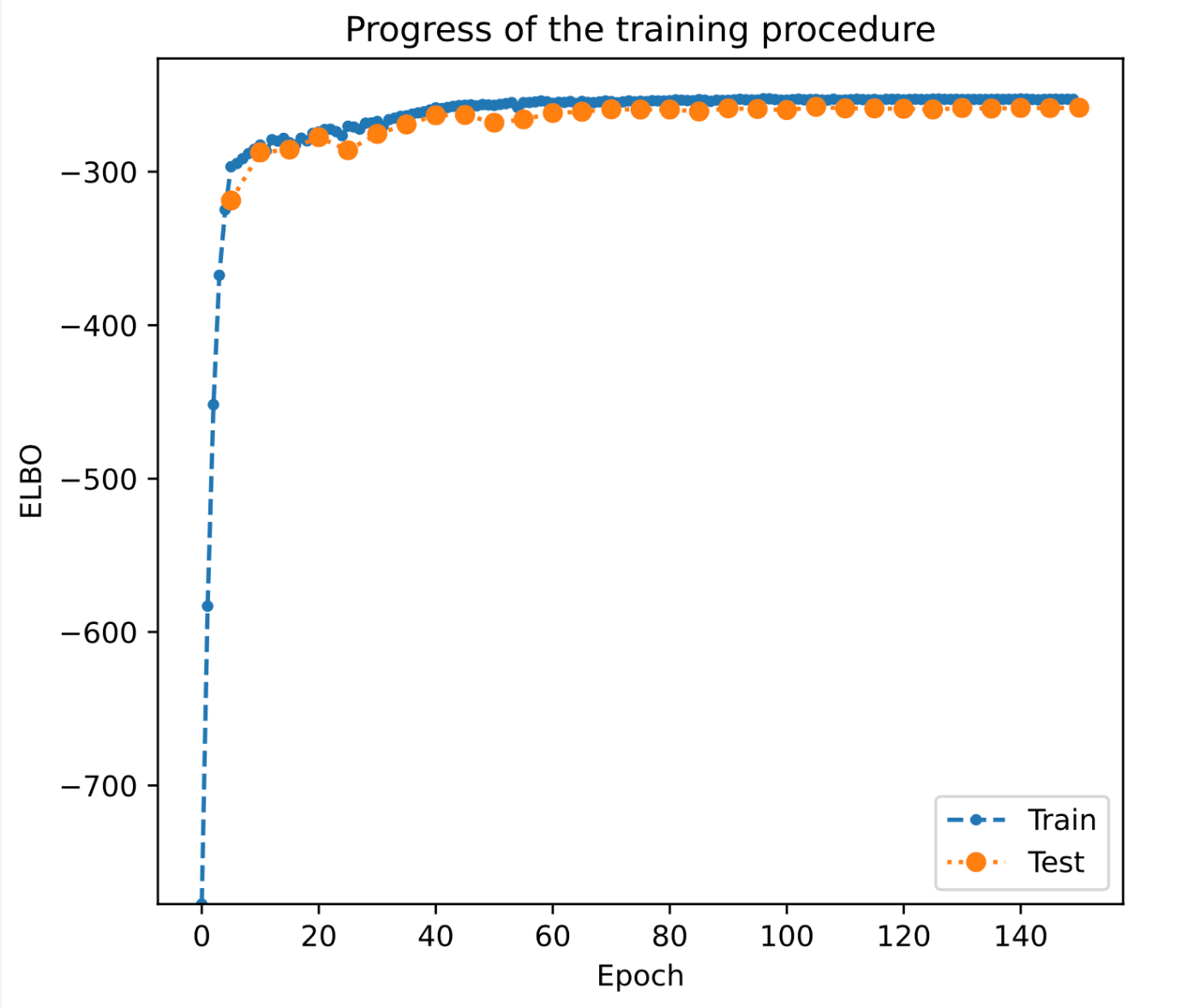
ELBO 和 Epoch 的解释¶
- ELBO 是“Evidence Lower Bound”的缩写,是贝叶斯统计学中的一种优化目标函数。如果 ELBO 没有增加或者大幅下降,可能表示学习率过高导致无法收敛。
- Epoch 指的是训练数据集被完整地传递给学习算法的次数。
如果 ELBO 值不是单调增加而是大幅下降(除去噪声),尝试将学习率减半。
如果 ELBO 值没有稳定收敛,建议通过 --epochs 选项增加 epoch 的数量。通常建议 epoch 数量不超过 300。
示例代码¶
以下是一个具体示例,将 tiny_10x_pbmc_filtered.h5 和 tiny_10x_pbmc.h5 分别转换为 Seurat 对象,并输出它们的样本数量:
# 加载必要的库
library(Seurat)
install.packages("hdf5r")
library(hdf5r)
# 从过滤后的h5文件加载数据
data.data_filtered <- Read10X_h5(filename = "tiny_10x_pbmc_filtered.h5", use.names = TRUE)
data.data <- Read10X_h5(filename = "tiny_10x_pbmc.h5", use.names = TRUE)
# 使用原始(未标准化)数据初始化Seurat对象
obj_filtered <- CreateSeuratObject(counts = data.data_filtered)
obj <- CreateSeuratObject(counts = data.data)
# 输出Seurat对象信息
print(obj_filtered)
print(obj)
成功运行后,您将看到过滤前后细胞数量的对比,进一步了解数据过滤的效果和质量。
查看 tiny_10x_pbmc.pdf¶
在分析过程中,您还可以参考 tiny_10x_pbmc.pdf 文件以获取更多的分析细节和结果。该 PDF 文件包括训练过程中的损失函数演变、UMI 图和后验细胞概率的排名等信息。
通过这些步骤,您可以有效地使用 CellBender 和 Seurat 进行 scRNA-seq 数据的分析和质量控制。
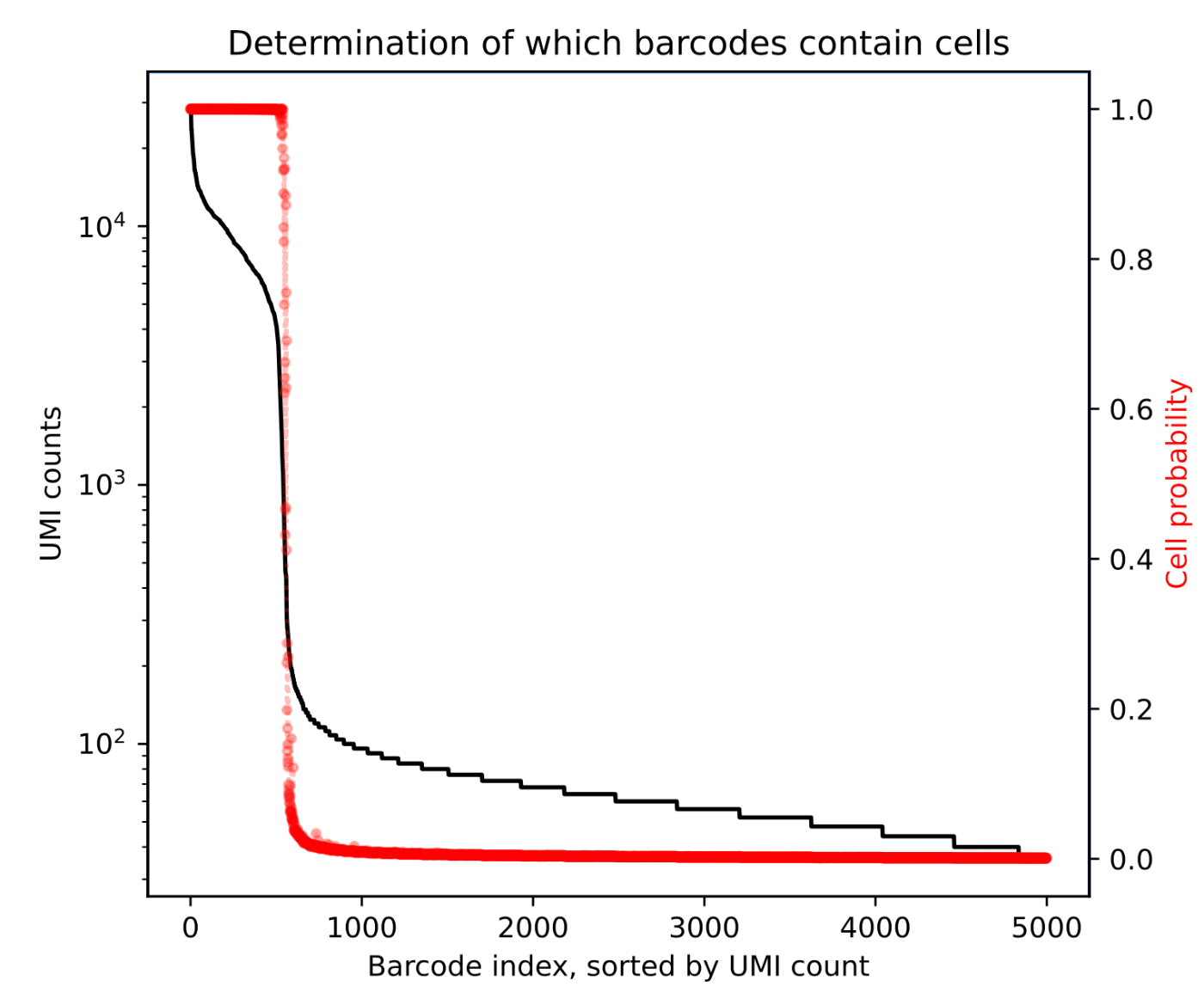
从下图中可以看到,当样本数超过 500 个时,校正后的细胞概率(红线)将趋向于 0。这就明确区分了样本和空液滴。
这是对编码基因表达的潜变量进行 PCA 分析。如果在这一阶段能看到一些聚类,则趋势良好。缺乏聚类可能表明存在质量控制问题,例如数据集只有一种细胞类型。
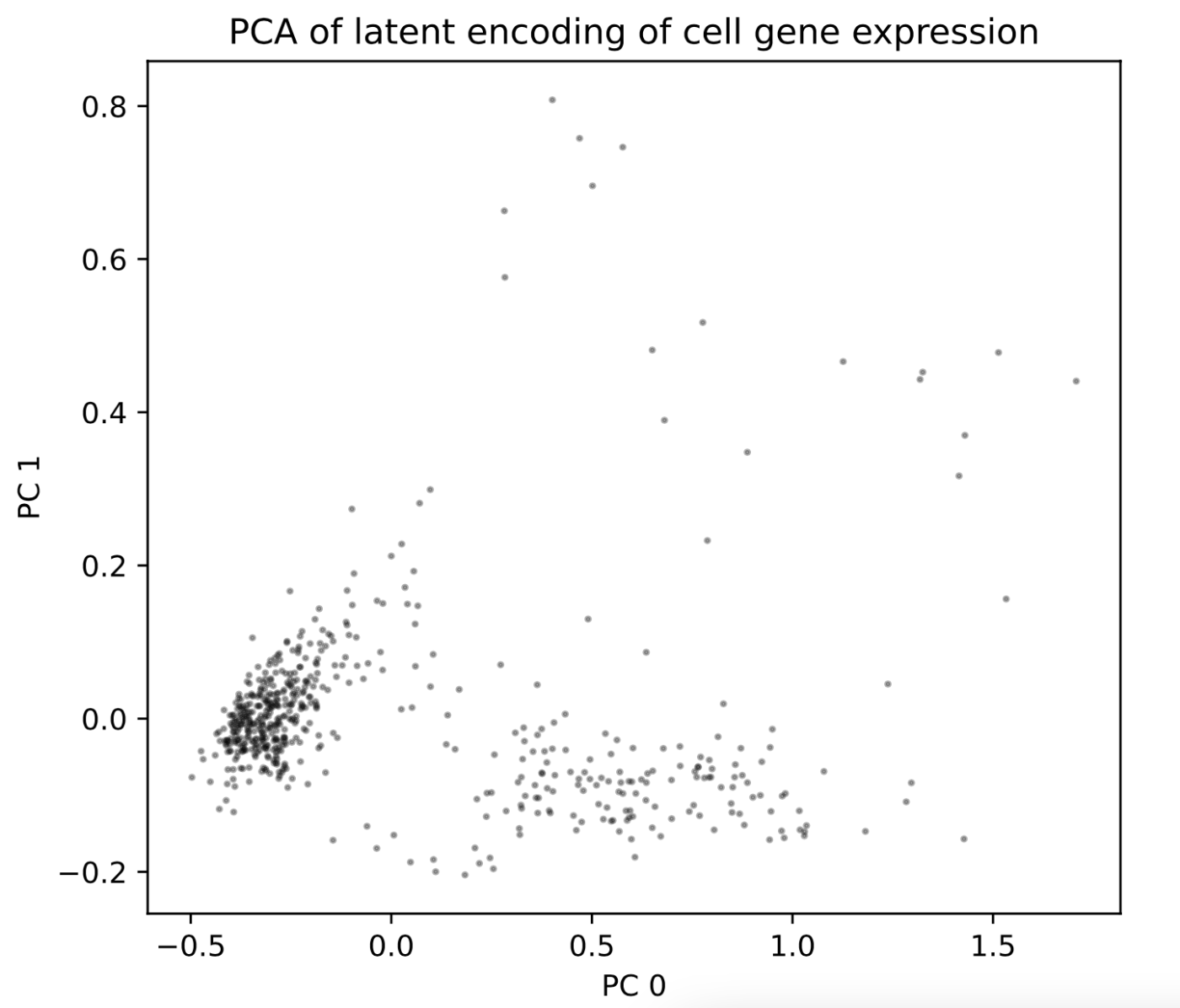
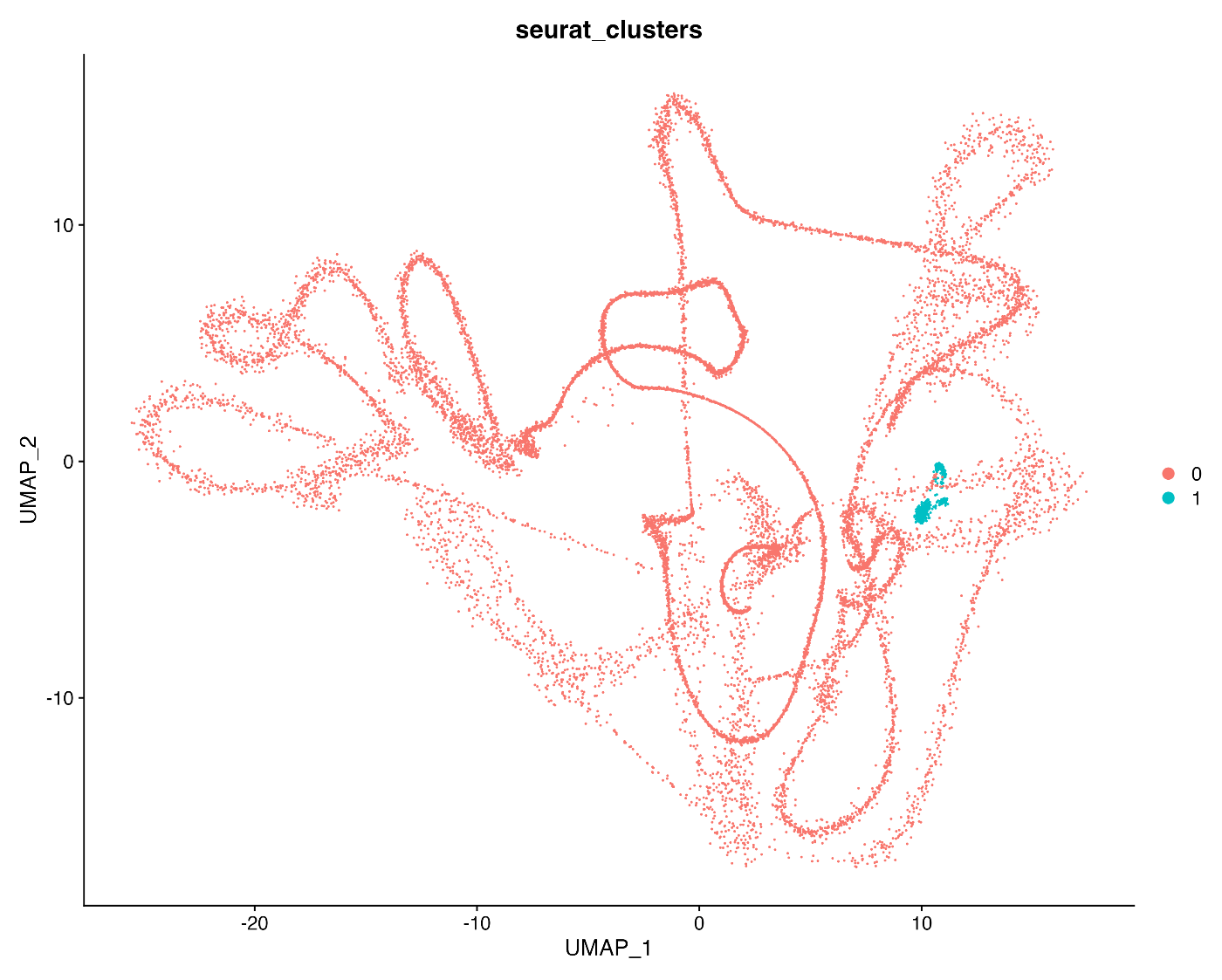
如果您分析 tiny_10x_pbmc.h5 并将其用于 UMAP 图,它不会聚类,也无法进行正确分析,这说明质量控制是多么重要。
设置参数时的注意事项¶
如果 CellBender 的 QC 没有达到预期效果,需要调整选项。以下是根据文档翻译的最佳实践指南:
-
epochs:一般选择 150。查看输出 PDF 中的学习曲线,寻找适度收敛的 ELBO 值(即达到某个饱和值)。尽管可能想增加更多的 epoch,但过多的训练会增加过拟合的风险,因此不建议过度训练(尽管通过正则化可以防止过拟合,但超过 300 个 epoch 的训练是过度的)。
-
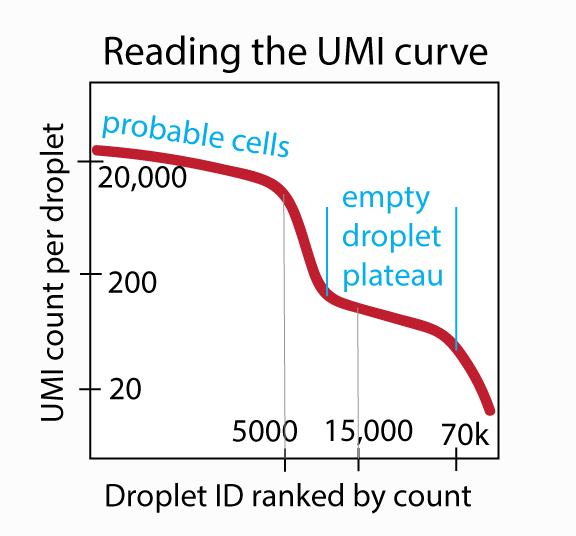
expected-cells:基于实验设计中的预期细胞数设置此参数。如果不清楚,可以根据 UMI 曲线来设置这个数值。选择一个合理确认左侧液滴都是实际细胞的数目。
-
total-droplets-included:选择一个数值,使数千个条形码进入“空滴平台”。但需注意,这个数值越大,算法运行时间越长(线性增加)。参考下面的 UMI 曲线,适当选择为 15,000。UMI 曲线中这个数值右侧的所有液滴应都是空的(可以在 cellranger count 的 web_summary.html 输出中查看 UMI 曲线)。
-
cuda:请包含此标志。该代码旨在在 GPU 上运行。
-
learning-rate:默认值 1e-4 通常没有问题,但如果在学习曲线质量检查(如上所述)中发现问题,可以调整此值。
-
fpr:0.01 的值通常是一个相当好的选择,但可以选择多个值生成输出计数矩阵进行比较:0.01、0.05、0.1。
通过这些设置,您可以优化 CellBender 的参数以获得最佳 QC 效果。