A lactate SREBP2 signaling axis drives tolerogenic dendritic cell maturation and promotes cancer progression
常规树突状细胞(DCs)是抗肿瘤免疫的关键介质。结果,癌症发展出一些尚未完全理解的机制,使 DCs 在肿瘤微环境(TME)中功能失调。在识别出 CD63 作为特异性表面标志物后,我们证明成熟调节性 DCs(mregDCs)迁移到肿瘤引流淋巴结组织,并通过促进 T 辅助细胞 2 和调节性 T 细胞分化来抑制 DCs 的抗原交叉呈递。转录和代谢研究表明,mregDC 的功能依赖于甲羟戊酸生物合成途径及其主转录因子 SREBP2。我们发现黑色素瘤来源的乳酸激活肿瘤 DCs 中的 SREBP2,并通过稳态或耐受性成熟驱动常规 DC 转变为 mregDC。特异性基因沉默和 SREBP2 的药理抑制促进了抗肿瘤 CD8+ T 细胞的活化,并抑制了黑色素瘤的进展。CD63+ mregDCs 在几个临床前肿瘤模型的淋巴结以及黑色素瘤患者的前哨淋巴结中被发现。总的来说,这项研究表明肿瘤乳酸刺激的依赖于 SREBP2 的程序促进了 CD63+ mregDC 的发育和功能,同时也为克服 TME 中的免疫耐受提供了有前途的治疗靶点。
引言¶
树突状细胞(DCs)通过呈递肿瘤抗原并在肿瘤引流淋巴结(TDLNs)中促进初始 T 细胞激活,在协调抗肿瘤免疫反应中发挥核心作用【1, 2】。DCs 启动适应性免疫反应的能力对于免疫检查点阻断的疗效至关重要【3-5】;然而,许多癌症中存在功能失调的 DCs,这些 DCs 无法激活 CD8+ T 细胞【6, 7】。多项研究表明,肿瘤相关的 DCs 可以表现出耐受性表型,特征是抗原交叉呈递减少,并且更容易诱导调节性 T 细胞(Treg)分化【8-13】。尽管耐受性 DCs 与癌症的关系已被广泛确立,但决定其发育和抑制功能的详细机制仍未完全了解。
常规 DCs(cDCs)由 cDC1s 和 cDC2s 组成,其中 cDC1s 因其交叉呈递抗原以刺激 CD8+ T 细胞反应的能力而被认可,cDC2s 则更善于激活各种类型的 CD4+ T 细胞反应。这两种 cDC 亚群都可以采用一种先前描述为“富含免疫调节分子的成熟 DCs”(mreg)cDCs 的替代程序,这些细胞表达高水平的 DC 成熟标志物和免疫调节基因【14】。尽管 mregDCs 被认为在调节肿瘤免疫中起作用,但 mregDCs 在肿瘤微环境(TME)中的作用及其对免疫逃逸的确切贡献仍不清楚【15, 16】。先前的研究描述了稳态或耐受性成熟,其中 cDCs 在稳态下经历一种成熟程序,使其能够迁移到引流淋巴结,这一过程对于维持外周耐受性至关重要【17-20】。这些迁移 DCs 的基因表达谱与最近报道的 mregDCs 非常相似【15】。肿瘤是否可以劫持 DC 耐受性成熟以抑制免疫监视尚不明确。建立导致 mregDCs 发育的机制及其如何影响免疫微环境,对于设计新的免疫治疗策略具有重要意义。
最近的研究揭示了代谢过程调节 DCs 的基本功能特性【21, 22】。具有高脂质含量的肿瘤浸润 DCs 表现出受损的免疫刺激特性【7, 23-25】。耐受性 DCs 使用脂肪酸氧化(FAO)满足其能量需求【26】。关键是,DCs 中的 FAO 促进原卟啉 IX 的合成和吲哚胺 -2,3- 双加氧酶 1(IDO1)的活性,随后驱动犬尿氨酸的产生和 Treg 分化【12】。由于 FAO 的重要性,脂质稳态受到复杂信号网络的严格调控,其中类固醇反应元件结合蛋白(SREBP)家族的转录因子是主要调节因子。在哺乳动物中,SREBF 家族由两个基因组成,SREBF1 和 SREBF2。SREBF1 调节脂质生成,而 SREBF2 及其编码的蛋白 SREBP2 控制胆固醇代谢【27】。这通过刺激甲羟戊酸(MVA)途径基因的表达来合成胆固醇,包括羟甲基戊二酸单酰辅酶 A 还原酶(HMG-CoA 还原酶),由 HMGCR 编码,以及低密度脂蛋白(LDL)受体编码基因 LDLR,负责 LDL 和细胞外胆固醇的摄取。MVA 途径先前已被证明与免疫有关,其中患有炎症性高免疫球蛋白 D 综合征的患者表现出 MVA 激酶功能丧失【28, 29】。此外,研究表明,抑制 MVA 途径促进抗原呈递并增强细胞毒性 T 细胞反应【30, 31】。在当前的研究中,我们描述了乳酸 -SREBP2 信号轴在 TME 中免疫耐受性 mregDCs 发育和功能中的作用。此外,我们展示了在 DCs 中靶向 SREBP2 和脂质代谢的治疗潜力,以克服肿瘤免疫耐受和免疫治疗抵抗。
结果¶
鉴定富含免疫调节和 MVA 通路基因表达的肿瘤相关 DC 群体¶
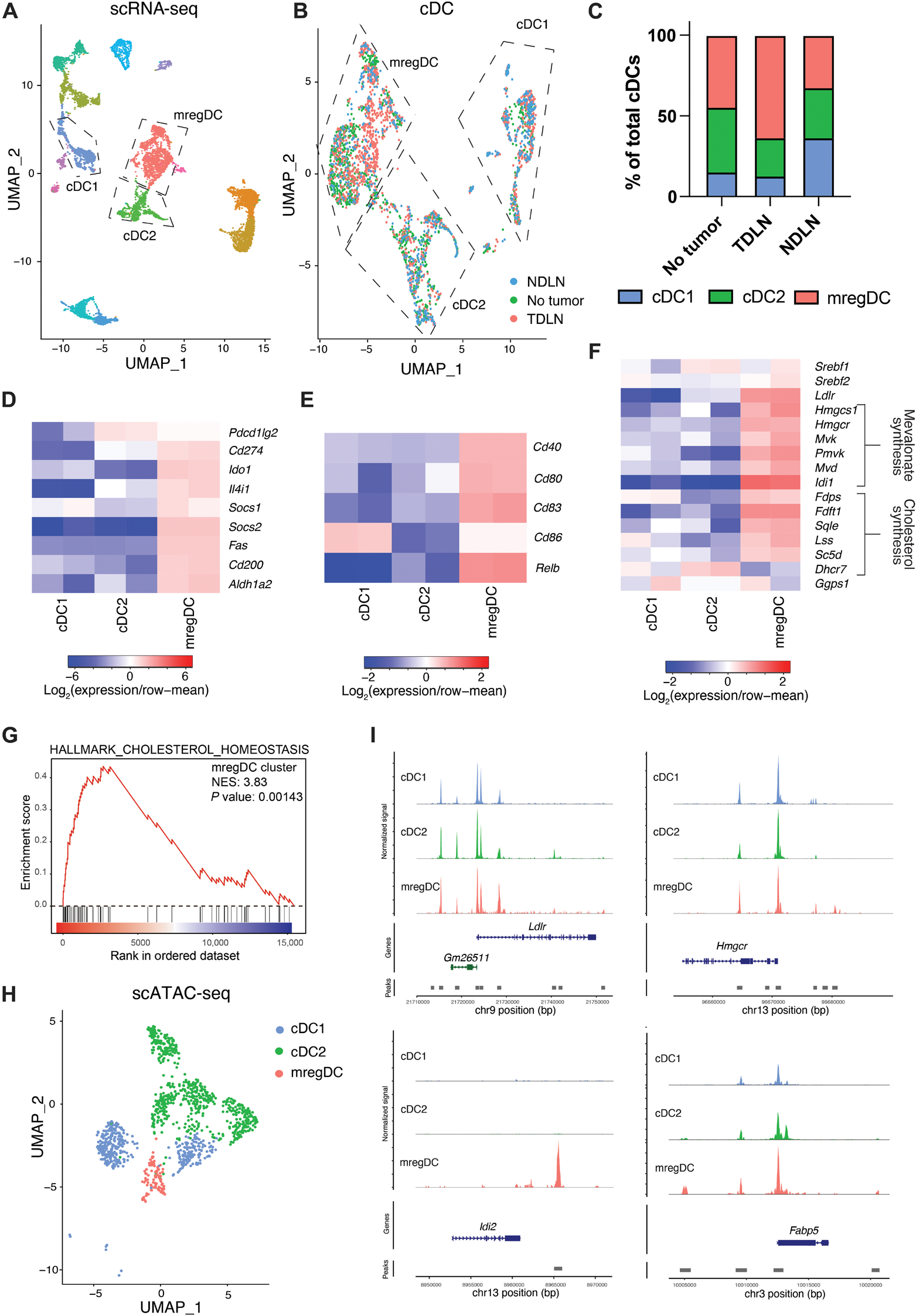
图 1. 单细胞转录组学鉴定了富含免疫抑制和 MVA 通路基因表达的 cDC 亚群。(A 到 G) 对在肿瘤发展 5 周时从 BRAFV600E PTEN−/−黑色素瘤小鼠的 TDLN 和 NDLN 以及未携带肿瘤对照小鼠的腹股沟 LN 中分选出的 CD11c+MHCIIhi DCs 进行 scRNA-seq 分析(n = 2 个独立实验,每个实验一只小鼠,每个样本约 3000 个细胞)。(A)UMAP 图显示 DC 亚群的聚类。注意其他簇代表 B 细胞和巨噬细胞群体。(B)cDCs 重新聚类后的 UMAP 图,显示每个样本中 DCs 的比例。(C)黑色素瘤小鼠的 TDLN 或 NDLN 中的 cDC1、cDC2 和 mregDC 簇占 cDCs 的百分比,以及未携带肿瘤对照小鼠的 LN 中的比例。(D 到 F)cDC 亚群差异基因表达的热图:(D)关键免疫抑制基因,(E)DC 成熟基因,以及(F)MVA 通路基因。(G)相对于总 DCs 的 mregDCs 中标志性胆固醇稳态基因集的 GSEA 分析。(H 和 I)在肿瘤发展 5 周时从 BRAFV600E PTEN−/−黑色素瘤小鼠的 TDLN 中分选出的 DCs 进行 scATAC-seq 分析(n = 2 个独立实验,每个实验一只小鼠,每个样本约 3000 个细胞)。(H)scATAC-seq UMAP 图显示 cDCs 的聚类。(I)cDCs 中 Ldlr、Hmgcr、Idi2 和 Fabp5 基因附近的染色质可及性轨迹。所有数据均代表两个或三个独立实验。NES,标准化富集评分。
DCs 的激活和向肿瘤引流淋巴结(TDLN)的迁移对于产生强大的抗肿瘤免疫力并支持检查点抑制剂免疫疗法的疗效是必要的【32, 33】。为了更好地理解肿瘤进展过程中 TDLN 内 DCs 的动态变化,我们对自发性 BRAFV600EPTEN−/−表达转基因黑色素瘤小鼠模型应用了单细胞 RNA 测序(scRNA-seq)【34】。在尾部基部产生原发性黑色素瘤后,从腹股沟 TDLN、远端非引流淋巴结(NDLN)和未携带肿瘤的对照小鼠的腹股沟淋巴结(LN)中分选出 DCs(CD45+CD11c+MHCIIhiF4/80−CD3−CD19−CD49b−B220−),并通过 scRNA-seq 分析其基因表达谱(图 S1A)。通过基于图的无监督聚类,基于 Itgax、Flt3 和 Zbtb46 的表达鉴定出 cDCs,并根据 Irf8、Irf4、Xcr1 和 Sirpa 的表达进一步细分为 cDC1 和 cDC2 亚群(图 1A 和图 S1B)。在 TDLN 中相对于 NDLN 和对照 LN 组织富集的第三个 cDC 簇(图 1,B 和 C,图 S1C)。与先前报道的 mregDC 和 LAMP3+ DC 亚群一致,这个 DC 簇表达高水平的免疫抑制基因,包括 Cd274、Ido1、Ili41 和 Socs2,以及与 DC 成熟相关的基因,如 Cd40、Cd80、Cd83 和 Cd86(图 1,D 和 E)【15, 16, 35-37】。我们还发现,mregDC 亚群上调了与 SREBP2 驱动的 MVA 生物合成途径相关的基因(图 1F)。此外,基因集富集分析(GSEA)显示,相对于其他 cDC 群体,mregDCs 中富集了标志性胆固醇稳态基因集(图 1G 和图 S1D)。这些 cDC 基因表达谱在独立的 scRNA-seq 实验重复中是一致的(图 S1E)。
然后,我们使用单细胞转座酶可及染色质测序(scATAC-seq)来鉴定这些 DC 亚群中差异可及的染色质区域(图 1H 和数据文件 S1)。该分析检测到 mregDCs 中富含 SREBP 结合位点的可及染色质峰,这些位点靠近几个关键的 MVA 基因,包括 Hmgcr、Ldlr、Idi2 和脂肪酸结合蛋白 Fabp5(图 1I 和图 S1F)。通过刺激 Toll 样受体 4(TLR4)和 TLR3 依赖性配体脂多糖和多肌苷酸:多胞苷酸 [poly(I:C)],脾脏 cDCs 的免疫原性成熟未显著诱导 MVA 通路相关基因表达的上调,这表明这一过程中存在替代途径(图 S1,G 和 H)。总体而言,这项研究支持我们之前的发现,即 DCs 中的脂质代谢增强促进了肿瘤进展期间的原耐受性 DC 表型【12】。这些数据还表明,TME 中的 cDCs 获得了类似于稳态成熟过程中生成的 DCs 的表型。总之,这些结果表明,mregDCs 在 TDLN 中积累,并表现出 SREBP2 依赖的 MVA 通路的增强激活。
CD63 标志 mregDCs 在肿瘤进展期间在 TDLN 中积累¶
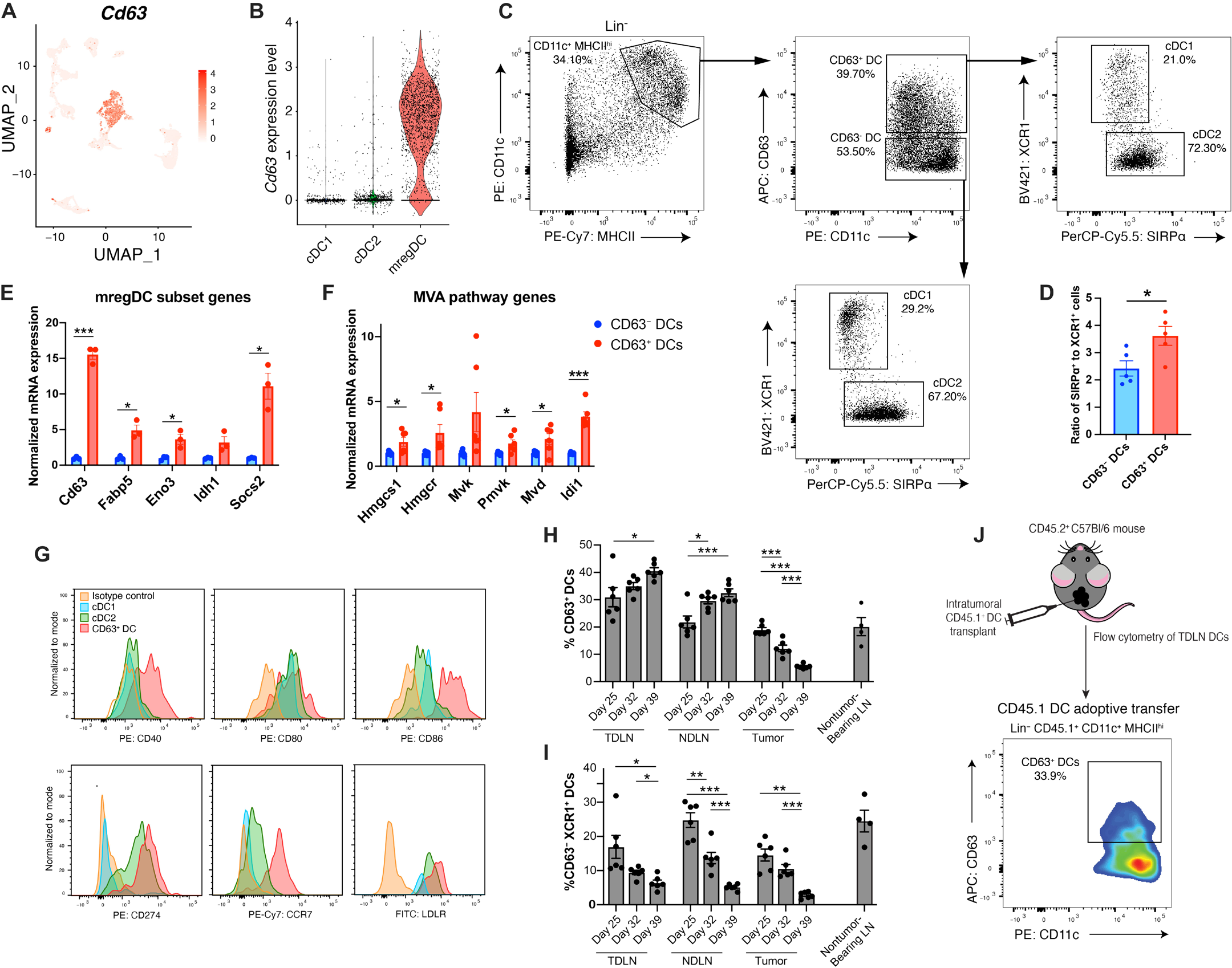
图 2. 四跨膜蛋白 CD63 是常规 mregDCs 的标志物。(A) 在图 1A 中生成的 UMAP 图上叠加的四跨膜蛋白 Cd63 基因表达(n = 2 个独立实验)。(B) 小提琴图显示 scRNA-seq 数据集中 cDC 亚群的 Cd63 基因表达(n = 2 个独立实验)。(C) 从携带肿瘤的小鼠的 LN 中分选 CD63+ DCs 的流式细胞术策略(n = 5 的代表性实验)。(D) 在肿瘤诱导后 5 周时,CD63−和 CD63+ DC 亚群中 SIRPα+ 细胞与 XCR1+ 细胞的比例(n = 4)。(E 和 F) 肿瘤诱导后 5 周时,FAC S 分选的 CD63+ 或 CD63− DCs 的 qPCR 分析:(E) 在 mregDC scRNA-seq 簇中高表达的关键基因和 (F) MVA 通路基因(n = 3)。(G) 流式细胞术直方图显示 mregDC scRNA-seq 簇中特定表面蛋白的表达(n = 4 的代表性实验)。(H 和 I) 流式细胞术定量:(H) BRAFV600E PTEN−/−黑色素瘤小鼠的 TDLN、NDLN 和肿瘤中的 CD63+ DCs 的百分比或 (I) XCR1+ cDC1s 的百分比。作为参考,显示肿瘤未感染小鼠的 LN 中 CD63+ DCs 的百分比(n = 6)。(J) 将分离的 CD45.1+CD63− DCs 注入 CD45.2+ 宿主的 BRAFV600EPTEN−/−黑色素瘤中的肿瘤内注射后,TDLN 中 CD45.1+ DCs 的流式细胞术分析(n = 4)。(D 到 F) 组间比较使用非配对 t 检验进行分析。(H 和 I) 统计分析通过单因素方差分析(ANOVA)并进行 Tukey 多重比较检验。所有数据以均值±SEM 表示。所有数据均代表两到三个独立实验。
为了帮助功能性表征 mregDC 亚群,我们鉴定了区分 mregDCs 与其他 cDCs 的表面标志物。在我们的 scRNA-seq 数据集中,Cd63 基因编码一个表面四跨膜蛋白,与 cDC1 和 cDC2 群体相比,mregDCs 中特异性表达(图 2,A 和 B)。我们使用流式细胞术确定 mregDC 亚群中 Cd63 基因表达增加是否对应于细胞表面 CD63 蛋白表达的增强。流式细胞术分析鉴定出一个 CD63hi 的 DC 群体,可以从其他 cDC 群体中分选出来(图 2C 和图 S2A)。与 CD63− DC 群体相比,CD63+ mregDCs 更高度表达 cDC2 标志物 CD172a(SIRPα),而不是 cDC1 标志物 XCR1。这与我们的 scRNA-seq 数据一致,其中 mregDC 簇包含更高比例表达 cDC2 基因 Irf4 和 Sirpa 的细胞,而不是 cDC1 基因 Irf8 和 Xcr1(图 2,C 和 D,图 S1B)。CD63+ mregDCs 中 cDC1 和 cDC2 基因的表达表明 CD63+ DCs 是一种可以由 cDC1 和 cDC2 发展而来的功能性 cDC 状态,而不是一个独立的谱系。这与先前发表的报告一致,显示人类表达 LAMP3 的 DCs,一种与 CD63 功能相似但不同的标志物,可以由 cDC1 和 cDC2 分化而来【37】。
为了确认 CD63+ DCs 代表 mregDC 程序,我们从携带 BRAFV600EPTEN−/−黑色素瘤的小鼠的 TDLN 中分选 CD63+ DCs,并进行定量实时聚合酶链反应(qRT-PCR)分析,以量化 mregDCs 差异表达的基因。我们观察到 CD63+ DCs 中 Cd63、Fabp5、Eno3 和 Socs2 的表达增加,并且重要的是,MVA 通路基因 Hmgcs1、Hmgcr、Pmvk、Mvd 和 Idi1 在 CD63+ DC 亚群中也显著上调(图 2,E 和 F)。为了进一步确认 CD63+ DCs 代表 mregDCs,我们通过流式细胞术分析其表面表型。与我们的 scRNA-seq 数据集(图 1,D 到 F,图 S1C)一致,CD63+ DCs 相对于其他 cDC 亚群,表达较高水平的 CCR7、LDLR、CD40、CD80 和 CD86(图 2G)。这一 CD63+ mregDC 群体还在转基因 p53Δ/Δ;KrasG12D 非小细胞肺癌模型的肿瘤组织以及正交 ApcΔ/Δ;KrasG12D/+;Tp53Δ/Δ;Smad4Δ/Δ(AKPS)结肠癌类器官模型的肿瘤和 TDLN 组织中被发现,表明 CD63+ mregDC 群体不仅限于 BRAFV600EPTEN−/−黑色素瘤模型(图 S2,B 到 D)【38, 39】。
利用 CD63 作为表面标志物,我们使用流式细胞术研究了转基因 BRAFV600EPTEN−/−小鼠模型中肿瘤进展过程中 CD63+ mregDCs 的动态变化。支持先前的 scRNA-seq 数据,流式细胞术显示相对于 NDLN 和未携带肿瘤的小鼠的 LN,在 TDLN 中 CD63+ mregDCs 增加(图 2H)。在肿瘤进展过程中,TDLN 中 CD63+ mregDC 群体的增加与肿瘤床中该群体的减少相一致。相对于原发性肿瘤,LN 中 CD63+ mregDCs 的比例增加,这与 mregDCs 的迁移表型一致(图 S2E)。此外,与 mregDCs 在肿瘤免疫中起免疫抑制作用一致,我们发现 TDLN 组织中 CD63+ mregDCs 的频率与 cDC1s 的频率负相关(图 2I)。在比较 TDLN 中 cDC 群体与未携带肿瘤的小鼠的 LN 中的 cDCs 时,基因表达谱是稳定的,这表明 mregDCs 可能不是在 LN 中发育,而是从其他组织迁移过来的(图 S2F)。为了测试这一假设,将 CD45.1+CD63− DCs 输送到携带 BRAFV600EPTEN−/−黑色素瘤的 CD45.2+ 宿主的肿瘤床中。流式细胞术分析显示 TDLN 中存在 CD45.1+CD63+ DCs,进一步表明肿瘤浸润的 DCs 可以在迁移到 TDLN 组织之前在肿瘤床中转变为 CD63+ DCs(图 2J 和图 S2G)。总之,这些发现表明 mregDCs 响应肿瘤床中的稳态成熟触发而发育,并且可以基于 CD63 表达进行识别。
CD63+ mregDCs 抑制 CD8+ T 细胞反应并促进 Treg 分化¶
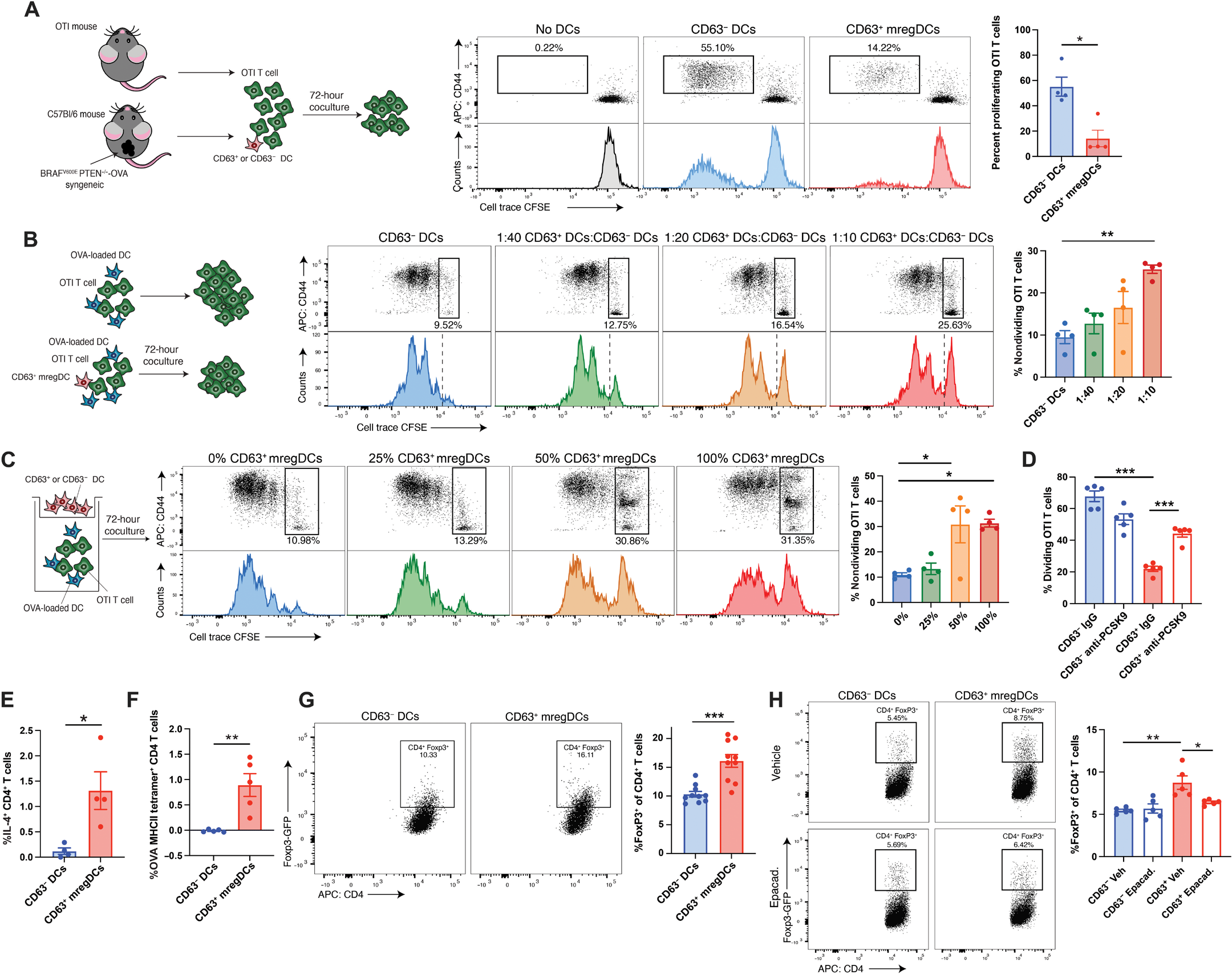
图 3. CD63+ DCs 抑制 CD8+ T 细胞并促进 Treg 分化。(A 到 D) 使用标记有羧基荧光素二乙酸琥珀酰亚胺酯(CFSE)的 OTI T 细胞与抗原脉冲的 DCs 共培养的 T 细胞增殖实验。(A) OTI CD8+ T 细胞与从表达 OVA 的 BRAFV600EPTEN−/−黑色素瘤小鼠的 TDLN 中分离的 CD63− DCs 或 CD63+ mregDCs 共培养,肿瘤植入 3 周后(5:1 T 细胞:DC)。左图:实验示意图。右图:流式细胞术图和定量分析(n = 4)。(B)OTI CD8+ T 细胞与 OVA 脉冲的 DCs 及增加数量的 CD63+ mregDCs 共培养。左图:实验示意图。右图:流式细胞术图和定量分析(n = 4)。(C) OTI CD8+ T 细胞与 OVA 脉冲的 DCs 在 0.4 微米 Transwell 底部室中共培养。在上部室中 CD63+ : CD63− DCs 的比例从 1 × 10^4:0 到 0:1 × 10^4,同时在各条件下保持相同的总细胞数。(D) cDCs 用 OVA 脉冲并在有或无 CD63+ mregDCs 或对照 CD63− DCs ± 抗 PCSK9 抗体的情况下与 OTI CD8+ T 细胞共同孵育。(E) 与 CD63+ 或 CD63− DCs 共培养后 CD4+ T 细胞的细胞内 IL-4 染色(T 细胞:DC 比例,5:1)(n = 4)。(F) 与 CD63+ 或 CD63− DCs 共培养后表达 MHCII-OVA323-329 四聚体的 CD4+ T 细胞染色,从表达 OVA 的黑色素瘤小鼠中分离的 DCs(T 细胞:DC 比例,5:1)(n = 5)。(G) 异基因 CD63−或 CD63+ DCs(H-2d)与从 Foxp3-GFP 小鼠(H-2b)中分离的初始 CD4+ T 细胞共培养后的流式细胞术图(左)和定量分析(右),显示 Foxp3 表达的 Tregs(T 细胞:DC 比例,5:1)(n = 10)。(H)流式细胞术图(左)和定量分析(右),显示 IDO1 抑制剂 epacadostat 对 CD63+ DC 诱导的 Treg 分化的影响(n = 5)。所有两组比较均使用非配对 t 检验分析。(B 和 C)统计分析通过单因素方差分析(ANOVA)并进行 Dunnett 多重比较检验。(D 和 H)统计分析通过双因素方差分析(ANOVA)并进行 Tukey 多重比较检验。所有数据以均值±SEM 表示。所有数据均代表两到三个独立实验。
在展示了 CD63+ mregDCs 在 TDLN 组织中积累后,我们接下来试图更好地理解它们在指导适应性免疫中的作用。为此,我们首先将从携带表达卵白蛋白(OVA)的同系 BRAFV600E PTEN−/−肿瘤小鼠的 TDLN 中分选的 CD63+ mregDCs 或 CD63− DCs 与从 OT-I T 细胞受体转基因小鼠中分离的初始 CD8+ T 细胞共培养。虽然我们的工作表明,CD63+ mregDCs 相对于 CD63− DCs 表现出更成熟的表型,但我们发现 CD63+ mregDCs 相对于 CD63− DCs 在刺激抗原特异性 CD8+ T 细胞反应方面表现出较低的能力(图 3A)。然而,与其成熟表型一致,我们还观察到相对于 cDC2s,CD63+ mregDCs 的抗原摄取能力受损,使用基于流式细胞术的偶联 OVA 测定(图 S3A)。CD63+ mregDCs 保持了直接呈递 SIINFEKL 肽和刺激 OT-I T 细胞反应的能力,这表明观察到的 T 细胞激活受损可能是由于抗原交叉呈递途径的缺陷(图 S3B)。
为了进一步验证 CD63+ mregDCs 是否主动抑制效应 T 细胞反应的产生,我们使用各种比例的 CD63+ mregDCs 与 OVA 脉冲的 CD63− DCs 进行额外的 CD8+ T 细胞反应测定。即使在较低比例的 CD63+ mregDCs 对 CD63− cDCs(1:10)下,CD63+ mregDCs 也能有效地通过转递抑制附近 cDCs 的抗原交叉呈递(图 3B)。这表明少量的 CD63+ mregDCs 可以强烈抑制局部 CD8+ T 细胞的激活。这种抑制效果似乎针对 DC 的抗原交叉呈递,因为 CD63+ mregDCs 无法抑制附近 cDCs 的 SIINFEKL 肽直接抗原呈递(图 S3C)。然后我们使用透析系统进行测定,将 CD63+ mregDCs 与 CD63− DCs 共培养,以测试 CD63+ mregDCs 衍生的可溶性分子对转递抗原交叉呈递的影响。在此测定中,我们观察到 OT-I CD8+ T 细胞反应的抑制,证实 CD63+ mregDCs 通过产生可溶性介质抑制附近 DCs 的抗原交叉呈递(图 3C)。SREBP2 靶向的前蛋白转化酶枯草杆菌蛋白酶 9(PCSK9)是胆固醇稳态调节的关键蛋白,先前已被证明可以诱导主要组织相容性复合体(MHC)I 类的降解【40】。为了检查 PCSK9 是否可以通过旁分泌方式损害抗原交叉呈递,我们测试了在我们的 CD63+ mregDC:CD63− DC:OT-I CD8+ T 细胞共培养测定中 PCSK9 阻断对 T 细胞增殖的影响。在用抗 PCSK9 抗体处理后,我们观察到 CD8+ T 细胞增殖增加,表明 PCSK9 可能通过 SREBP2 介导,是 CD63+ mregDCs 抑制 T 细胞激活的机制之一(图 3D)。
尽管 CD63+ mregDCs 促进 CD8+ T 细胞增殖的能力有限,但从 BRAFV600EPTEN−/−肿瘤小鼠中分选的 CD63+ mregDCs 保持了促进 OT-II CD4+ T 细胞增殖的强大能力(图 S3D)。额外的流式细胞术研究表明,这些扩增的 CD4+ T 细胞表现出 T 辅助细胞 2(TH2)表型(图 3E)。这一发现与我们 scRNA-seq 研究中 mregDC 簇中 TH2 相关基因的表达增加以及活化的 CD4+ T 细胞中 GATA 结合蛋白 3(GATA3)转录因子的表达增加一致(图 S3,E 和 F)。在 CD4+ T 细胞中未检测到 T 盒转录因子 21(T-Bet)或 RAR 相关孤儿受体γT(RORγT)的表达变化(图 S3F)。一致的是,使用从携带 OVA 表达肿瘤的小鼠中分离的 CD63+ mregDCs 的 MHC II 类四聚体共培养研究表明,CD63+ mregDCs 比 CD63−对照 DCs 更容易扩展肿瘤抗原特异性 CD4+ T 细胞(图 3F)。这些数据表明,CD63+ mregDC 群体还可以支持 FoxP3+ Tregs 的分化。已知在 TME 中的髓样细胞表达的色氨酸降解酶如 IDO1 和白介素 -4 诱导基因 1(IL-4I1)可以促进 Treg 分化【41-43】,而这些酶在 CD63+ mregDCs 中相对于 cDC1s 和 cDC2s 更为丰富(图 1D)。与这些发现一致,CD63+ mregDCs 与异基因初始 CD4+ T 细胞的共培养显示,与 CD63− DCs 相比,CD63+ mregDCs 在依赖 IDO1 的方式下更能促进 CD4+FoxP3+ Treg 分化(图 3,G 和 H)。这些数据共同表明,累积在 TDLN 组织中的 CD63+ mregDCs 促进了免疫耐受性 T 细胞反应的产生。
CD63+ mregDCs 可以分解为类似 cDC1 和 cDC2 的亚群¶
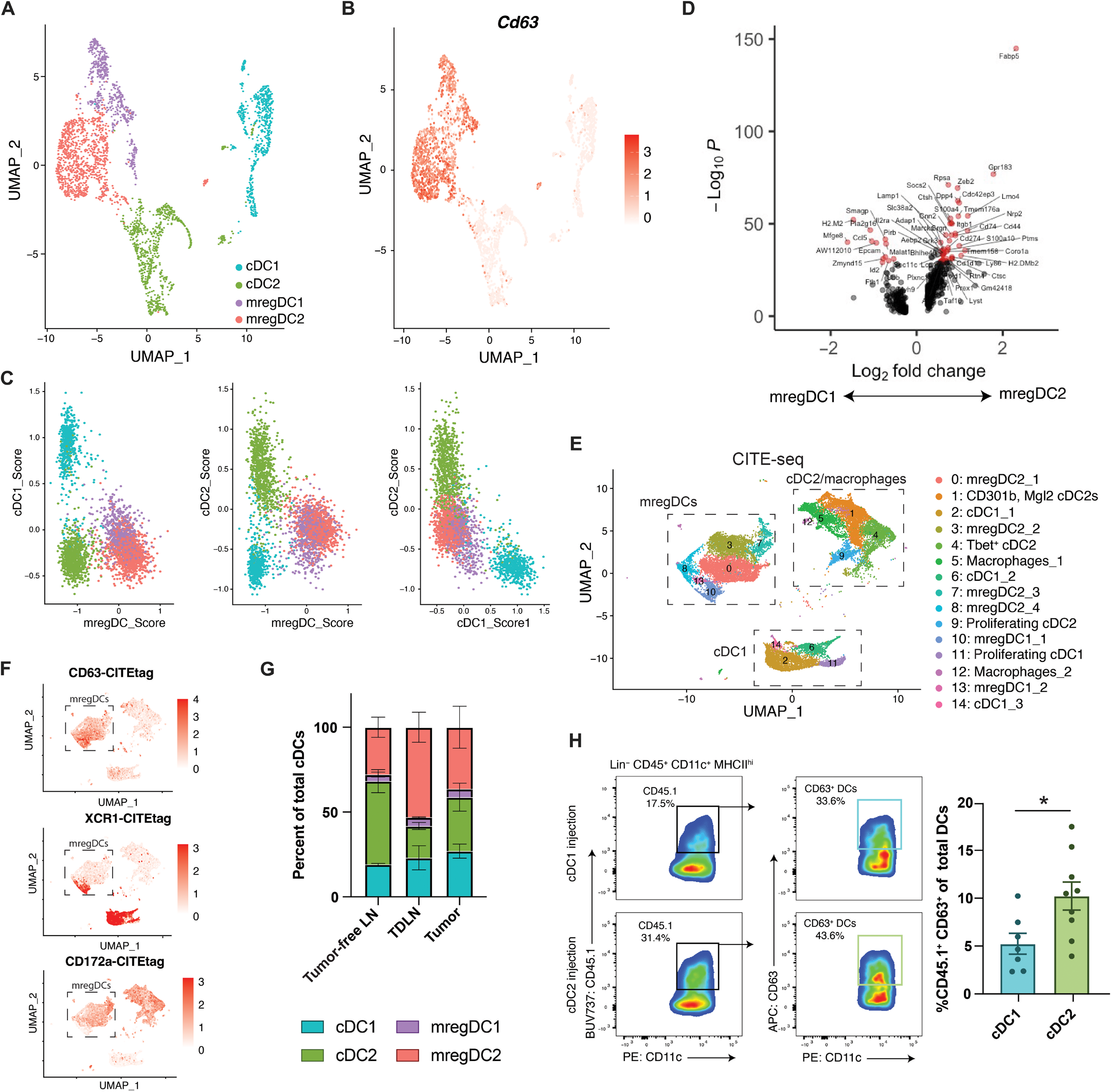
图 4. CD63+ mregDCs 表达独特的基因特征并可分为两个亚群。 (A) UMAP 图显示来自图 1 的 scRNA-seq 数据子集,仅显示 cDC 簇。(B) 在 UMAP 图上叠加基因表达,显示 (A) 中的 cDC 簇中 Cd63 的表达。(C) 从图 1 中的原始簇中差异表达最高的基因生成 DC 亚群评分,并应用于不同的 DC 亚群,比较 mregDC 亚群与 cDC1 和 cDC2。(D) 火山图显示 mregDC1 和 mregDC2 亚群之间的差异表达基因。(E) UMAP 图显示肿瘤、TDLN 和对照 LN 中分选的 DCs 的 CITE-seq 无监督聚类,肿瘤诱导 5 周后进行。样本在降维前整合。每个簇的识别在右侧报告(n = 2,每个实验两只小鼠,每个样本约 5000 个细胞)。(F) 通过 CITE-seq 鉴定的 CD63、XCR1 和 CD172a (SIRPa) 的细胞表面蛋白表达。方框代表 mregDC 群体。(G) 量化 (E) 中识别的 DC 亚群占每种条件下总 DCs 的百分比。(H) 左图:将 CD45.1+ cDC1s (上) 或 CD45.1+ cDC2s (下) 注入 CD45.2+ 宿主肿瘤内后,流式细胞术图显示 CD45.1+ CD63+ DCs。右图:DC 转移后 3 天,肿瘤内注射 CD45.1+ cDC1 或 cDC2 后的总 DCs 中 CD45.1+CD63+ DCs 的百分比定量分析(n = 8)。数据使用非配对 t 检验分析。所有数据均以均值±SEM 表示。所有数据均代表两个独立实验。
接下来,我们试图理解 mregDCs 与其他 cDC 亚群之间的发育关系。因此,我们重新聚类 cDC 群体,并通过选择在我们的 scRNA-seq 数据集中每种 DC 亚型差异表达的前 20 个基因,生成 cDC 签名评分。在高分辨率分析中,mregDCs 分离为两个亚群(图 4,A 和 B)。我们通过选择 cDC1s、cDC2s 或 mregDCs 差异表达的前 20 个基因生成 DC 表达签名,并将这些基因表达评分应用于两个 mregDC 簇及其各自的 cDC1 或 cDC2 细胞群体(图 4C 和数据文件 S2)。根据它们与 cDC1s 和 cDC2s 的相似性,我们现在将这些独立的簇分别称为 mregDC1s 和 mregDC2s。mregDC2 簇富含脂质伴侣 Fabp5 和氧化固醇受体 Gpr183 的表达,与 cDC2 基因程序更紧密相关(图 4D 和数据文件 S2)。为了进一步支持 CD63+ DC 簇由具有 cDC1 或 cDC2 转录状态的成熟 mregDCs 组成,我们对从肿瘤、TDLNs 和肿瘤未感染小鼠的 LNs 中分选的 DCs 进行了单细胞转录组和表位索引测序(CITE-seq),包括条形码抗体以更好地识别细胞表面蛋白并促进 DC 亚群的表征(图 4E)。这有助于鉴定出表达 cDC1 标志物 XCR1 或 cDC2 标志物 SIRPa(CD172a)的 CD63+ mregDC 亚群(图 4F)。如上所述,CITE-seq 还显示,相对于未携带肿瘤的小鼠的 LNs,mregDCs 在 TDLN 组织中富集,并显示这些 mregDCs 表达 CD83、IA-IE(MHC II 类)、CD197(CCR7)和 CD274(PD-L1)(图 4G 和图 S4,A 到 C)。接下来,我们比较了从肿瘤、TDLN 和对照 LNs 中分离的 mregDC 簇的平均基因表达,显示 TDLN 和肿瘤内的 mregDCs 在转录上是相似的(图 S4D)。这些发现表明,CD63+ mregDCs 可以由 cDC2 或 cDC1 转录状态组成,并且可以在肿瘤和 TDLN 组织中识别出来。
为了确认 CD63+ mregDCs 可以由 cDC1s 和 cDC2s 发育而来,我们从 CD45.1+ 小鼠的脾脏中分选 cDC2s 和 cDC1s,并将这些细胞直接注射到 CD45.2+ 宿主的 BRAFV600EPTEN−/−肿瘤中。3 天后,我们观察到,虽然 cDC1s 和 cDC2s 都能在 TDLN 中产生 CD63hi mregDCs,但 cDC2s 比 cDC1s 更容易分化为 CD63hi mregDCs(图 4H)。总体而言,我们的研究结果表明,CD63+ mregDCs 通过一种类似于稳态或耐受性成熟的过程,在 TME 中由 cDC1s 和 cDC2s 发育而来【17, 20】。
mregDCs 在代谢上与 cDCs 不同并表现出较高水平的 FAO¶
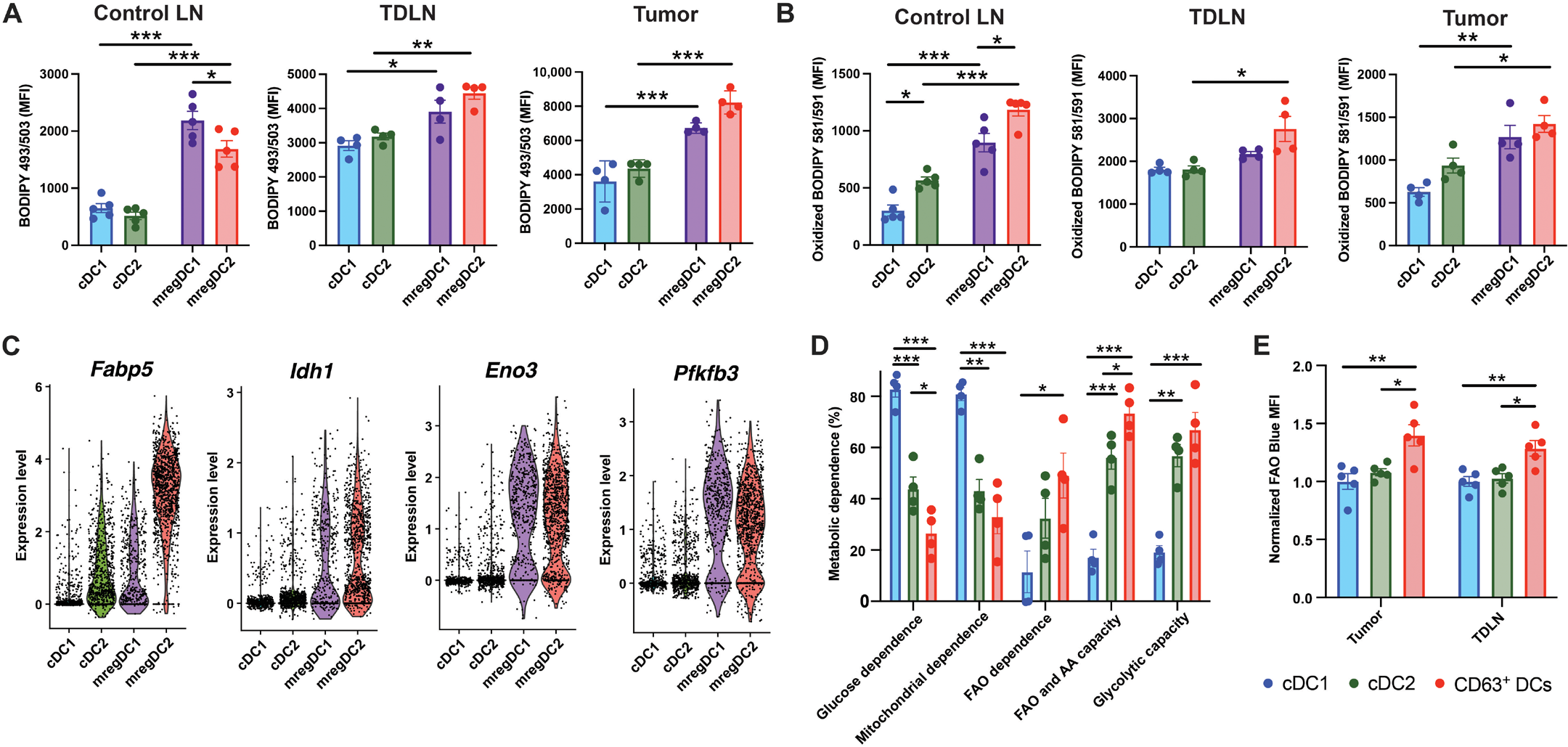
图 5. CD63+ mregDCs 在代谢上与其他 DCs 不同。
(A) 肿瘤诱导 5 周后,基于 BODIPY 493/503 流式细胞术定量 cDC 中性脂质含量(对照 LN n = 5,TDLN 和肿瘤 n = 4)。
(B) 基于 BODIPY 581/591 C11 流式细胞术定量脂质过氧化(对照 LN n = 5,TDLN 和肿瘤 n = 4)。
(C) 从图 1 中的 scRNA-seq 分析中得出的 cDC 亚群关键代谢基因的转录分析(n = 2 个独立实验)。
(D) cDC 亚群的 SCENITH 测定分析(n = 4)。
(E) cDCs 中 FAO 蓝色染色的流式细胞术定量分析。数据归一化为 cDC1 FAO 蓝色 MFI(n = 5)。统计分析使用单因素方差分析(ANOVA)并进行 Tukey 多重比较检验。数据以均值±SEM 表示。所有数据均代表至少两个独立实验。
如上所述,mregDCs 富含由转录因子 SREBP2 驱动的 MVA 代谢途径中的多种酶的表达(图 1F)。先前的研究表明,肿瘤浸润的 DCs 具有增加的脂质体形成和增加的脂质过氧化,最终可能干扰 DC 抗原交叉呈递【7, 23, 44】。此外,稳态 DC 成熟与胆固醇含量的增加一致【20】。相对于 cDC1s 和 cDC2s,我们发现 CD63+ mregDCs 在未携带肿瘤的对照小鼠的 LNs、TDLNs 和肿瘤中含有增加的中性和氧化脂质含量(图 5,A 和 B)。这一发现与我们对 CD63+ mregDCs 的功能性数据一致,因为先前的研究表明,氧化脂质干扰抗原加工和肽 MHC I 类的形成【23】。
MVA 途径将糖酵解或 FAO 产生的乙酰辅酶 A(acetyl-CoA)转化为胆固醇和异戊二烯类化合物【45】。由于 CD63+ mregDCs 的 MVA 途径酶的表达增加(图 1F)和 mregDC 脂质含量的变化(图 5,A 和 B),我们使用京都基因和基因组百科全书数据库生成代谢途径基因表达评分,并比较 CD63+ mregDCs 与 cDC1s 和 cDC2s 的中心代谢途径基因表达。CD63+ mregDCs 表现出几种限速酶的表达增加,包括催化三羧酸循环(TCA 循环)限速步骤的 Idh1,两种限速糖酵解酶 Eno3 和 Pfkfb3,以及脂肪酸结合蛋白 Fabp5(图 5C 和图 S5,A 到 D)。为了确定 CD63+ mregDCs 使用的主要代谢途径,我们使用基于流式细胞术的单细胞能量代谢分析(SCENITH)方法,通过蛋白质合成/嘌呤霉素掺入测量作为腺苷三磷酸(ATP)生成的替代指标【46】。这种方法显示,CD63+ mregDCs 相对于其他 cDC 群体更依赖于 FAO,并表现出更高的 FAO/氨基酸氧化能力(图 5D 和图 S5,E 和 F)。进一步测试,我们使用荧光脂肪酸衍生物(FAOblue),显示相对于 cDC1s 和 cDC2s,CD63+ mregDCs 在肿瘤和 TDLN 中的 FAO 增加(图 5E)。这些实验表明,mregDCs 在代谢上与其他 cDC 群体不同,并表现出增强的 FAO 水平。这些发现与我们之前的工作一致,连接了 DC 介导的 Treg 分化和免疫耐受的诱导与 FAO 水平的增加【12】。
SREBP 抑制逆转 CD63+ mregDC 的耐受性并抑制肿瘤进展¶
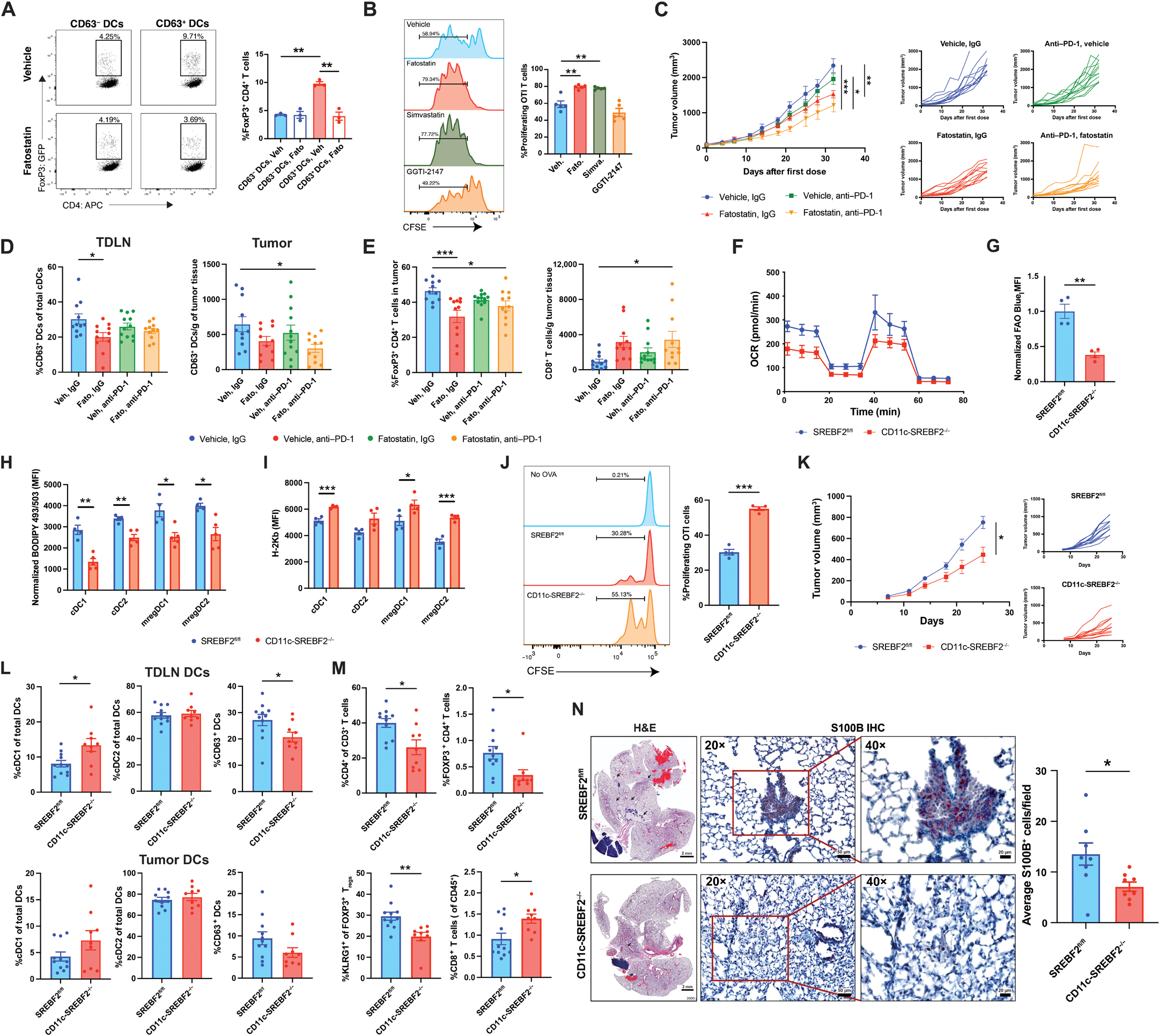
图 6. SREBP2 支持 CD63+ mregDCs 的发育并促进肿瘤生长
(A) 流式细胞术分析与 FoxP3-GFP 初始 CD4+ T 细胞共培养的来自 TDLN 的 CD63+ 或 CD63− DCs,预先用 fatostatin 或车辆对照处理(n = 3)。(B) OVA 脉冲后从 TDLN 中分选出的 CD63+ DC 与初始 OTI CD8+ T 细胞共培养,±fatostatin、simvastatin 或 GGTI-2147 的流式细胞术 CFSE 增殖实验(n = 5)。(C) 用 fatostatin、抗 PD-1 抗体、联合治疗或车辆对照处理的 BRAFV600EPTEN−/−黑色素瘤小鼠的肿瘤生长曲线(每组 n = 11 或 12)。(D 和 E) 在 (C) 中治疗开始 32 天后,流式细胞术定量分析小鼠 TDLN/肿瘤组织中的免疫细胞群。 (D) TDLN 和肿瘤中 CD63+ mregDCs 的定量。(E) 肿瘤内 Foxp3+ Tregs 和 CD8+ T 细胞的定量。(F) 测量由 CD11c-SREBF2−/−和对照 SREBF2fl/fl 宿主来源的骨髓 DC 的氧消耗率(OCR)的细胞外流量分析(n = 5)。(G) 来自 CD11c-SREBF2−/−和对照 SREBF2fl/fl 小鼠的 TDLN 中分离的 DC 的 FAO blue 染色的流式细胞术定量(n = 4)。(H) 肿瘤植入 4 周后,从 CD11c-SREBF2−/−和对照 SREBF2fl/fl 宿主中分离的 TDLN cDCs 的 BODIPY493/503 染色的流式细胞术定量(n = 5)。(I) 肿瘤植入 4 周后,从 CD11c-SREBF2−/−和 SREBF2fl/fl 对照小鼠的 TDLNs 中分离的 cDC 的 H-2Kb MHC I 类表面表达的流式细胞术定量(n = 4)。(J) 肿瘤植入 3 周后,从携带 OVA 表达的 BRAFV600EPTEN−/−黑色素瘤的小鼠 TDLNs 中分离的 OVA 脉冲 DC,与初始 OTI CD8+ T 细胞共培养的流式细胞术 CFSE 增殖实验(n = 4)。(K) 在 CD11c-SREBF2−/−和 SREBF2fl/fl 宿主中的 BRAFV600EPTEN−/−黑色素瘤的肿瘤生长曲线(n = 12)。(L 和 M) 肿瘤植入 25 天后,从 CD11c-SREBF2−/−和 SREBF2fl/fl 宿主中分离的肿瘤和 TDLN 免疫细胞群的流式细胞术定量分析。(L) TDLN 和肿瘤中 DC 亚型的定量。(M) 肿瘤浸润的 CD4+ T 细胞、Foxp3+CD4+ Tregs、KLRG1+ Tregs 和 CD8+ T 细胞的定量(n = 9 到 11)。(N) CD11c-SREBF2−/−小鼠及其 SREBF2fl/fl 对照被植入 BRAFV600EPTEN−/−CDKN2A−/−黑色素瘤细胞。肿瘤植入 4 周后,通过 H&E 和 S100β IHC 显微镜量化微转移。(A 和 C 到 E) 统计分析通过双因素方差分析(ANOVA),然后是 Tukey 或Šidák 多重比较检验进行。(B) 统计分析通过单因素方差分析(ANOVA),然后是 Dunnett 多重比较检验进行。(G 到 N) 所有两组比较均使用未配对 t 检验分析。数据以平均值±标准误(SEM)表示。
鉴于脂质代谢在 DC 稳态中的重要作用以及我们在 mregDCs 中观察到的中性脂质储存增加,我们假设 SREBP2 抑制将干扰 CD63+ mregDCs 调节 T 细胞反应的能力。为了初步测试这一假设,我们用 SREBP 抑制剂 fatostatin 处理从携带自发性 BRAFV600EPTEN−/−黑色素瘤小鼠的肿瘤引流淋巴结中分选出的 CD63+ mregDCs 或 CD63− DCs,并将它们与同种异体初始 CD4+ T 细胞共培养。Fatostatin 处理减少了 CD63+ mregDCs 相对于未经处理的 CD63+ mregDCs 促进 Treg 分化的能力,同时不影响其活力(图 6A 和图 S6A)。此外,fatostatin 和 HMG-CoA 还原酶抑制剂 simvastatin 相对于车辆处理对照,均诱导了 CD63+ mregDCs 的 CD8+ T 细胞交叉引发能力的增加(图 6B)。然而,香叶基香叶基转移酶抑制剂 GGTI-2147 未能增加交叉引发,这表明下游的异戊二烯生物合成途径不调节 CD63+ mregDCs 激活效应 T 细胞的能力(图 6B)。这些结果使我们假设 SREBP2 抑制可以缓解 CD63+ mregDCs 的免疫抑制并在体内促进抗肿瘤免疫反应。
为了测试这一假设,携带自发性 BRAFV600EPTEN−/−黑色素瘤的小鼠接受单独 fatostatin(30 mg/kg,每 2 天一次)或联合抗 PD-1 抗体(抗 PD-1,200 μg,每 3 天一次)的治疗。Fatostatin 单独治疗减少了肿瘤组织中 CD63+ mregDCs 的 SREBP2 靶基因的表达,并相对于车辆处理对照抑制了肿瘤生长(图 6C 和图 S6B)。虽然抗 PD-1 单药治疗对肿瘤生长无影响,但 fatostatin/抗 PD-1 联合治疗相对于单独使用 fatostatin 更能抑制原发性肿瘤生长(图 6C)。Fatostatin 减少了 TDLN 中的 CD63+ mregDCs 并增加了 cDC1s,同时在与抗 PD-1 联合使用时减少了肿瘤中的 CD63+ mregDCs 的总数(图 6D 和图 S6,C 和 D)。这些发现与 Treg 分化的抑制和肿瘤内 CD8+ T 细胞数量的增加相关(图 6E 和图 S6,D 和 E)。尽管 fatostatin 在体外抑制了 BRAFV600EPTEN−/−黑色素瘤细胞的增殖,但它无法减缓非肥胖型糖尿病严重联合免疫缺陷(NSG)小鼠中的肿瘤生长(图 S6,F 和 G),表明 fatostatin 的作用主要通过宿主免疫系统介导。总体而言,这些发现表明全身性 SREBP2 抑制产生了支持增强抗肿瘤免疫的 TME。
接下来,为了确定 DC 特异性 SREBP2 对肿瘤进展的重要性,我们使用 CD11c 启动子(Itgax)驱动的 Cre 重组酶小鼠生成了 DC 特异性 Srebf2 敲除小鼠模型(图 S7,A 到 C)。这些 CD11c-SREBF2−/−小鼠的 DC 显示氧消耗率降低,如细胞外流分析所示,并基于 SCENITH 测定显示嘌呤霉素掺入减少,与 ATP 产量减少相关(图 6F 和图 S7D)。与这些发现一致的是,SREBF2−/− DC 相对于对照 DC 表现出 FAO 减少(图 6G)。此外,SREBF2−/− DC 的中性脂质含量显著低于对照 SREBF2fl/fl 同窝小鼠的 DC(图 6H 和图 S7E)。SREBP2 靶基因 Pcsk9 调节 MHC I 类表达,Pcsk9 的丧失导致几种肿瘤类型中 MHC I 类表面表达增加。此外,我们的研究表明 PCSK9 阻断逆转了 CD63+ mregDCs 抑制依赖 DC 的 CD8+ T 细胞刺激的能力。因此,我们假设 SREBP2 的丧失将导致 DC 表面 MHC I 类表达增加。流式细胞术显示,与对照同窝小鼠相比,从 CD11c-SREBF2−/−小鼠的 TDLNs 中分离的 DC 具有更高的 MHC I 类表达(图 6I)。然后,我们从 CD11c-SREBF2−/−或对照 SREBF2fl/fl 小鼠的 LN 和骨髓中分离 cDC1s,并用 OVA 脉冲后与初始 OTI CD8+ T 细胞共培养,结果表明 SREBP2−/− DC 表现出促进 CD8+ T 细胞增殖的能力增加(图 6J 和图 S7F)。这些发现表明,SREBF2−/− DC 表现出增强的 T 细胞刺激能力。
为了测试 SREBF2−/− DC 是否能抑制肿瘤进展,我们将同种自发性 BRAFV600EPTEN−/−黑色素瘤植入 CD11c-SREBF2−/−和对照 SREBF2fl/fl 同窝小鼠,并监测肿瘤生长。我们观察到,相对于 SREBF2fl/fl 对照小鼠,CD11c-SREBF2−/−小鼠的肿瘤生长受抑,CD63+ mregDCs 减少,TME 中的 cDC1s 增加(图 6,K 和 L)。这些改变与肿瘤浸润的 CD4+ TH 细胞和 Tregs 的减少以及肿瘤内 CD8+ T 细胞的增加相关(图 6M)。此外,在将 BRAFV600E PTEN−/−黑色素瘤细胞移植到 CD11c-SREBF2−/−宿主后,相对于对照 SREBF2fl/fl 同窝小鼠,我们观察到切除的肺组织中的微转移减少(图 6N)。总体而言,这些发现表明,SREBP2 作为 CD63+ mregDC 介导的免疫抑制和肿瘤免疫逃逸的关键调节因子。
肿瘤来源的乳酸促进 TME 中 DC SREBP2 的激活¶
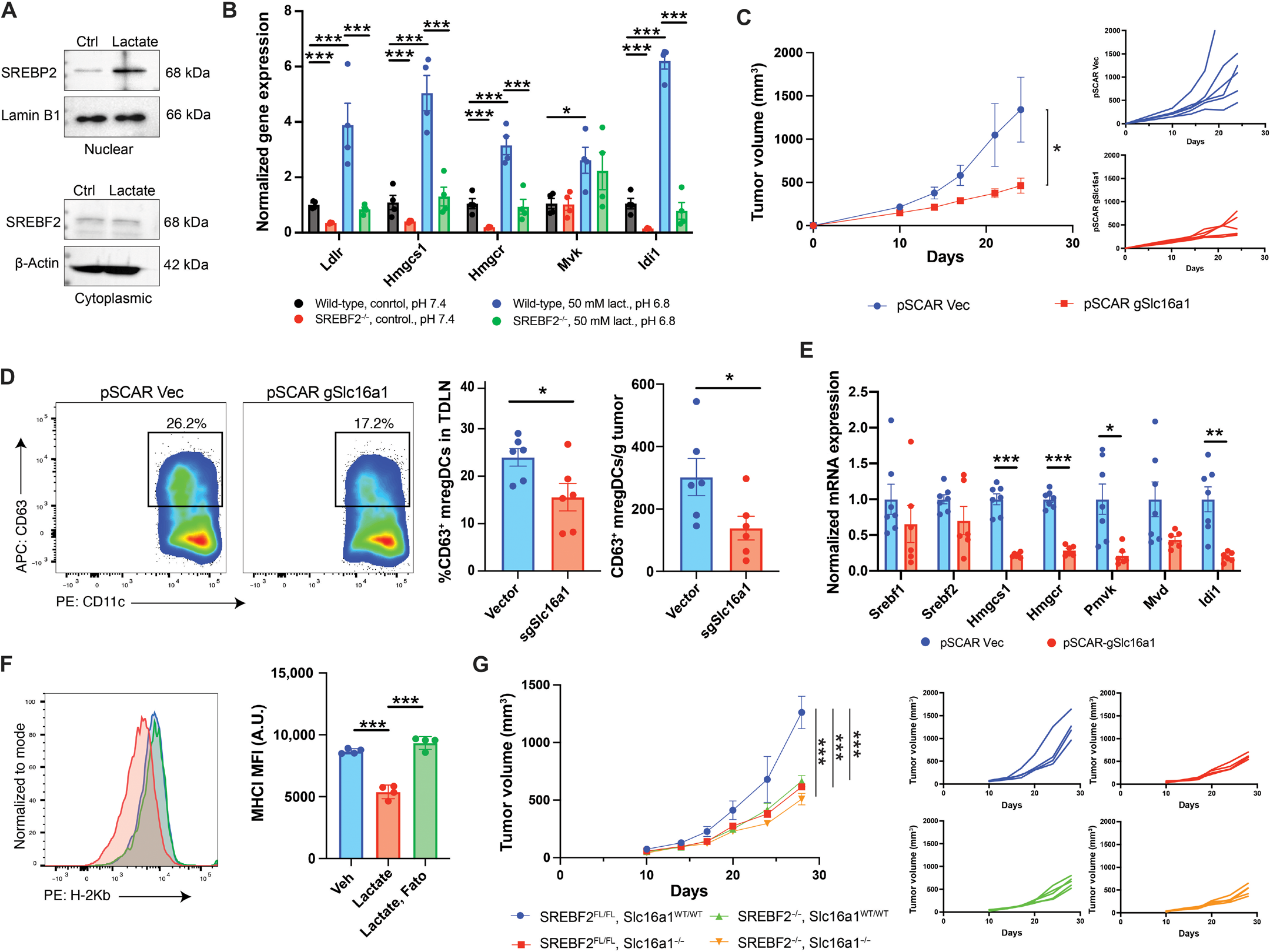
图 7. 肿瘤来源的乳酸在 TME 中促进 SREBP2 的激活。(A)Western blot 显示骨髓来源 DCs 在处理乳酸 24 小时后(pH 6.8)SREBP2 的激活和核定位。(B)qRT-PCR 显示 CD11c-SREBF2−/−小鼠或同窝 SREBF2fl/fl 对照组在处理乳酸(pH 6.8)或正常条件(pH 7.4)下分离的骨髓来源 DCs 中 SREBP2 靶基因 Ldlr、Hmgcs1、Hmgcr、Mvk 和 Idi1 的表达(n = 4)。(C)左图:CRISPR-Cas9 介导的 Slc16a1 敲除或未编辑(Vec)对照的 BRAFV600EPTEN−/−黑色素瘤的肿瘤生长曲线。右图:显示个体肿瘤进展的生长曲线(n = 6)。(D)左图:在肿瘤植入 24 天后,流式细胞术检测携带 BRAFV600EPTEN−/−Slc16a1−/−或未编辑对照 BRAFV600EPTEN−/−黑色素瘤肿瘤的小鼠中浸润 TDLN 的 CD63+ mregDCs。右图:TDLN 中 CD63+ mregDCs 的百分比和肿瘤中 CD63+ mregDCs 的数量的量化(n = 6)。(E)qRT-PCR 显示从 BRAFV600EPTEN−/−Slc16a1−/−或未编辑对照 BRAFV600EPTEN−/−黑色素瘤肿瘤中分离的 CD63+ mregDCs 中 SREBP2 靶基因的表达(pSCA R-scr:n = 7,pSCA R-Slc16a1:n = 6)。(F)流式细胞术显示脾脏 cDCs 在体外处理乳酸(pH 6.8)或乳酸和 fatostatin 后的 MHC I 类表面表达。(G)在 CD11c-SREBF2−/−小鼠或同窝 SREBF2fl/fl 对照中,植入 BRAFV600EPTEN−/−Slc16a1−/−或载体对照细胞,Slc16a1WT/WT 的肿瘤生长曲线(n = 5)。所有两组比较均使用未配对 t 检验分析。(B、F 和 G)统计分析使用双向 ANOVA,随后进行 Tukey 多重比较检验。数据以平均值 ± SEM 表示,并显示个体数据点。所有数据均代表两到三次独立实验。
在我们的实验中,我们发现相对于 cDC1 和 cDC2,CD63+ mregDCs 中富含中性脂质。通常情况下,在高固醇状态下,SREBP2 被束缚在内质网,防止其激活其靶基因。尽管如此,我们观察到相对于 cDC1 和 cDC2,CD63+ mregDCs 中富集了 SREBP2 靶基因的特征,表明可能存在 SREBP2 激活的替代机制。酸性 pH(pH 6.8)可以促进 SREBP2 的激活和核转位,导致参与胆固醇稳态的 SREBP2 靶基因的表达【48】。黑色素瘤具有显著高的有氧糖酵解率,通过生成乳酸导致 TME 的酸化【49, 50】。因此,我们推测黑色素瘤产生的乳酸可能促进 DCs 中 SREBP2 的激活。为了验证这一假设,我们用乳酸(50 mM,pH 6.8)处理 BMDCs 并测量 SREBP2 的核转位,结果显示核 SREBP2 增加,表明 SREBP2 激活(图 7A)。接下来,将 CD11c-SREBF2−/−小鼠或对照 SREBF2fl/fl 同窝小鼠的 BMDCs 分别用乳酸(50 mM,pH 6.8)处理或不处理(pH 7.4),并分析基因表达。乳酸处理仅在对照 SREBF2fl/fl DCs 中导致了关键 SREBP2 靶基因如 Ldlr、Hmgcs1、Hmgcr 和 Idi1 的表达增加(图 7B)。这些实验表明乳酸促进 DCs 中 SREBP2 依赖的胆固醇稳态基因的表达。
基于这些结果,我们进一步检查了 TME 中的肿瘤来源乳酸是否可以促进 SREBP2 的激活和 CD63+ mregDCs 的发展。为了解决这个问题,我们使用 CRISPR-Cas9 删除了 BRAFV600EPTEN−/−黑色素瘤细胞系中的乳酸出口蛋白 MCT1(Slc16a1)(图 S7G)。从 BRAFV600EPTEN−/−Slc16a1−/−细胞中收集的条件培养基中的乳酸分泌显著减少(图 S7H)。我们将 BRAFV600EPTEN−/−Slc16a1−/−黑色素瘤细胞植入同种基因小鼠中,并监测肿瘤生长和 CD63+ mregDC 的发展。与对照肿瘤相比,BRAFV600EPTEN−/−Slc16a1−/−肿瘤的生长以及 TDLN 和肿瘤组织中的 CD63+ mregDCs 的数量显著减少,这种效果仅在免疫健全宿主中观察到(图 7C 和 D,图 S7I)。此外,黑色素瘤 Slc16a1 的基因沉默导致从 TME 中分选的 DCs 中 SREBP2 靶基因的抑制(图 7E)。我们还观察到通过对脾脏 DCs 进行体外 fatostatin 处理,可以逆转乳酸介导的 MHC I 类分子的下调(图 7F)。
为了确认观察到的黑色素瘤细胞乳酸释放的抑制不会通过与 DCs 中 SREBP2 通路无关的方式调节抗肿瘤免疫,我们将 BRAFV600EPTEN−/−Slc16a1−/−黑色素瘤细胞植入 CD11c-SREBF2−/−宿主中。肿瘤生长曲线显示,与对照 BRAFV600EPTEN−/−黑色素瘤相比,BRAFV600EPTEN−/−Slc16a1−/−黑色素瘤在 CD11c-SREBF2−/−宿主中生长无差异,表明 SREBP2 在 DCs 中的激活是乳酸促进免疫功能障碍的重要机制(图 7G)。总的来说,这些数据表明,肿瘤来源的乳酸导致 TME 中 DCs 中的 SREBP2 激活,驱动了原位耐受 CD63+ mregDCs 的发展。
CD63+ mregDCs 在黑色素瘤患者的前哨淋巴结中被保留¶
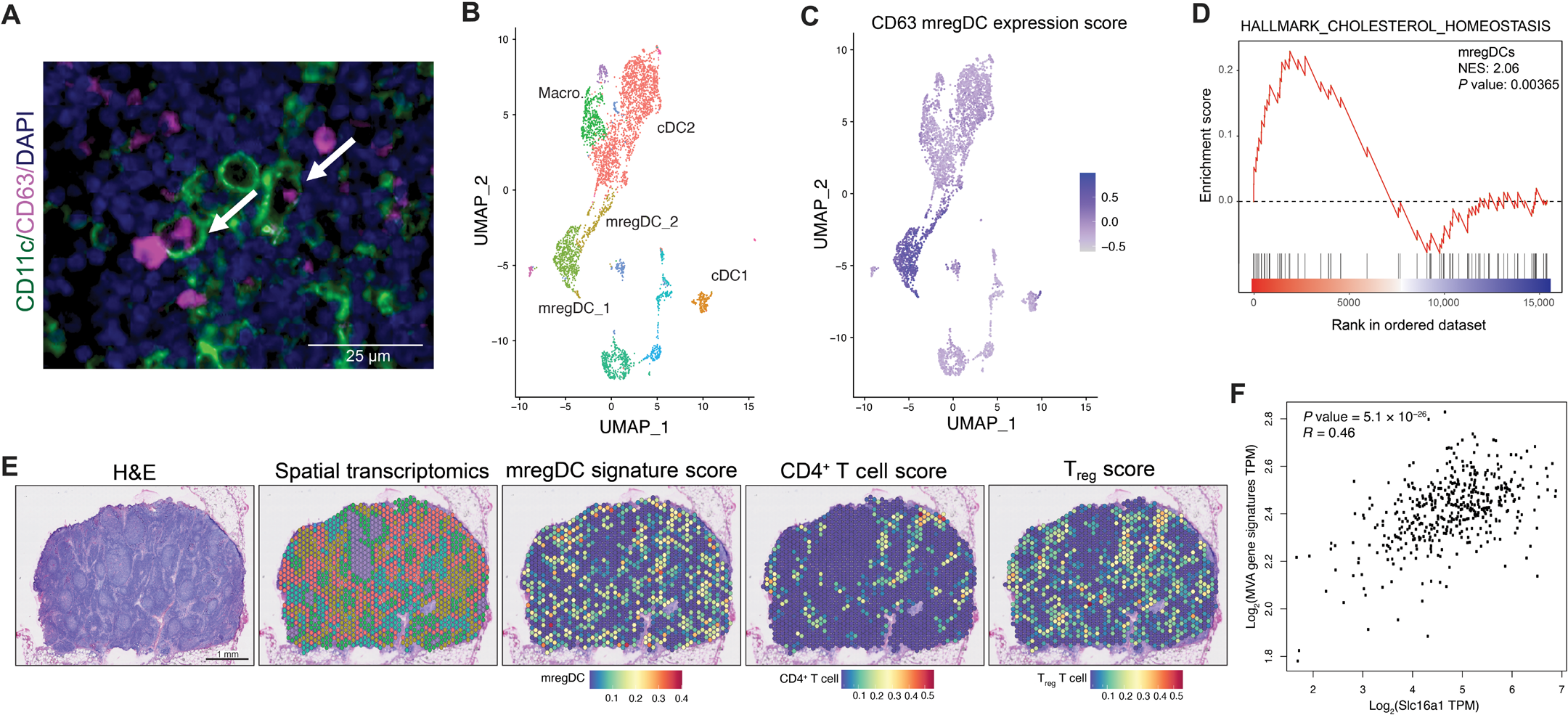
图 8. CD63+ DCs 存在于人类黑色素瘤肿瘤中。(A)对从黑色素瘤患者中分离出的前哨淋巴结切片进行 CD63(品红色,细胞质)和 CD11c(绿色,膜表面)的免疫荧光染色(n = 3 个独立实验,来自 12 名患者)。白色箭头,CD63+CD11c+ 细胞。(B)scRNA-seq UMAP 图显示从黑色素瘤患者的前哨淋巴结中分离出的 DC 亚群的聚类情况(n = 3 个独立实验,来自三名患者)。(C)图 4 中生成的 CD63+ mregDC 表达评分应用于人类黑色素瘤 scRNA-seq UMAP 图。(D)相对于总 DCs 的 Hallmark 胆固醇稳态基因集合在人体 mregDCs 中的 GSEA。(E)从黑色素瘤患者中收集的前哨淋巴结组织的 Visium 空间转录组分析。左图:前哨淋巴结组织的 H&E 染色。中图:覆盖在 H&E 图像上的 Visium 点。右图:mregDC 表达评分、CD4+ T 细胞评分和 Treg 评分覆盖在 Visium 空间转录组点图上,显示黑色素瘤患者中存在 mregDCs(n = 3 个独立实验,来自三名患者)。(F)TCGA 数据集中黑色素瘤患者的 Sl16a1(MCT 1)与 MVA 基因的相关性。显著性通过 Spearman 相关性确定。DAPI,4′,6- 二脒基 -2- 苯基吲哚;TPM,每百万转录本。
鉴于我们的临床前数据表明 CD63+ mregDCs 群体支持免疫耐受 TME 的发展,我们调查了最近确诊的黑色素瘤患者中收集的人类前哨淋巴结组织中是否存在这种 DC 群体。我们进行了免疫荧光显微镜分析,并在所有检查的人类淋巴结组织标本中(12/12)鉴定出了 CD63+CD11c+ DCs(图 8A)。然后我们进行了 scRNA-seq,并应用我们的 CD63+ mregDC 表达评分,以确认人类前哨淋巴结组织中存在 CD63+ mregDCs(3/3)(图 8B 和 C)。此外,人类 CD63+ mregDCs 的胆固醇稳态基因表达也如同其小鼠对应物一样富集(图 8D)。
接着,我们进行了空间转录分析,以评估 CD63+ mregDCs 在额外的人类淋巴结标本组织结构中的解剖位置(图 8E)。CD63+ mregDCs 位于人类淋巴结组织的副皮质区,邻近 CD4+ T 细胞和 Tregs(图 8E 和图 S8A、B)。一项先前的研究表明,SLC16A1 表达的增加与黑色素瘤患者的不良生存率相关【51】。癌症基因组图谱(TCGA)数据库分析显示,SLC16A1 表达与黑色素瘤组织中 SREBP2-MVA 信号轴中基因的表达相关,这表明肿瘤来源的乳酸可能在人体 TME 中触发 SREBP2 的激活(图 8F)。总之,我们的结果表明,黑色素瘤患者的前哨淋巴结中存在 CD63+ mregDCs。
讨论¶
尽管肿瘤学领域近年来取得了进展,但大多数癌症患者仍对免疫检查点抑制剂治疗无反应。已有多种肿瘤依赖性特征被报道会影响免疫治疗的效果。然而,我们对肿瘤介导的免疫逃逸机制的理解仍不完全,克服抗 PD-1 免疫治疗耐药性的能力也有限。尽管树突状细胞(DC)被认为是抗肿瘤免疫的重要介质,但越来越多的研究表明,肿瘤可以通过转化 DC 来产生耐受的肿瘤微环境(TME)。
最近有报道提到一种被称为 mregDCs 的独特 DC 程序,显示其能够驱动 TH2 极化的免疫反应,从而抑制肿瘤免疫。在此基础上,我们通过表征 mregDCs 在 TME 中的发育以及支持 mregDCs 抑制抗肿瘤免疫的生化信号传导和代谢途径来进一步扩展这一研究。我们发现,MVA 生物合成途径及其主要转录因子 SREBP2 支持 CD63+ mregDCs 的发展和功能。通过识别 CD63 作为 mregDC 亚群的特定表面标志物,我们进一步展示了 mregDCs 的免疫调节功能,使其能够驱动 Treg 发育,同时在高 cDC:CD63+ mregDC 比率下,仍能在同源和异源条件下抑制 DC 抗原交叉呈递。我们证明,药理学 SREBP2 抑制和 DC 特异性 Srebf2 沉默均可逆转 mregDC 依赖的免疫逃逸,并在体内抑制肿瘤进展。额外的研究显示,CD63+ mregDCs 存在于黑色素瘤患者的引流淋巴结组织中。鉴于在几项大型泛癌转录组研究中已识别出 mregDC/LAMP3+ DCs,这些发现可能对多种癌症具有相关性。这些发现支持了 SREBP2 依赖的 mregDCs 在建立免疫耐受 TME 中的作用,并表明这一途径是克服免疫治疗耐药性的一个有前途的靶点。
鉴于代谢在调节 DC 功能中的重要作用,我们开始研究 CD63+ mregDCs 相对于其他 DC 亚群的独特代谢特征。我们发现,CD63+ mregDCs 富含多种 MVA 酶的表达,并激活其主要转录因子 SREBP2。相对于 cDC1 和 cDC2,CD63+ mregDCs 储存了更多的脂质,并表现出较高水平的 FAO。mregDCs 中改变的中性脂质含量可能通过从周围 TME 中增加脂质摄取或通过 MVA 途径增加胆固醇合成来支持,这两者均可由 SREBP2 驱动。这些结果与先前将 MVA 合成途径与炎症调节联系起来的研究一致。我们的数据表明,SREBP2 和 HMG-CoA 还原酶抑制均可增强 CD63+ mregDC 介导的 CD8+ T 细胞反应激活。这与其他研究表明使用他汀类药物与接受抗 PD-1 免疫治疗的癌症患者的临床结果改善有关。我们的工作表明,这些代谢途径对 CD63+ mregDCs 具有功能意义,因为 SREBP2 抑制剂 fatostatin 和 DC 特异性 Srebf2 基因沉默均能增强 CD8+ T 细胞的激活,减少 Treg 分化,并抑制体内 BRAFV600EPTEN−/−黑色素瘤的进展。整体结果与先前的数据一致,表明 FAO 是肿瘤介导的 DC 耐受化过程中分子机械的重要组成部分。
调节 mregDCs 在 TME 中发育的潜在机制也一直不明确。我们的数据表明,随着肿瘤进展,CD63+ mregDCs 在肿瘤床和局部引流淋巴结组织中扩展和迁移。CD63+ mregDCs 的迁移行为与先前描述的在维持外周耐受中的 DC 稳态成熟一致。我们的工作现在表明,肿瘤可以劫持这一过程,从而在 TME 中消除和转化免疫刺激性的 cDC 群体。我们的数据表明,CD63+ mregDCs 通过抑制效应 CD8+ T 细胞反应,同时促进 Treg 和 TH2 CD4+ T 细胞分化,从而形成耐受的免疫微环境。此外,我们的研究证实了先前的观察,即 mregDCs 可以从 cDC1 和 cDC2 发育而来。
我们随后询问了肿瘤如何诱导 SREBP2 信号并增加 CD63+ mregDCs 中的胆固醇含量。基于先前研究暗示低 pH 在 SREBP2 激活中的作用,我们在黑色素瘤细胞中沉默了乳酸转运蛋白 MCT1,发现肿瘤依赖的乳酸产生通过激活 SREBP2 依赖的信号传导途径,促进了 CD63+ mregDCs 的发展。因此,这项工作有助于以往文献中将乳酸视为 TME 中免疫抑制的重要介质的观点。尽管我们的数据表明肿瘤来源的乳酸可以驱动 SREBP2 激活,但也可能有其他机制也有助于增加 DC 脂质含量。最近有报道称,非肿瘤模型中的 cDC1 通过摄取凋亡细胞可以增加脂质含量,并通过激活肝 X 受体信号通路最终导致耐受性 DC 成熟。这支持了我们的工作,即 DC 脂质稳态在癌症进展过程中调节稳态 DC 成熟中起着重要作用。先前的一项研究描述了在小肠固有层中,凋亡细胞摄取刺激了驱动 Treg 的 CD103+ DC 群体的发展。
先前的研究已经说明了耐受性 DC 在抑制抗肿瘤免疫中的效力,表明仅少量耐受性 DC 就足以抑制效应 T 细胞反应。我们的研究表明,通过释放可溶性介质,抑制附近 DC 的抗原交叉呈递可能有助于这种现象。考虑到 DC 依赖的抗原交叉呈递在抗 PD-1 免疫治疗中的关键作用,我们认为这一机制可能是增强传统上难治肿瘤的免疫治疗反应的一个关键靶点。与最近的一份报告一致,我们的研究进一步表明,PCSK9 抑制 DC 介导的 CD8+ T 细胞激活。使用已经在临床上用于管理难治性高胆固醇血症的单克隆抗体靶向 PCSK9,现在正在探索其与抗 PD-1 免疫治疗在实体瘤中的联合应用。
这项工作存在一些局限性。虽然条件性 Srebf2 删除显示了 SREBP2 对 mregDC 发展的重要性,但这项工作使用了 ITGAX 启动子驱动的 Cre 重组酶,导致了 Srebf2 在包括 cDC1、cDC2 和浆细胞样 DC 在内的各种 DC 群体中的表达丧失。我们无法使用 ZBTB46 驱动的 Cre,因为这些小鼠中的 Srebf2 丧失在胚胎阶段意外致死。考虑到 SREBP1 在脂质生成中的作用,未来的研究需要区分其在指导 CD63+ mregDC 功能中的角色。需要进一步研究以确定 CD63+ mregDCs 释放的其他可溶性因子是否也会导致观察到的 DC 依赖抗原交叉呈递的抑制。最后,需要进行实验以检验 CD63+ mregDCs 对 TME 中 CD8+ T 细胞动态的影响,同时需要对临床标本进行研究以调查 CD63+ mregDCs 群体对癌症患者临床结果的影响。
在这里,我们提供了 TME 中耐受性 DC 成熟的生化途径以及 mregDC 群体在抑制抗肿瘤免疫和支持肿瘤进展中的作用的见解。总体而言,这项工作为未来研究利用 mregDCs 增强抗肿瘤免疫和克服现有检查点抑制剂免疫治疗耐药性提供了基础。