A systematic pan cancer study on deep learning based prediction of multi omic biomarkers from routine pathology images
摘要¶
背景¶
这项综合泛癌研究的目标是评估深度学习(DL)在从苏木精和伊红(H&E)染色的全切片图像中直接进行多组学生物标志物分子分析的潜力。
方法¶
我们训练和验证了总计 12,093 个 DL 模型,这些模型预测了 32 种癌症类型中的 4,031 种多组学生物标志物。研究包括广泛的遗传学、转录组学和蛋白质组学生物标志物,以及已建立的预后标志物、分子亚型和临床结果。
结果¶
研究表明,50% 的模型的曲线下面积(AUC)达到 0.644 或更高。25% 的模型的 AUC 至少为 0.719,前 5% 模型的 AUC 超过 0.834。基于图像的组织形态学特征进行分子分析对于大多数被调查的生物标志物和不同的癌症类型总体上是可行的。性能似乎与肿瘤纯度、样本大小和类别比例(患病率)无关,表明组织形态学中存在一定程度的内在可预测性。
结论¶
结果表明,DL 有望仅使用 H&E 染色的实体瘤组织切片来预测广泛的组学谱系生物标志物。这为加速诊断和开发更精确的癌症治疗方法铺平了道路。
通俗语言总结¶
分子分析测试用于检查癌症中某些基因、蛋白质或其他分子的变化。这些测试结果可用于确定对癌症患者最有效的治疗方法。需要比标准测试更快且更易获得的替代方法来改善癌症护理。本研究调查了深度学习(DL)能否直接从常规收集的用于诊断目的的肿瘤标本图像中进行分子分析。利用超过 12,000 个 DL 模型,使用统计方法评估了数千种生物标志物。结果表明,DL 可以有效地从这些图像中检测肿瘤的分子变化,适用于许多生物标志物和肿瘤类型。研究表明,基于 DL 的图像分子分析是可能的。将这种方法引入常规临床工作流程中,可能会加速治疗决策并改善结果。
引言¶
研究分子景观不同层次的改变有助于更好地理解肿瘤发生和癌症进展【1,2】。深入分析分子异常与肿瘤微环境之间的关系已促进了多种癌症类型中靶向治疗的发展【3,4,5】。基因组分析已成为一项重要工具【6】,尤其是对于那些因遗传因素而具有较高癌症风险的个体【7】。此外,标准的分子和基因组分析方法通常因需要时间进行准备、处理和分析而导致实验室延迟,这在日常临床工作流程中是一个常见的问题【8,9】。昂贵的测试可能无法普遍适用于所有患者【10】。
与此同时,越来越多的证据表明,用苏木精和伊红(H&E)染色的诊断组织图像可能包含可以直接从组织切片中推断分子特征的信息【11,12】。深度学习(DL)可以有效揭示恶性肿瘤中形态表型的差异,从而能够直接从 H&E 染色的全切片图像(WSIs)预测分子特征【11,12,13,14,15】。DL 方法已被用于推断多种癌症类型的分子改变,包括乳腺癌【9,16,17,18】、结直肠癌【13,15,19,20】、肺癌【12,21】、胃癌【22】、前列腺癌【23】、皮肤癌【24】和甲状腺癌【25】(参见 Echle 等人对 DL 应用于生物标志物分析的广泛综述【26】)。最近,泛癌研究探索了 H&E 图像中的遗传/分子改变与组织形态特征之间的联系。这些研究表明,几乎所有恶性肿瘤中,DL 方法都可以用于直接从常规组织学中推断大量生物标志物【11,14,15,27,28,29】。
在以往研究的基础上,我们开展了一项大规模研究,以评估使用 DL 从常规诊断切片中分析生物标志物的可行性。所研究的生物标志物包括广泛的基因组、转录组和蛋白质组改变以及各种临床相关的下游生物标志物(例如,标准护理特征、分子亚型、基因表达、临床结果和治疗反应)。我们系统地评估了由癌症基因组图谱(TCGA)项目研究的所有实体癌症的可预测性。我们的 DL 方法利用自编码器网络作为特征提取器,使得学习与分析任务相关的组织图像表示成为可能。与以往的研究不同,我们扩大了 DL 在泛癌预测中的评估范围,涵盖了更多的癌症类型(n = 32)和跨分子生物学中心法则的数千种生物标志物(n = 4031)。许多这些生物标志物的系统可预测性尚未在大规模上进行评估,包括某些表型和临床结果,如药物反应。
我们的研究结果表明,多组学生物标志物可以直接从组织形态学中预测。对大多数测试的基因来说,从组织形态中预测突变是可行的。像 TP53 这样的频繁突变基因在多种癌症类型中都是可预测的。转录组和蛋白质的低表达/高表达状态在一定程度上是可预测的。DL 可以检测到形态视觉特征,从而能够直接从 WSIs 中预测分子亚型和已建立的临床生物标志物。我们的实验在基于 TCGA 数据集的某些癌症类型和具有等效生物标志物的外部数据集上重复时,获得了类似的结果,进一步证实了从 H&E 染色切片中预测泛癌生物标志物的总体可行性。考虑到可能影响可预测性的各种因素,如肿瘤纯度、样本大小和生物标志物状态的流行率,我们得出结论,组织形态学可能存在一定程度的真实可预测性。
方法¶
外部数据集¶
为了评估生物标志物在外部数据集上的预测可行性,我们使用了通过癌症成像档案(https://wiki.cancerimagingarchive.net/display/Public/CPTAC+Imaging+Proteomics)获取的临床蛋白质组肿瘤分析联盟(CPTAC)数据重复了我们的实验。CPTAC 数据集包含 3481 张 H&E 染色图像,涉及 1329 名患者,这些图像来自以下七项不同的研究:LUAD(肺腺癌)、COAD(结肠腺癌)、HNSCC(头颈癌)、LSCC(肺鳞状细胞癌)、PDA(胰腺导管腺癌)、GBM(胶质母细胞瘤)和 UCEC(子宫内膜癌)。LUAD、GBM 和 COAD 来自冷冻组织,而其余数据集包含 FFPE 切片。分辨率不同于 COAD 的 0.25 MPP 和其他数据集的 0.5 MPP 的图像被丢弃,以确保每个癌症队列中的分辨率一致。CPTAC 队列中包含的最终图像和患者详细信息见补充表 2。
生物标志物获取¶
获取可操作的驱动基因¶
临床相关的驱动基因从 https://cancervariants.org 获取【30】。我们只考虑了已知与(1)美国食品和药物管理局(FDA)批准的疾病特异性治疗相关和(2)根据专业指南和/或基于专家共识的有力研究显示的治疗反应或抗性相关的驱动基因【31】。驱动突变和药物相关数据从以下来源获取:BRCA Exchange、癌症基因组解读器癌症生物标志物数据库、癌症变异的临床解释、Jackson 实验室临床知识库(JAX-CKB)、精准医学知识库。这些源文件已经包含 SNP 突变与表型之间的关联,使得专家病理学家能够将它们映射到 TCGA 研究中。最后,通过使用这些映射和每种表型的驱动突变数据,我们为每个 TCGA 研究创建了一组驱动基因,并随后用于过滤转录组、蛋白质组和基因组数据中的可操作生物标志物。
TCGA 基因组生物标志物谱¶
使用 cBioPortal Web API 和 GDC API 收集基因组生物标志物数据。对于每个 TCGA 研究,我们检索了所有与诊断切片相关的样本。未进行全基因组或全外显子测序数据的样本被排除在外,使我们能够假设剩余样本中的所有感兴趣基因都已被分析。虽然存在一些没有 WGS 或 WXS 数据的样本有突变,但无法假设没有突变的基因处于野生型,因为它们可能只是没有被测序。对于 cBioPortal 上列出的所有 TCGA 研究,我们获取了“MUTATION_EXTENDED”变异类型的分子谱,并检索了这些分子谱中的所有突变,并存储为中间格式。最后,我们使用这些突变数据为所有驱动基因创建了分子谱。如果样本包含至少一个单核苷酸变异(SNV),则认为该样本对驱动基因呈阳性。SNV 通常是插入、替换或删除一个碱基,但在少数情况下可以是多个碱基(例如,“T”被替换为“CGC”)。结果谱被过滤以排除在给定癌症中阳性样本少于十个的驱动基因。
转录组和蛋白质组谱¶
通过 cBioPortal API 检索 TCGA 数据集的转录组和蛋白质组数据。cBioPortal 提供了从基因表达的原始 FPKM 计数计算的 z-score 以及相对于表达值均值的标准差数。这些 z-score 是为每个编码基因和每个在 TCGA 研究中具有相关组织切片的样本获取的。cBioPortal 将转录组 z-score 的计算限制在肿瘤中包含二倍体细胞的样本。蛋白质组 z-score 是在给定癌症的所有可用样本中计算的。z-score 基于以下阈值为每个基因和样本二值化:对于每个样本,z-score 小于或等于 t_under 的基因被认为是低表达,而 z-score 大于或等于 t_over 的基因被认为是高表达。我们将{t_under, t_over}分别设置为{-2, 2}和{-1.5, 1.5},以便在所有感兴趣基因的 z-score 分布中创建平衡的类别。这些阈值随后用于生成蛋白质组和转录组基因的低表达/高表达谱。在低表达谱中,所有被认为低表达的样本被标记为阳性,而所有其他样本被标记为阴性。类似地,在高表达谱中,高表达样本被标记为阳性,而其余样本被认为是阴性。最后,为了减少目标生物标志物的数量,我们将高表达和低表达谱限制为每个研究中的驱动基因(参见获取可操作的驱动基因)。此外,不包含足够阳性样本的谱也被排除。蛋白质组基因的最小阳性样本数设置为 20。对于转录组谱,只保留至少 10% 阳性比例且至少有十个阳性样本的谱。
标准护理特征、基因表达签名和分子亚型¶
使用相关研究提供的公开数据集来获取基因表达签名、分子亚型和标准临床生物标志物的谱【11】。该数据集最初是从系统研究的 TCGA 数据结果中整理而来的(https://portal.gdc.cancer.gov/)【32,33,34】,包含 17 个 TCGA 数据集的谱(请参阅原始研究【11】了解生物标志物的描述和获取协议的其他详细信息)。对于某些生物标志物,我们使用共识意见将分子状态映射为二进制标签。例如,考虑到微卫星不稳定性(MSI),所有定义为 MSI-H 的患者被纳入阳性类别,而微卫星稳定(MSS)和 MSI-L 患者被标记为阴性。具有多个分类值的谱被用 one-hot 编码二值化,其中为每个类别创建一个谱,只有该类别的样本被设为阳性。具有连续值的非分类谱在消除 NaN 值后被在均值处二值化【11】。
临床结果和治疗反应¶
从 TCGA 泛癌临床数据资源(TCGA-CDR)获取生存数据,该资源是一个公开的数据集,提供四个主要的临床结果终点【35】,即总生存期(OS)、疾病特异性生存期(DSS)、无病生存期(DFI)和无进展间隔期(PFI)。这些终点通过考虑从 TCGA 常规收集的临床数据(https://portal.gdc.cancer.gov/)中获取的多个临床和预后特征系统地二值化为可操作事件,例如生命状态、肿瘤状态、死亡原因、新肿瘤事件、局部复发和远处转移。将临床数据整合为可操作的生存结果的详细信息见原始研究【35】,并在补充表 3 中总结。此外,我们还添加了从 TCGA 临床文件中获取的残留肿瘤状态作为另一个预后目标。具有显微镜或肉眼可见残留肿瘤(R1 或 R2)的患者被归类为阳性,而没有残留肿瘤(R0)的患者被归类为阴性【36】。由于 TCGA-BRCA 没有残留肿瘤信息,我们使用“margin_status”属性。同样,“treatment_outcome_first_course”被用来创建表示治疗反应的二值目标。为此,任何被标记为“完全缓解/反应”的患者都被归类为阳性,而“疾病稳定”、“部分缓解”和“疾病进展”被认为是阴性。最后,使用 TCGA 数据集中的临床药物文件识别药物反应。这是通过首先基于另一项研究提供的数据统一药物名称来实现的【37】,然后识别具有足够样本的药物 - 研究对。最后,“treatment_best_response”属性被用来将药物反应映射到二进制类别,其中“完全反应”构成阳性类别,其他则为阴性。对于治疗和药物反应,我们只关注评估组织学中完全反应的可预测性,并将包括部分反应在内的其他结果分类为阴性。
CPTAC 基因组生物标志物谱¶
使用 GDC¶
API 收集分子分析数据。我们重点关注“单核苷酸变异”(SNV)文件,其中包含与单个 DNA 碱基的替换或插入和删除相关的信息。使用 GDC API 的 files 端点检索 SNV 文件,筛选条件为:files.data_category=[“Simple Nucleotide Variation”],files.data_type=[“Masked Somatic Mutation”] 和 files.experimental_strategy=[“WXS”]。获取 SNV 文件后,可以获得 CPTAC 数据集中每个样本的 SNV 基因突变列表。由于突变数据基于全外显子测序,如果给定样本的 SNV 文件中没有列出某个基因,则认为该基因为野生型。最后,生物标志物谱被限制为仅包含驱动基因(参见获取可操作的驱动基因)。如果样本包含至少一个驱动基因的突变,则认为该样本为阳性。结果谱被过滤以排除在给定数据集中阳性样本少于十个的基因。
实验设置¶
我们在 3 折交叉验证设置中评估每个生物标志物的预测性能,其中每个数据集中具有有效生物标志物状态的病例被随机分成三个分区(折),每个分区大约具有相同比例的阳性样本。我们为每个生物标志物训练和测试三个模型,每次保留不同的折进行验证,并使用其余的进行训练。此设置确保了可多次在不同的(保留)验证集上评估可预测性,从而使我们能够评估模型性能的变异性。图像在患者级别分区,因此任何患者都不能出现在多个折中。阳性患者少于十个的生物标志物谱被排除在研究之外。
预处理流程和训练细节¶
卷积神经网络(CNN)用于从 H&E 图像预测分子谱,如图 1 所示。为每个生物标志物和折端到端从头开始训练单个 CNN,共产生 12,093 个独特模型,用于获得本研究中展示的结果。每个模型在从 H&E 染色的 WSIs 中获取的一组 256×256 瓦片上训练,考虑到整个组织材料。标准偏差过滤器用于消除不包含任何相关信息的瓦片,使我们能够从图像的其余部分提取组织。如果过滤过程后图像包含的瓦片少于十个,则该图像被丢弃。对剩余的瓦片应用 Macenko 颜色和亮度归一化【38】,然后分配一个真实的分子谱。
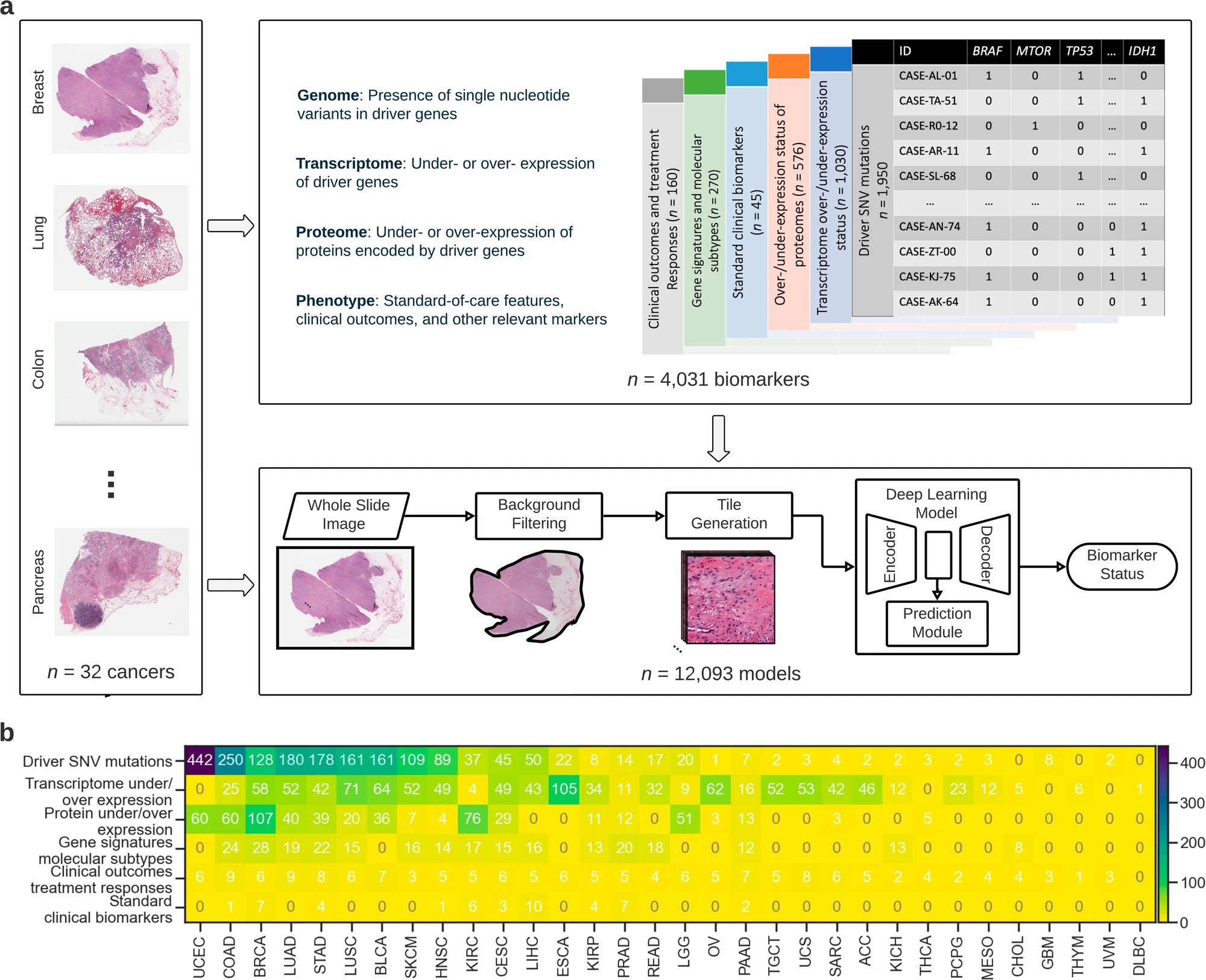
a. 预处理和训练流程概述 图示展示了用于评估从 H&E 染色全切片图像中通过深度学习预测多种基因组、转录组和蛋白质组生物标志物以及各种临床相关生物标志物(例如,标准护理特征、分子亚型、临床结果、治疗反应)的可行性的预处理和训练流程。使用了一个卷积神经网络(CNN),该网络包括编码器(即特征提取器)、解码器和分类模块,用于从 H&E 图像预测分子谱(参见方法:预处理流程和训练细节)。为每个生物标志物从头开始端到端训练单个 CNN 模型。每张切片被分割成一组 256×256 的瓦片,并自动丢弃不包含任何组织的瓦片。剩余的瓦片被分配一个真实的分子谱(参见方法:生物标志物获取)。 b. 每种癌症类型的生物标志物数量 以热图形式显示每种癌症类型的生物标志物数量。生物标志物按组学类别和癌症类型分组。SNV 指单核苷酸变异。癌症缩写定义见补充表 1。
卷积神经网络(CNN)模型架构和训练流程¶
该 CNN 模型由特征提取器(编码器)、解码器和分类模块组成。编码器通过在不同深度层应用卷积滤波器捕捉瓦片内的组织属性,有效地将高级视觉特征编码为 d 维特征向量,其中 d 取决于 CNN 的架构。这些向量被视为瓦片的指纹,提交给解码器和分类模块。解码器模块将 d 维嵌入作为输入,并返回与嵌入表示的原始瓦片形状相同的输出。它由一系列转置卷积和上采样层组成,用于从潜在向量重建原始瓦片,以实现每个不包含无关特征的瓦片的更好表示。解码器的输出与原始瓦片进行比较,使用均方误差(MSE)或重建损失。同时,编码器的输出,即 d 维特征向量,提交给分类模块,该模块由具有 softmax 非线性的全连接层组成,并执行实际的分类任务。该模块的输出,即分类概率,与 WSI 相关的真实标签进行比较,并产生交叉熵(CE)损失。CE 损失最终加上 MSE 损失以获得总损失。通过在该组合损失函数上反向传播,我们训练模型以输出更接近瓦片级目标的分类分数,同时实现独立于图像噪声(即无关特征)的每个瓦片的表示。
模型超参数和 CNN 架构是基于一个为 BRCA 分子谱分析开发的 DL 模型的临床验证研究中的相关基准分析确定的。我们采用了最佳性能模型的特征提取网络(基于“resnet34”架构)和超参数,为当前泛癌研究中的每个生物标志物配置 CNN 模型。最近的一项基准研究也进一步支持了我们的模型选择,该研究比较了计算病理学中用于生物标志物分析的弱监督 DL 方法,其中基于瓦片的 DL 模型在分类任务中可能优于新架构(如基于多实例学习的架构)。每个模型使用 Adam 优化器以 0.0001 的学习率训练 10 个 epoch。从每张训练切片中随机采样 200 个瓦片,并对来自未表示类的瓦片进行过采样,以确保在训练期间每个类的大致 50-50 的表示。在验证期间,平均所有瓦片的预测结果以确定切片级预测。训练期间,以验证 AUC 作为选择最终模型的目标指标。平均同一患者的图像分类分数以计算患者级的 AUC 值。
肿瘤纯度实验¶
TCGA 肿瘤纯度数据¶
我们从名为“nationwidechildrens.orgbiospecimen_slide${study_name}.txt”的生物样本切片文件中获取肿瘤成分数据。这些文件是 TCGA 元数据的一部分,可以从 TCGA GDC 门户 下载。我们提取了“percent_tumor_cells”属性,该属性测量组织图像中肿瘤细胞的比例,并将其用作肿瘤纯度的估计值。对于有多个组织切片的病例,通过平均相应的“percent_tumor_cells”测量值计算单个肿瘤纯度值。缺少此属性的病例被排除在肿瘤纯度实验之外。
实验细节¶
为了评估肿瘤纯度预测终点的能力(补充图 1),我们采用了用于 H&E 驱动的生物标志物分析的实验设置。然而,代替基于图像的特征预测(XH&E),我们选择使用肿瘤纯度(XTP)来确定生物标志物状态(y)。肿瘤纯度定义为组织切片中肿瘤细胞的百分比,该数据在 TCGA 元数据中提供(参见 TCGA 肿瘤纯度数据获取详细信息)。使用相同的 3 折交叉验证设置(参见实验设置),我们为每个生物标志物训练和测试三个随机森林(fRF)分类器,每次保留一个不同的折进行验证,并使用剩余的折进行训练。总共训练了 12,093 个 fRF 分类器,跨越 4031 个不同的终点(生物标志物)和 32 种癌症类型,使用 AUC 指标评估分类性能。
按照相同的实验设置建立了所有研究生物标志物的图像驱动 DL 模型(fDL)和基于肿瘤纯度的分类器(fRF)之间的一对一对应关系。这便于直接比较定义为 ƒDL(XH&E) = y 和 ƒRF(XTP) = y 的预测任务,其中 XTP 和 XH&E 表示分类器的输入,y 对应生物标志物状态。使用双侧 t 检验评估 fDL 和 fRF 分类器性能之间差异的显著性(参见性能特征和统计程序)。该比较分别对每个生物标志物子组进行,以提供更高的粒度。
此外,我们还探讨了肿瘤纯度与可预测性(由模型性能指示)之间的线性关系。肿瘤纯度的百分比在每个生物标志物的样本中取平均值,并使用皮尔逊相关系数(PCC)与 DL 模型的 AUC 值进行比较。这种相关性分析分别对每个生物标志物子组进行。
性能特征和统计程序¶
模型性能通过受试者工作特征曲线下面积(AUC)衡量,该曲线绘制了不同预测阈值下的真阳性率和假阳性率之间的关系。AUC 为 0.5 表示随机模型,而能够正确预测所有样本的完美模型则产生 AUC 为 1。对于每个生物标志物,我们报告了三个模型的平均 AUC 性能(除非另有说明),并附有标准偏差(适当时用 ± 表示)。
使用 python scipy-stats 库中的“ttest_ind”函数通过双侧 t 检验确定结果的统计显著性。这是一个零假设检验,假设两个独立样本具有相同的预期值和方差。为了测试组级的显著性,测量的 AUC 值与具有相同底层方差的随机值集合进行比较。例如,为了测量特定癌症和生物标志物类型的 AUC 值差异的统计显著性,将该组中的所有 AUC 值与从均值为 0.5(类似于随机性能)的分布中随机采样的一组 AUC 值进行比较,并比较组的标准偏差。为了确定生物标志物级的可预测性的统计显著性,我们在真实的阴性和阳性病例中获得的预测分数上应用相同的检验。为此,回顾性地结合了一个生物标志物的所有三个折的分类分数,并将阳性病例的分数与阴性类的分数进行比较,以双侧 t 检验确定阴性和阳性预测之间的差异是否具有统计显著性。使用 Benjamini-Hochberg 程序以 0.05 的错误发现率(FDR)对结果的 p 值进行多重检验校正。所有调整后的 p 值小于 0.05 的生物标志物被视为具有统计显著性。
伦理监督¶
只使用了回顾性和公开的数据。作者在参与者招募中没有任何角色。因此,不需要伦理批准。
报告摘要¶
有关研究设计的进一步信息,请参见链接到本文的 Nature Portfolio 报告摘要。
结果¶
深度学习用于常规组织学图像的分子谱分析¶
我们使用卷积神经网络(CNN)从 H&E 图像中预测分子谱,如图 1a 所示,并在方法部分详细说明。我们的 CNN 由编码器(即特征提取器)、解码器和用于分类的谱分析模块组成。在传统的 CNN 中,从组织学图像获取的形态特征直接与目标分子谱相关联。在我们的方法中,结合分类和编码器 - 解码器架构可以学习更好的表示(即编码),这种表示能消除诸如图像噪声等无关特征。我们研究的生物标志物包括驱动基因的遗传改变、驱动基因和相关蛋白质的过表达和低表达、在临床管理中常规使用的已建立的生物标志物、临床结果(如总体生存期(OS)和治疗反应)、与预后和靶向治疗高度相关的生物标志物(包括分子亚型和基因表达特征)(方法:生物标志物获取)。我们在 3 折交叉验证设置中评估了每个生物标志物的预测性能,每个队列中具有有效生物标志物状态的病例被分成三个随机分区(折),每个分区具有相似比例的阳性/阴性样本。我们为每个生物标志物训练和测试了三个模型,每次保留一个不同的折进行测试,使用其余的进行训练。这种设置确保了每个患者可以获得测试预测,并允许我们评估生物标志物性能的差异。生物标志物的可预测性通过受试者工作特征曲线下面积(AUC)进行衡量。对于每个生物标志物,我们报告了三个模型的平均 AUC 性能(除非另有说明),并附有标准偏差(适当时用 ± 表示),以衡量标记物内的变异性。总共有 12,093 个模型跨越 4031 个不同的生物标志物和 32 种癌症类型进行训练,具体分类如下(图 1b):1950 个驱动基因中的 SNV 突变、1030 个转录组表达水平标志物、576 个蛋白质表达水平标志物、270 个基因特征和分子亚型标志物、160 个与临床结果和治疗反应相关的标志物和 45 个标准护理标志物。
使用 DL 预测常规组织学图像中的多组学生物标志物的泛癌症可预测性¶
我们评估了使用标准 H&E 染色的全切片图像的组织形态学特征来分析生物学上不同的生物标志物的整体可行性。50% 的模型的 AUC 达到或高于 0.644。25% 的模型的 AUC 大于或等于 0.719,5% 的模型的 AUC 超过 0.834。前 1% 的模型(n = 122)的 AUC 至少为 0.909(图 2a,b)。对于我们调查的大多数生物标志物,其可预测性大多一致,标准偏差小于 0.1 AUC,最小和最大性能之间的差异小于 0.2 AUC(图 2c)。调查的大多数生物标志物在所有组学/标志物类型中表现出优于随机的性能(图 2d,表 1)。在预测驱动基因中的 SNV 方面,平均表现最低(AUC 0.636 ± 0.117),而标准临床生物标志物的表现最高(AUC 0.742 ± 0.120)。相同生物标志物类型的不同模型之间的变异性显示出与总体分布相似的趋势,所有组学的标准偏差约为 0.2 AUC,随着最小 - 最大性能范围的增加,方差增加(图 2e)。
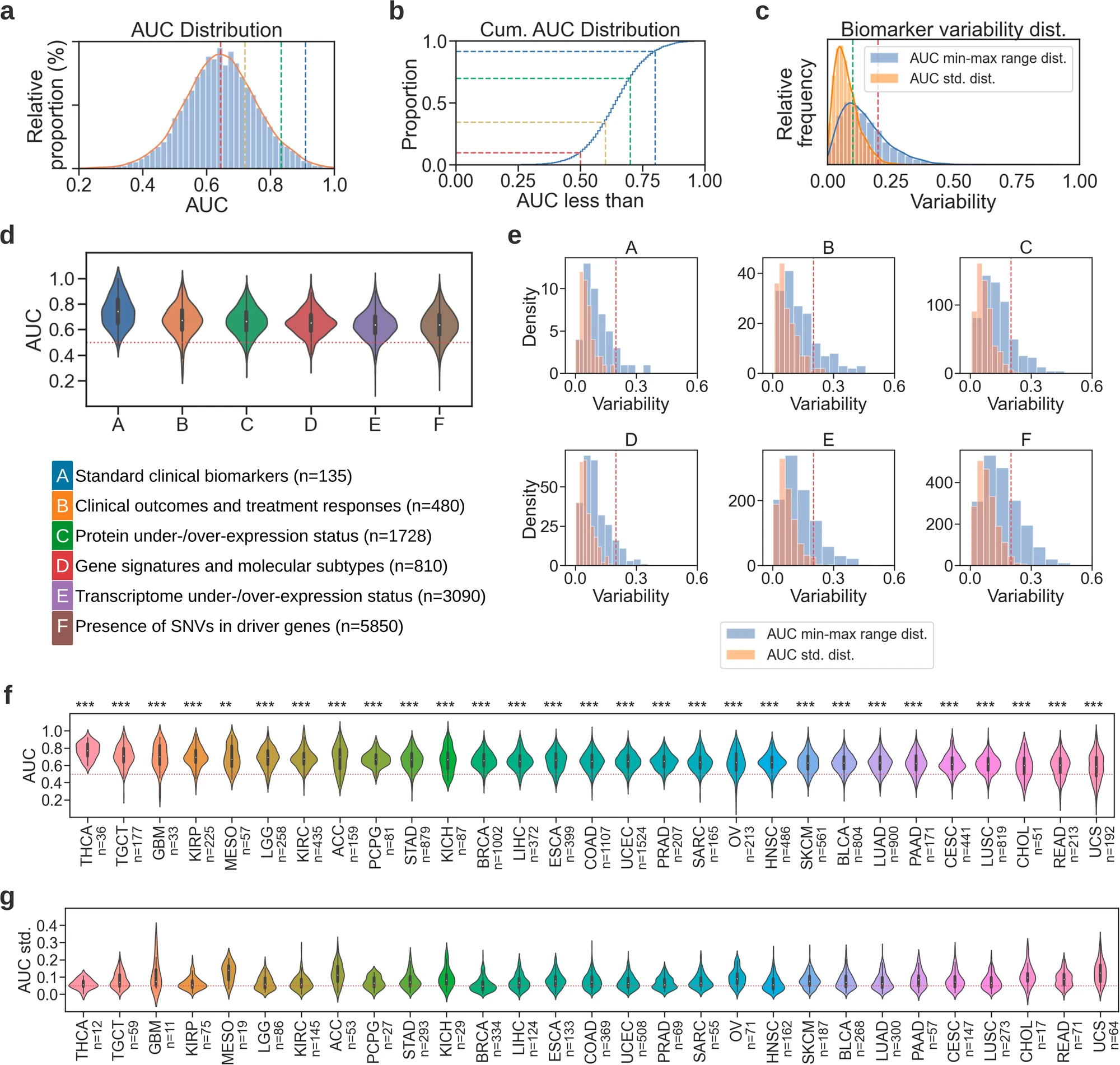
a. 所有模型 (n = 12,093) 的曲线下面积 (AUC) 值的直方图分布和核密度估计,其中标记表示模型在 50%、75%、95% 和 99% 的比例。 b. 累积 AUC 分布显示 AUC 小于所示标记(0.5、0.6、0.7 和 0.8)的模型比例。 c. 模型性能在 AUC 中的标准差和最小 - 最大范围分布。 d. 小提琴图显示每种生物标志物类型的 AUC 分布。小提琴图中使用了表 1 中的相同编码来缩写生物标志物类型。箱线图用于表示小提琴图中的数据点,须线表示 1.5 倍四分位距,白点表示中位数值。 e. 在交叉验证折中的模型性能的标准差(橙色直方图)和最小 - 最大范围分布(蓝色直方图)。 f. 小提琴图显示每种癌症类型的 AUC 分布(参见补充表 1 中的癌症缩写)。图表按研究内平均 AUC 排序。由于在所有生物标志物类型中仅有一到七个有效目标,淋巴瘤弥漫性大 B 细胞淋巴瘤 (DLBC)、脉络膜黑色素瘤 (UVM) 和胸腺瘤 (THYM) 被排除在外。每种癌症类型的模型数量在括号中给出。 g. 小提琴图显示每种生物标志物在不同折中的模型性能标准差,顺序与 (f) 中的相同。每种癌症类型的标准差值数量在括号中给出。
为了更细致地展示各癌种中生物标志物性能,我们绘制了所有具有足够样本量的恶性肿瘤的 AUC 值分布图(图 2f),并在补充表 4 中提供了平均性能和标准差。总体而言,所有癌种的表现均显著优于随机模型(即平均 AUC > 0.5 且 p < 1e-05,其中统计显著性通过对比癌种所有模型的 AUC 值与一组均值为 0.5 且标准差相同的随机 AUC 值的双侧 t 检验确定)。最低的整体性能出现在 UCS 中,平均 AUC 为 0.585(±0.158),最高的模型出现在 THCA 中,平均 AUC 为 0.768(±0.091)。每个生物标志物在交叉验证折中的变异性大多稳定,标准差集中在 0.05 AUC 左右(图 2g)。每种恶性肿瘤的总体可预测性性能按生物标志物类型的细分见补充图 2。
从组织学预测遗传变异的可行性¶
最近的泛癌研究表明,可以通过 DL 从组织形态特征中检测到突变 11,14。在我们的研究中,我们将之前的预测突变状态的工作扩展到 1950 个基因组生物标志物。我们专注于预测与 FDA 批准的特定疾病治疗相关的驱动基因中的 SNVs,或根据临床指南、强有力的研究证据和专家共识已知对特定治疗有重要意义的基因 31。在我们的实验中,我们使用了 TCGA 项目提供的基因组数据。
在大多数研究的癌种中,遗传变异均显著可预测(图 3a),平均 AUC 为 0.636(±0.117)。超过 40% 的突变可以达到至少 0.65 的 AUC,考虑每种癌症类型中表现最好的突变,几乎所有主要恶性肿瘤都有至少 10 个基因的突变可达到 0.70 或更高的 AUC。其中,子宫内膜癌具有最多的可预测突变状态(在 442 个基因中有 112 个)。其次是结肠癌(在 250 个基因中有 62 个)、胃癌(在 178 个基因中有 58 个)、皮肤黑色素瘤(在 109 个基因中有 29 个)、LUAD(在 180 个基因中有 28 个)和 BRCA(在 128 个基因中有 26 个)。在所有测试的基因中,表现最好的基因是 KIRC 中的 NUMA1 和 JAK1,肺癌中的 PDGFRB 和 BCL6,子宫内膜癌中的 IRS2 和 BRCA 中的 GNAS,每个基因的 AUC 均至少为 0.89。许多基因在多种癌症类型中高度可预测(补充图 3a)。特别是,TP53 的 SNV 变异在许多恶性肿瘤中可检测到,22 种测试癌症中有 7 种 AUC 至少为 0.7,有 14 种 AUC 超过 0.65,最高达 0.841(脑 LGG),BRCA 达 0.785,子宫内膜癌达 0.771。
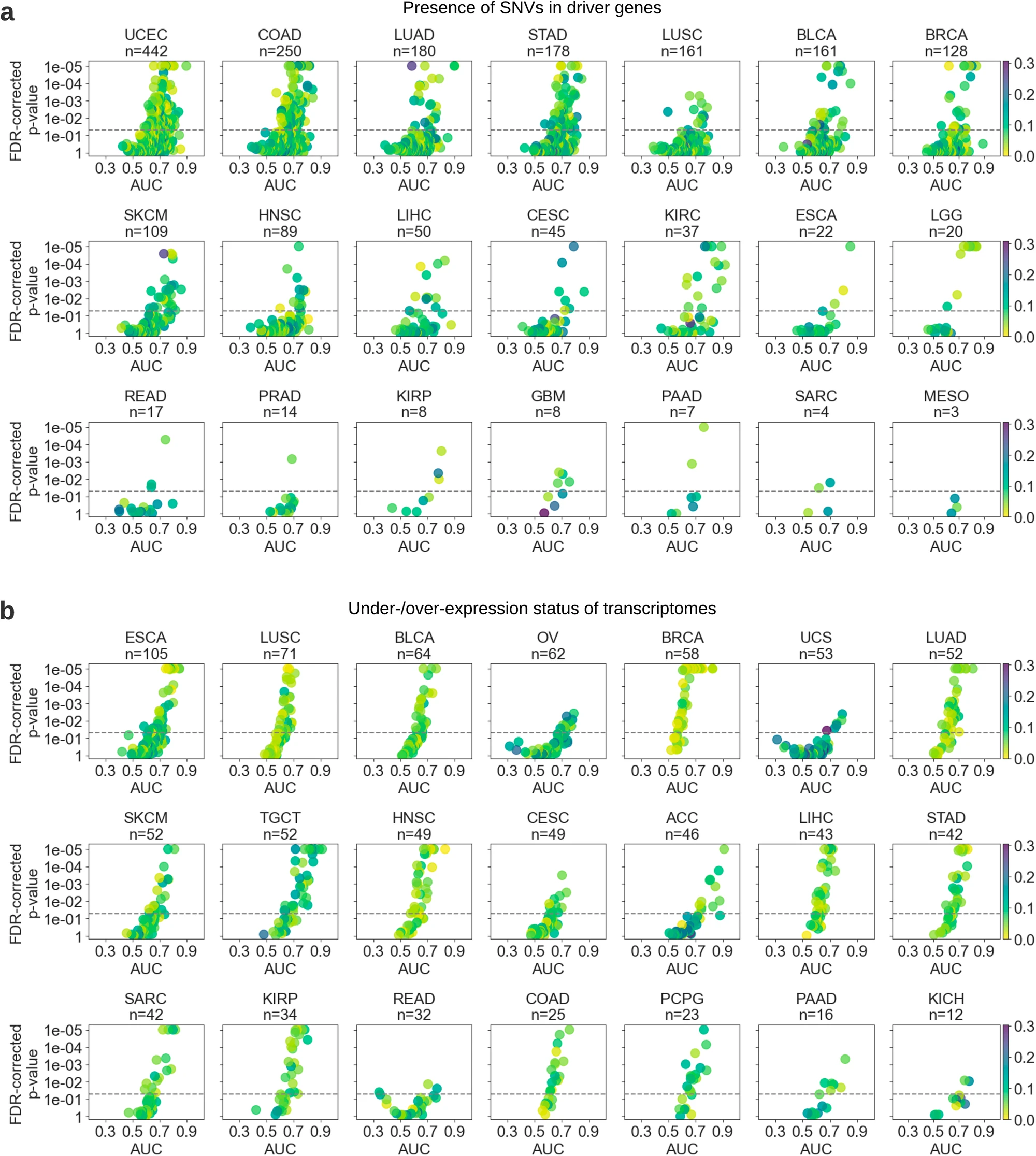
散点图展示了每个模型预测的平均测试 AUC,用于预测 (a) 驱动基因中单核苷酸变异(SNVs)的存在和 (b) 跨选定癌症类型的转录组低/过度表达状态。每个散点图中的标记代表一个测试的生物标志物。对每个模型的预测分数应用双侧 t 检验,以评估统计显著性,并对相应的 p 值进行错误发现率(FDR)校正。每个图的 y 轴反转,且 p 值经过对数变换以便可视化。p 值小于 1e-05 的设置为 1e-05,以避免转换过程中的数值误差。统计显著性阈值 0.05 用虚线标出。标记的颜色阴影表示每个生物标志物的可预测性能的标准差。图按每行测试的生物标志物数量排序。由于空间限制,本图未展示具有较少生物标志物的癌症类型,但在补充图 9 中提供。癌症缩写定义见补充表 1。
从诊断组织切片推断转录组过表达/低表达的可行性¶
对基因表达的分析对更好地理解癌症机制至关重要,并有望改善癌症诊断和促进药物发现 42。虽然已知基因突变可以通过深度学习(DL)从组织形态学中检测到,但对于转录组和蛋白质水平的预测能力的研究相对有限。最近的研究表明,转录组谱与 DL 检测的组织形态学特征相关,并且无需注释 15。我们的研究采用了更直接和全面的方法,训练 DL 模型预测选定驱动基因的过表达和/或低表达状态,使用来自 TCGA 的转录组谱。我们确定了 97 个基因用于研究低表达的可预测性,933 个基因用于研究不同癌症中转录水平的过表达可预测性。
在大多数癌症类型中,所研究基因的过表达/低表达状态大多是可预测的(图 3b),平均 AUC 为 0.637(±0.108)。低表达基因的平均表现稍低(平均 AUC 0.633 ± 0.115)。至少 40%的基因的表达状态可预测性达到或超过 0.65 AUC。食管癌(ESCA)和睾丸癌是具有最多可预测表达状态基因的恶性肿瘤(分别总计 105 个和 52 个基因),定义为 AUC 水平至少为 0.70。其次是卵巢癌(62 个基因中有 18 个)和肾上腺皮质癌(ACC,46 个基因中有 16 个)。几乎所有表现最好的基因都是过表达的,例如在胸腺瘤中的 PMS2;睾丸癌中的 CARD11、LASP1、STIL、POLE、KMT2C 和 CLIP1;ACC 中的 ERC1、WRN、OLIG2、FANCC 和 ACSL6;以及食管癌中的 SOX2 和 NDRG1,表现最好的 AUC 范围为 0.832-0.911。在低表达基因的可预测性中,最显著的是胸腺瘤中的 RHOA(AUC 0.908±0.05),脑低级别胶质瘤中的 LSM14A、THRAP3 和 MTOR(AUC 范围为 0.785 到 0.818)和间皮瘤中的 BAP1(AUC 0.818±0.084)。许多基因的表达状态在多种癌症类型中是可预测的(补充图 3b)。
使用 DL 预测蛋白质表达水平状态的可行性¶
在我们多组学泛癌研究的下一步中,我们评估了 DL 检测与蛋白质表达变化相关的组织形态学变化的能力。为此,我们训练了模型以预测基于 TCGA 提供的蛋白质组谱的某些驱动基因相关蛋白质的过表达和/或低表达状态。值得注意的是,在本文中,基因的关联指的是该基因编码的蛋白质,我们在整篇文章中交替使用关联和编码。总共有 267 个和 309 个驱动基因分别符合评估其相应蛋白质水平低表达和过表达状态的可预测性。
我们取得了平均 AUC 为 0.666(±0.107)的结果,低表达状态的平均可预测性略低(平均 AUC 0.662 ± 0.105)与其过表达状态相比(平均 AUC 0.669 ± 0.109)。几乎所有研究中的基因的表达状态预测都表现优于随机预测(图 4a),其中超过一半的基因可预测性达到或超过 0.65 AUC,超过 30% 的基因可预测性达到或超过 0.70 AUC。BRCA 的可预测性最高,其中 107 个基因中的 37 个基因的表达状态可预测性达到或超过 0.7 AUC。其次是肾透明细胞癌(KIRC)和低级别脑胶质瘤(分别为 51 个和 76 个基因)。大量由驱动基因编码的蛋白质表达水平状态高度可预测,例如低级别脑胶质瘤中的 TFRC、ATM 和 PIK3CA;乳头状肾细胞癌中的 NRAS、FOXO3、MYC 和 TP53;头颈癌中的 CDKN1B;和肉瘤中的 MYC,表现最佳的 AUC 范围为 0.835-0.974。多个低表达蛋白质也具有很大的可预测性,表现最好的包括 BRCA 中的 CASP8、MET、BCL2 和 SETD2;胃癌中的 AR 和 TFRC;以及肺癌中的 VHL,AUC 范围为 0.814-0.866。我们发现 TP53 编码的 p53 蛋白的过表达在测试的八种癌症中有六种是一致可预测的,包括肾细胞癌、低级别脑胶质瘤和子宫内膜癌,AUC 范围为 0.672 到 0.835。许多其他蛋白质的表达状态也可以在多种癌症类型中检测到(补充图 3c)。
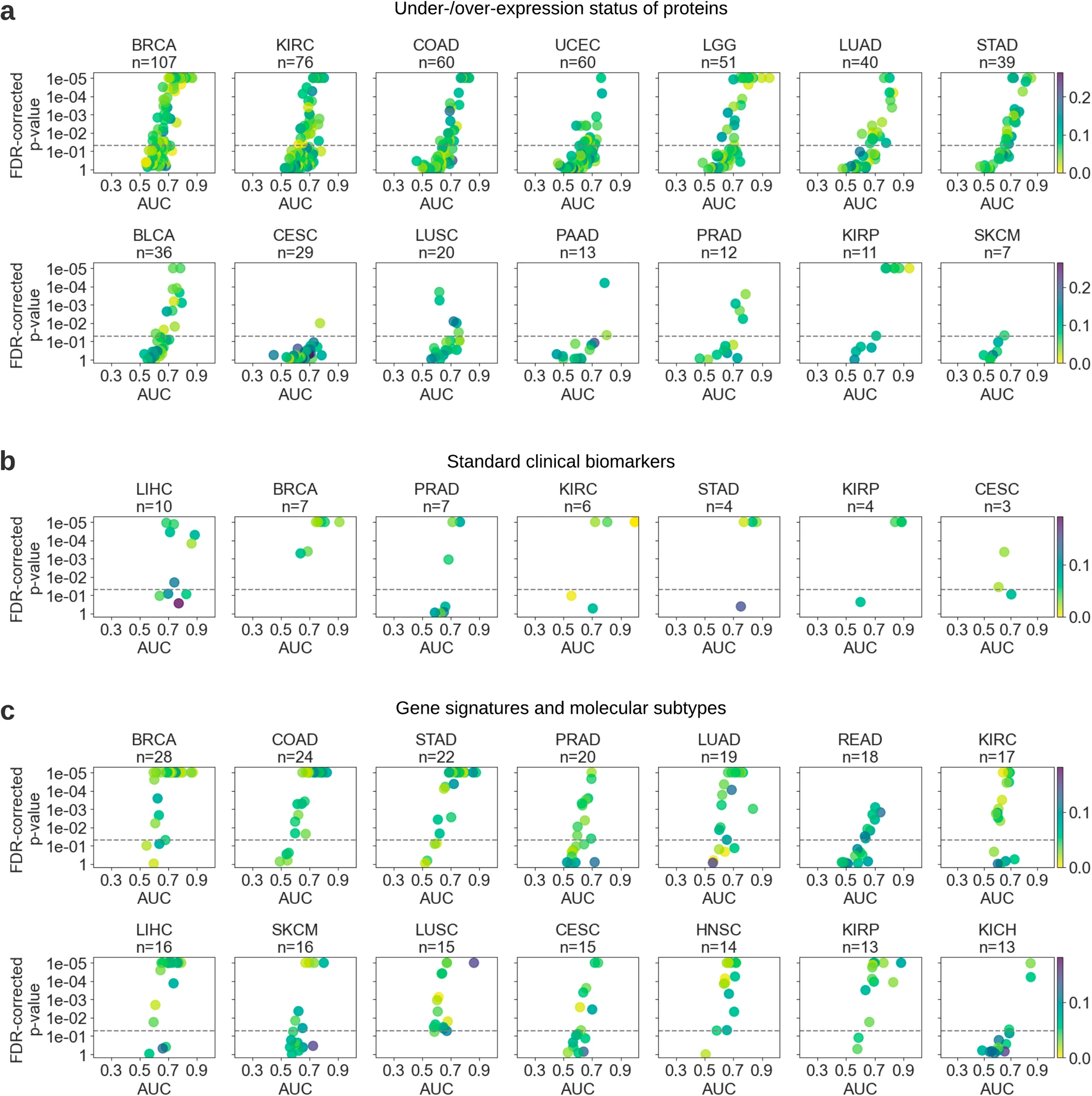
散点图显示了各模型在测试中预测的平均曲线下面积(AUC),用于预测以下项目: (a) 蛋白质的过表达/低表达状态; (b) 不同癌症类型的标准临床生物标志物; (c) 基因特征和分子亚型。
图表按每种癌症类型中测试的生物标志物数量排序。由于空间限制,生物标志物较少的癌症类型未在此图中显示,但在补充图 9 中提供。有关详细的可视化解释,请参阅图 3 的说明。
转录组和蛋白质组生物标志物的可预测性呈正相关¶
尽管我们调查的目标中蛋白质表达状态的平均可预测性高于转录组(表 1),但这两种组学类型都有大约 200 个高度可预测的基因(即 AUC > 0.7)。转录组表达预测的整体表现略低,可能是由于其内在的可预测性较蛋白质组差。考虑到分子景观中的可预测性变化(补充图 4),我们测量了转录组和蛋白质组生物标志物之间的正相关性,转录组过表达状态的 PCC 为 0.227(p < 1e–05)。基因突变和转录组低表达状态(PCC: 0.131, p < 0.01)以及其蛋白质组对应物(PCC: 0.068, p < 0.01)之间也存在正线性关系。
利用 DL 预测标准临床生物标志物的可行性¶
我们测试了 DL 预测临床管理中常用的已建立生物标志物的可行性。为此,我们通过参考文献 11 中的生物标志物获取方法,编制了一组标准护理特征,包括肿瘤分级、微卫星不稳定性(MSI)状态(在结直肠癌和胃癌中)、组织学亚型、激素受体状态(在 BRCA 中)和 Gleason 评分(在前列腺癌中)。
标准病理生物标志物表现出相对较高的可预测性,平均 AUC 为 0.742(±0.120)。没有一个生物标志物的表现比随机更差(即所有 AUC > 0.5),其中近 30% 的生物标志物的 AUC 超过 0.8,显示出高度可预测性(图 4b)。如预期,组织学亚型在总体上具有高度可预测性,尤其是对于 BRCA、肾细胞癌、肝细胞癌和胃癌。肾细胞癌的透明细胞和嗜酸性细胞亚型的分子特征预测表现最好,AUC 达到了 0.999。BRCA 中的浸润性导管癌(IDC)和浸润性小叶癌(ILC)亚型也能从 WSIs 中很好地检测到,AUC 范围为 0.759-0.908。我们的模型能够预测 BRCA 中的激素受体状态,雌激素(ER)和孕激素(PR)受体的 AUC 分别为 0.806 和 0.744。值得注意的是,多种对肝细胞癌重要的临床生物标志物也可以从组织学中准确推断出来,包括生长模式(AUC 达 0.862)和非酒精性脂肪肝病(NAFLD)的病因状态(AUC 0.826 ± 0.054)。另一个高度可预测的生物标志物是 MSI 状态,在结肠癌和胃癌中分别具有平均 AUC 为 0.716 和 0.773。
从常规图像推断分子亚型和基因表达特征的可行性¶
为了评估 DL 从 WSIs 检测癌症分子亚型和基因表达特征的能力,我们通过严格遵循前人研究的实验细节 11,编制了一组具有临床和/或生物学意义的特征。这些特征包括与预后和靶向治疗相关的分子亚型和簇、免疫相关基因表达、同源重组缺陷、细胞增殖、干扰素 -γ 信号传导和巨噬细胞调节以及超甲基化/突变 32,33,34。鉴于它们与较高层次功能的关联,这些生物标志物可能对形态学的影响比之前评估的变化更大,尤其是相比单一突变 11。
总体而言,分子亚型和基因特征具有相当可预测性,平均 AUC 为 0.653(±0.097)。其中几乎一半的生物标志物在 AUC 水平大于 0.65(图 4c)。BRCA(28 个生物标志物中有 18 个)和胃腺癌(22 个生物标志物中有 16 个)以及结肠腺癌(24 个生物标志物中有 14 个)中的许多生物标志物具有高 AUC。我们的方法能够在多种癌症类型中推断 TCGA 分子亚型,包括 KIRP(AUC 高达 0.884 ± 0.085)、胃癌(AUC 高达 0.875 ± 0.048)、LUSC(AUC 高达 0.861 ± 0.015)和 BRCA(AUC 高达 0.859 ± 0.028)。值得注意的是,BRCA 中 PAM50 亚型的平均 AUC 为 0.752(±0.080),其中 Basal 亚型达到了 0.871(±0.015)。结肠癌中的共识分子亚型(CMS1、CMS2、CMS3、CMS4)也有可能被检测到,亚型间平均 AUC 为 0.763(±0.068),CMS1 的 AUC 高达 0.821(±0.083)。细胞增殖和超甲基化在乳腺癌、胃癌、结肠癌和肺癌中特别是相对较好预测的生物标志物,AUC 高达 0.854。
从诊断组织切片推断临床结果和治疗反应的可行性¶
准确估计预后对于临床管理操作至关重要。以前的工作主要集中在基于常规临床数据、标准护理特征、组织病理评估、分子分析和更近期的通过 DL 获得的形态学特征来开发预后模型 43,44,45,46,47。也有尝试使用机器学习方法和基于图像的特征来预测不同癌症的临床终点,如黑色素瘤和非小细胞肺癌 48,49。在我们的研究中,我们通过将临床结果终点如 OS、DSS、DFI 和 PFI 视为潜在的预后生物标志物,探讨了直接从组织学中推断预后结果的端到端可预测性。我们进一步扩展了分析范围,评估了是否可以直接从 WSIs 检测治疗反应,以评估 DL 模型是否能够将形态学特征与治疗或药物的结果相关联。据我们所知,这项研究是第一次系统地评估跨多种癌症类型的药物反应的 DL 可预测性。
总体而言,临床结果和治疗反应的预测表现相当高,平均 AUC 为 0.671(±0.12)。几乎 40% 的测试目标在 AUC 水平达到 0.70 或以上(图 5)。我们在 GBM、ACC 和 KICH 中获得了最好的总体表现,平均 AUC 为 0.77。它们之后是肾乳头状细胞癌、MESO、THCA、前列腺癌、肾透明细胞癌和食管癌,总体 AUC 范围为 0.731 到 0.76。KICH、GBM、THCA 和 ACC 中的 OS;食管癌和肾透明细胞癌中的 DFI;GBM 中的 DSS;子宫内膜癌中的残余肿瘤状态;以及乳头状肾细胞癌中的治疗反应是表现最佳的目标,AUC 范围为 0.815 到 0.924。
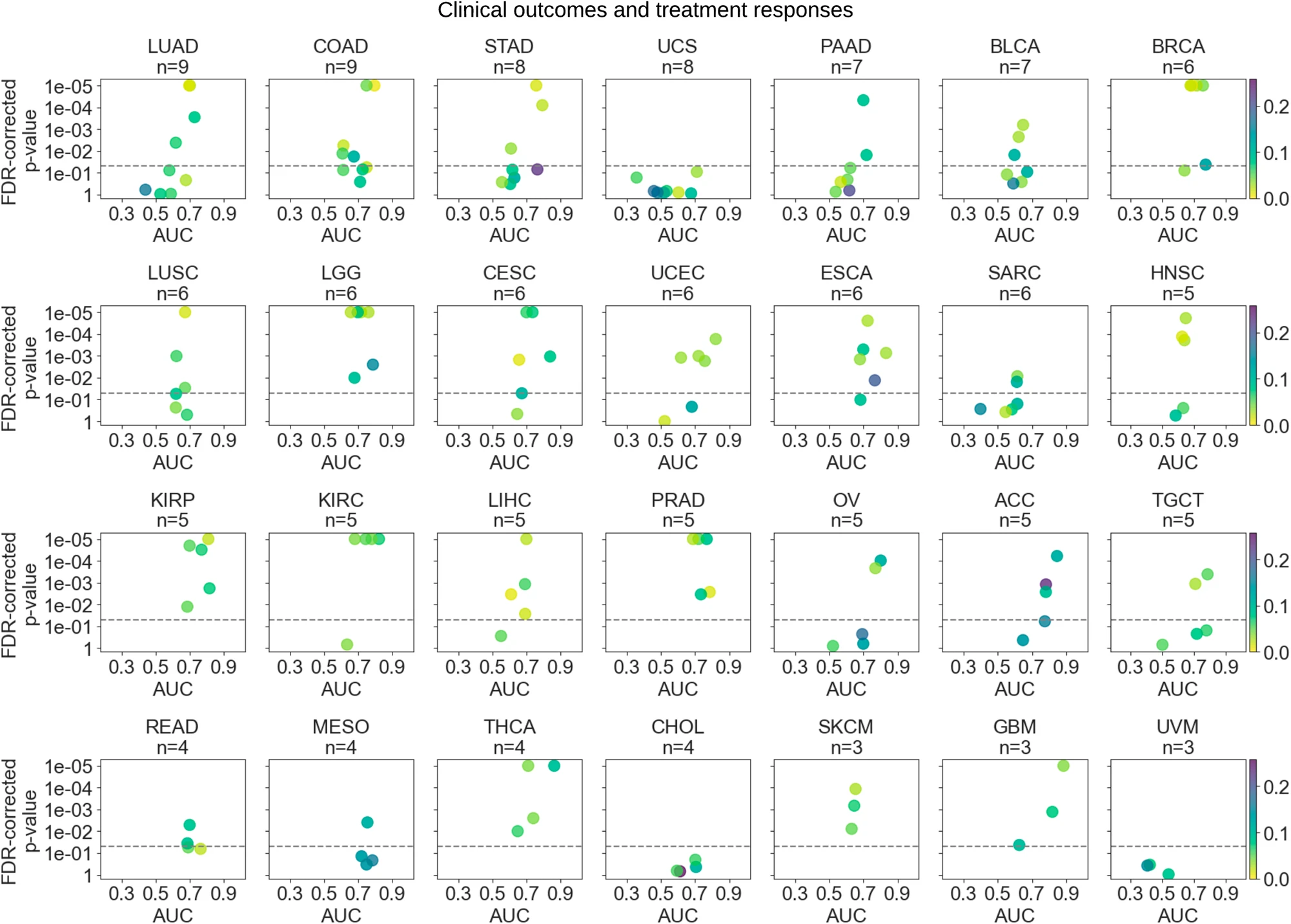
散点图显示了在各选定癌症类型中,每个模型预测临床结果和治疗反应的平均测试 AUC。图表按每种癌症类型中测试的生物标志物数量排序。由于空间限制,生物标志物较少的癌症类型未在此图中显示,但在补充图 9 中提供。请参阅图 3 的说明以获取详细的可视化解释。
在我们研究的 20 种药物中,深度学习(DL)能够在其中一半的药物中预测响应,AUC 至少为 0.7。顺铂是最显著的药物,在宫颈癌、睾丸癌和胃癌中的 AUC 范围为 0.763–0.837。其他可预测的药物包括用于 LGG 的替莫唑胺,用于 BRCA 的紫杉醇,用于结肠癌的亚叶酸、奥沙利铂和氟尿嘧啶,用于睾丸癌的依托泊苷,以及用于胰腺癌的吉西他滨。
不同数据集之间的泛癌症预测一致性¶
为了展示在不同数据集之间可以实现的可比性能,我们使用 CPTAC 的公开数据重复了我们的实验。由于 TCGA 和 CPTAC 依赖于同一组驱动基因,因此我们将分析范围限制为预测驱动基因中的 SNV。我们确定了在两个数据集中都有突变数据的 176 个驱动基因,这些基因跨越了七种癌症类型。所研究的癌症包括子宫内膜癌(n = 61),PDA(n = 4),LUSC(n = 21),LUAD(n = 14),头颈癌(n = 12),GBM(n = 4),和结肠腺癌(n = 60)。几乎所有测试的癌症类型在两个数据集上的整体性能相当(补充图 5 中,除了 COAD 外,所有其他癌症的 p > 0.05,双侧 t 检验),在每种癌症类型内的平均 AUC 范围为 0.578–0.655(TCGA)和 0.567–0.672(CPTAC)。CPTAC 队列的生物标志物之间的性能变化更大,这从小提琴图的形状可以看出。总体而言,我们的结果表明生物标志物的预测性是一致的且独立于数据集。
讨论¶
本研究从泛癌症的角度评估了使用 DL 和从 H&E 染色的诊断切片中提取的组织形态特征预测大量生物标志物的总体可行性。通过编码 - 解码 DL 模型从组织学中捕捉的形态学特征用于推断跨越多组学谱系的生物标志物。我们观察到某些基因突变(例如 TP53)和在多个癌症类型中的临床相关标志物(例如标准护理特征和分子亚型)具有显著的性能。生物标志物级别的性能表现出一定程度的一致性,因为一个生物标志物的交叉验证模型通常表现相似。当在一个独立的数据集上重复实验时,我们获得了相当的预测性能,进一步显示了 DL 在多个癌症类型中进行分子谱分析的总体能力。
生物标志物的性能似乎不依赖于测试人群的内在因素,例如病例数量(即人口规模)和全人群中阳性 - 阴性样本的比例(补充图 6a,补充图 7)。我们认为数据集的大小对结果的稳定性更为重要,而不是性能本身,即拥有更大的训练子集可能会产生性能更一致的模型。此外,我们的生物标志物通常具有不平衡的分布,因为大多数生物标志物的阳性病例数量远小于阴性病例数量。在训练时,我们通过从欠代表的类别中进行过采样来解决这个问题,并在稍后评估其对性能稳定性的影响。通过将样本数量和类别比率与每个生物标志物的交叉验证折叠中的 AUC 范围(通过标准偏差衡量)进行比较,我们的观察结果显示了这两个因素之间的负相关关系(补充图 6b)。这可能表明随着样本数量的增加和数据集的更加平衡,性能可能会变得更不变。此外,我们进行了一个实验以评估生物标志物可预测性与肿瘤纯度之间的关系(补充图 1,方法:肿瘤纯度实验)。我们的分析表明,仅根据肿瘤纯度作为独立变量预测生物标志物状态是不可行的。然而,我们观察到某些生物标志物的预测性能与肿瘤纯度之间存在一定的相关性。这种相关性可能归因于较大的肿瘤组成倾向于在某种程度上提高性能。
我们的研究发现,在大多数研究的基因中,从组织学检测不同层次的组学景观的变化在很大程度上是可行的。某些可能与不良临床结果相关的突变在多种癌症类型中表现出良好的性能,显著的例子包括 TP53、BAP1、MTOR 和 GNAS。TP53 表现出的相对较高且一致的性能可能归因于具有 TP53 突变的肿瘤可能分化不良,并表现出明显的高等级细胞变化 15。识别具有某些变异的患者对于精准治疗至关重要,并且可以为开发靶向疗法铺平道路。例如,最近的研究表明,检测 BAP1 突变可能对开发 KIRC 的靶向治疗策略有潜在用处 50。类似地,MTOR 突变可以作为预测肿瘤对 mTOR 抑制剂反应的生物标志物,这些抑制剂已经用于治疗人类癌症 51。一个高度预测的基因 GNAS 在高表达时被认为在 BRCA 中促进细胞增殖和迁移,因此,可能被用作治疗靶点 52。
识别肿瘤中的下游变化及其与正常细胞的区分有助于揭示影响抗癌药物反应的复杂机制,并可能增强对治疗结果的预测 53,54,55,56。尽管有许多研究针对肿瘤代谢,但评估从 WSIs 中检测转录组和蛋白质组变化的尝试非常有限 15,57,58。在我们的研究中,我们观察到在某种程度上预测转录组和蛋白质的下 -/过表达状态的可能性。例如,我们发现 p53 过表达在多种癌症类型中具有一致的可预测性。
我们的研究结果表明,DL 可以从组织病理学图像中推断出已知的标准护理临床生物标志物、基因表达特征和分子亚型。这些高度可预测的靶点中的一些已经被采用为临床实践中的可操作生物标志物。这种相关性是合理的,因为从根本上影响肿瘤生物学的改变也通常伴随着形态的变化,使这些生物标志物成为有效治疗策略的有价值靶点。我们的发现在很大程度上与最近由 ref. 12 进行的泛癌症研究所得结果一致。两项研究之间的相似性使我们能够证实他们的发现,而我们的结果提供了更多证据,证明从组织学中检测分子生物标志物的可行性。
残留肿瘤的分类是治疗过程中的关键阶段,被认为是一个重要的预后生物标志物 36。在我们的分析中,我们发现 DL 可能能够在多种癌症中检测到残留肿瘤的发生(或缺乏),这可能表明在诊断时,某些与治疗后完全缓解相关的视觉线索可能存在于组织形态中。虽然其他临床结果,如 OS 和疾病特异性生存(DSS)在一定程度上也是可预测的,但需要注意的是,临床结果的定义可能并不总是准确的,尤其是对于需要较长随访时间、队列规模较小或事件数量有限的癌症类型 35。本研究评估了从 H&E 染色图像中预测药物反应的可行性,结果表明,某些药物如顺铂、替莫唑胺和紫杉醇的完全反应可能通过 DL 检测出来。这些发现表明,DL 方法在精准医学方面具有前景,能够通过分析常规组织学切片帮助肿瘤学家选择可能对患者最有效的治疗方案。
我们的研究存在一些限制,为了保持研究范围的可控性,我们对生物标志物获取设定了特定的限制。例如,我们将多组学生物标志物配置计算限制为驱动基因,并在考虑基因组变异时将分析范围限制为单一变异突变。因此,比对生物学上不同的靶标的可预测性是不可能的,因为分析只集中在部分变异上,而不是检查所有可能的变异。研究的另一个限制直接来自数据本身。虽然我们比较了多种癌症和组学类型的可预测性,但所有生物标志物在不同的样本大小和流行率条件下进行了测试。许多潜在的生物标志物在数据获取期间被简单地丢弃,因为它们没有包含足够的阳性样本。需要承认的是,TCGA 的数字切片中固有的特定于站点的指纹可能会偏倚预测模型的准确性。为缓解这一担忧,可以考虑更复杂的分裂技术 59。我们使用 AUC 评估模型性能,这是在类别不平衡情况下最常用的评估指标。然而,在只有少数样本用于少数类别(如罕见突变)的场景中,AUC 值可能不太可靠。AUC 估计的性能可能会根据少数类别的正确和错误预测显著变化。在解释非常小样本和低流行率的生物标志物结果时应注意这一固有缺陷。所有模型的 AUC 值、类别流行率、验证样本大小、校正后的 p 值和其他相关信息在补充数据 1 中提供。
探索 DL 模型的黑盒表示可以有助于揭示可能与某些变异或表型结果相关的形态学模式 11,14。可视化推断生物标志物状态的空间区域的一种方法是将切片级预测分数(即概率)叠加到 WSIs 上以创建空间热图(图 6)。这些热图中的最高排名的切片代表了 DL 模型为解决当前预测任务而学习到的视觉特征。例如,不同类型的乳腺肿瘤可能显示出形态上的显著差异,这些差异可以通过 DL 识别,并用于区分特定亚型(图 6a,b)。同样,结肠癌 CMS 的最高排名切片(补充图 8a)显示出与之前研究显示的 CMS 亚类的组织病理学一致的形态模式 11,19。此外,与 MSI 不稳定病例相关的形态特征,例如包含大量肿瘤浸润淋巴细胞,可以在结肠和胃癌患者的最高排名 MSI 切片中看到(补充图 8b-c)。虽然 DL 可以识别临床相关的形态特征,但它也可能有助于追踪与分子变异相关的视觉模式。例如,在乳头状甲状腺癌中突变的 BRAF 病例中,高度预测的切片显示出与其野生型对照相比,明显不同的组织学特征(补充图 8d)。此外,DL 可能是探索与组织形态学未知联系的视觉特征的有用工具。例如,目前没有已知特征可以区分乳腺癌中突变的 TP53 和其野生型,但 DL 仍然可以选择显示两类不同视觉特征的肿瘤切片(补充图 8e)。这可能为更好地理解这种变异对癌细胞形态的潜在影响提供了见解。
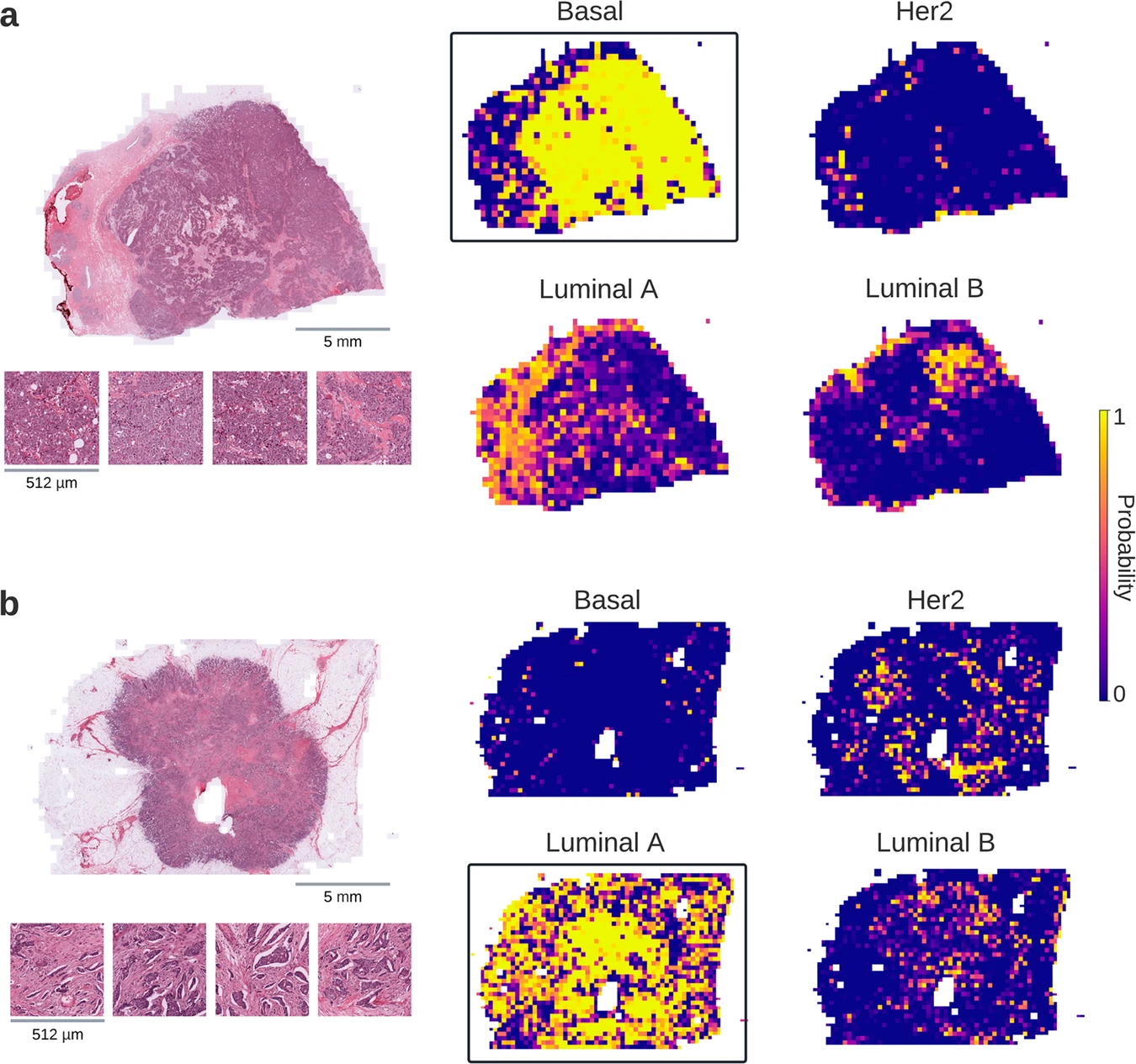
使用深度学习(DL)对乳腺癌分子亚型(即 Basal、HER2、Luminal A 和 Luminal B)的预测结果可视化为两个选定患者的热图。在每个病例中,正确预测的亚型用矩形框住,并将该类中的最高排名的切片与原始全切片图像(WSI)一起显示。Basal 型(a)显示没有任何可辨别腺体形成的肿瘤细胞片状区域,而 Luminal A 患者的肿瘤(b)由形成良好的腺体组成。考虑到两种情况下的热图,可以注意到 DL 模型能够识别与目标类别相关的空间区域。WSIs 的比例尺:5 mm。切片的比例尺:512 µm。
展示通过深度学习(DL)预测多组学生物标志物的整体可行性,标志着实现从组织学进行端到端检测系统的重要一步,这可能有助于临床医生的患者管理,加快诊断,并帮助开发更以患者为中心的治疗方法。该方法还提供了在快速和低资源环境中应用多组学生物标志物的机会,无需耗时且昂贵的生物测试。虽然本研究阐明了决定生物标志物可预测性因素的早期观察,但在日常临床设置中采用基于 DL 的方法进行多组学生物标志物分析之前,还需要进一步理解这些因素 。
系统分析模型的普遍性对于理解可预测性的真正范围至关重要。此外,还需要进一步研究生物标志物可检测性的具体机制。本研究结果为识别具有临床应用潜力的生物标志物以及需要重新评估的生物标志物提供了机会。例如,对于 AUC 较低的生物标志物,可以使用更大的数据集和定制模型重新训练,以确认其表现不佳是由于生物信号有限还是由于模型配置不佳无法捕捉这些信号。相反,AUC 较高的生物标志物可以进行严格验证,以进一步评估其临床应用的紧迫性。这可能包括在多个外部数据集上的评估、与标准护理方法的比较、与治疗反应的一致性、前瞻性评估以及临床实施的成本效益分析,考虑潜在的收益和资源影响 。
未来的工作将探索预测模型的内部表示,以揭示组学、肿瘤形态和模型预测之间的潜在关联。这一探索可能有助于理解不同组学之间可预测性表现的差异。虽然本研究将从 H&E 染色图像中预测生物标志物状态的问题表述为单任务分类问题,但未来的研究应探讨明确的多任务方法。考虑到某些生物标志物可能相互关联,采用这些方法可能会潜在地提高可预测性表现。
数据可用性¶
TCGA 全切片图像可在 https://portal.gdc.cancer.gov/获取。用于生成 TCGA 队列中病例的生物标志物谱的遗传、转录组、蛋白质组和临床数据可在 https://portal.gdc.cancer.gov/和 https://cbioportal.org/获取。临床相关驱动基因可在 https://cancervariants.org/获取。CPTAC 全切片图像可在 https://wiki.cancerimagingarchive.net/display/Public/CPTAC+Imaging+Proteomics/获取。用于生成 CPTAC 队列中病例的生物标志物谱的遗传数据可在 https://portal.gdc.cancer.gov/获取。用于生成图形的源数据,包括 AUC 值、类的普遍性、验证样本大小、校正的 p 值和本研究中评估模型的其他相关信息,见补充数据 1。
代码可用性¶
用于分析的代码可在以下 URL 获取:https://github.com/Panakeia-Technologies/Multiomics-PANCancer (https://doi.org/10.5281/zenodo.6566146) 。本工作中的实验和分析是使用 python 3.6 和以下 python 库进行的:torch (1.9.0), torchvision (0.10.0), google-cloud-storage (1.32.0), openslide-python (1.1.1), pillow (6.0.0), opencv (3.4.2), tensorboard (1.15.0), numpy (1.18.1), pandas (1.3.5), seaborn (0.11.0), scipy (1.4.1), scikit-learn (0.22.1), statsmodels (0.11.0) 和 matplotlib (3.5.1)。