Best practices for single cell analysis across modalities
摘要¶
单细胞技术的最新进展使得跨模态和位置的细胞高通量分子分析成为可能。单细胞转录组数据现在可以与染色质可及性、表面蛋白表达、自适应免疫受体库分析以及空间信息相结合。随着跨模态单细胞数据的可用性不断增加,促使了新型计算方法的发展,以帮助分析人员获得生物学洞见。随着该领域的发展,导航庞大的工具和分析步骤变得越来越困难。在本文中,我们总结了独立的单模态和多模态单细胞分析的基准研究,以建议最常见分析步骤的全面最佳实践工作流程。在没有独立基准的情况下,我们回顾并对比了流行的方法。我们的文章为单细胞(多)组学分析领域的新手提供了入门点,并为高级用户提供了最新的最佳实践指南。
引言¶
单细胞 RNA 测序(scRNA-seq)技术通过在前所未有的规模和分辨率下测量转录组谱,彻底改变了分子生物学。实验技术的进步推动了计算方法的大规模创新,目前已有超过 1400 种工具可用于分析 scRNA-seq 数据 1。计算框架和软件库,如 Bioconductor2、Seurat3 和 Scanpy4,以及方法基准和最佳实践工作流程 2,5,6,使数据分析人员能够在这一领域中导航并构建分析管道。这种实验与计算创新的相互作用,使得揭示组织细胞异质性的生物学里程碑式发现成为可能 7,8。
然而,scRNA-seq 仅捕捉了调控细胞功能和信号传导的复杂调控机制中的一层。为了补充这一点,已经做出了大量努力以在单细胞分辨率上测量其他模态,包括染色质可及性 9、表面蛋白 10、T 细胞受体(TCR)/B 细胞受体(BCR)库 11 和空间位置 12,从而实现了一些重要发现,例如 2 型糖尿病的调控特征 13、先天 14 和适应性 15 免疫系统对严重急性呼吸综合征冠状病毒 2(SARS-CoV-2)失调的响应,以及对肿瘤微环境免疫抑制效应的空间分辨理解 16。实验创新导致了许多新型计算工具的开发,以处理各种单细胞组学模态,但缺乏最佳实践工作流程使得导航这一新工具的广阔领域变得具有挑战性。此外,尽管之前已经为 scRNA-seq2,5,6,17 制定了计算最佳实践和工具推荐,但它们要么过时,要么不完整。
在本文中,我们将引导读者完成单模态和多模态单细胞数据分析的各个步骤,并讨论分析中的陷阱和建议(图 1)。由于工具的新颖性或缺乏独立基准,无法确定最佳实践的情况下,我们列出了流行的工具和社区推荐。我们将文章组织成特定模态部分和分析步骤组,而不是单一的工作流程,因为在现代单细胞分析中,由于任务的多样性,单一工作流程几乎不再存在。欲了解更多信息,我们提供了一个更加全面且定期更新(但未经同行评审)的单细胞最佳实践在线书籍,其中包含超过 50 个章节,包括详细的代码示例、分析模板以及计算要求的评估。
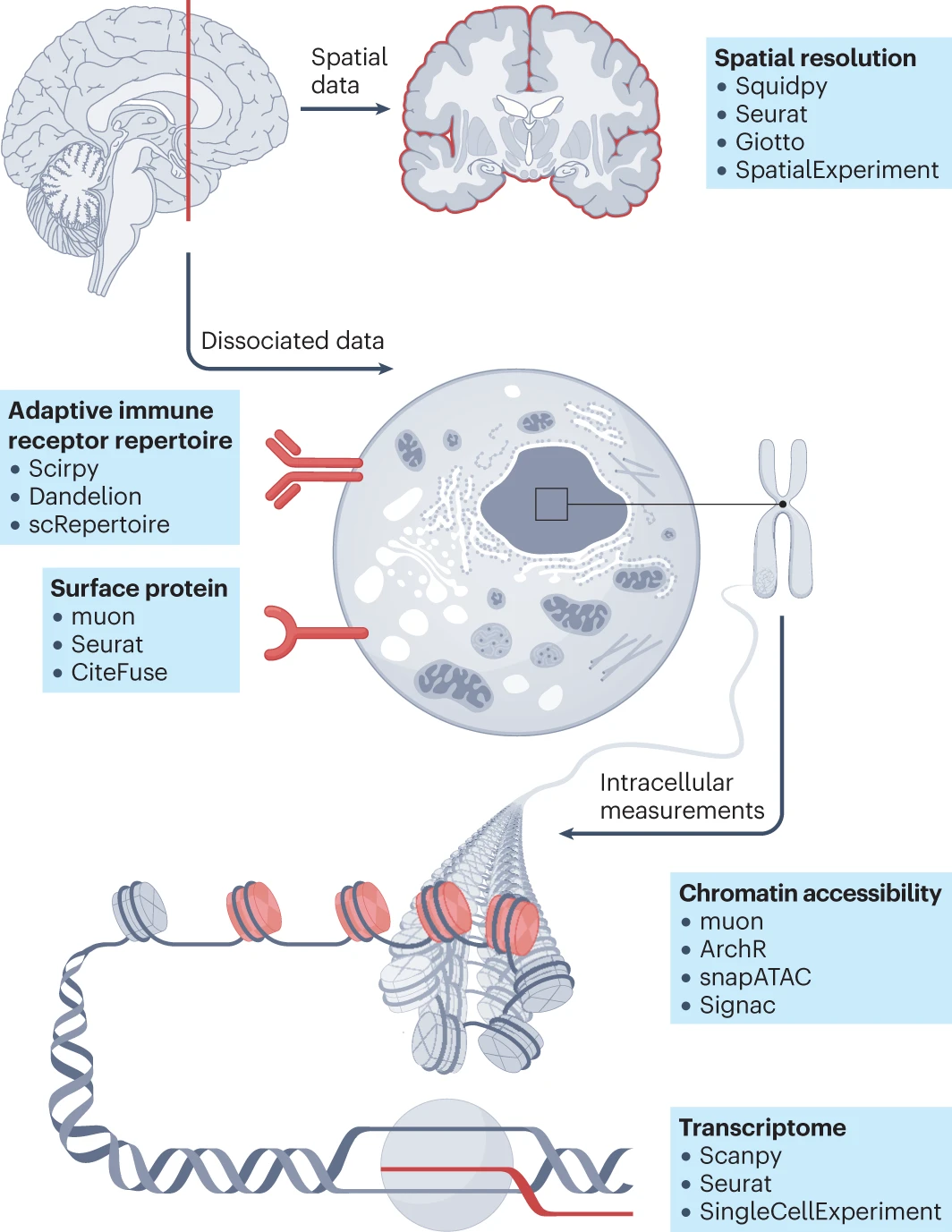
细胞状态由多种模态表征,包括但不限于 RNA 转录、染色质可及性、表面蛋白(包括 T 细胞受体(TCR)和 B 细胞受体(BCR))以及空间位置。涵盖最重要分析步骤的各种框架已经被开发出来。转录组数据可以使用 Scanpy4、Seurat36 和基于 Bioconductor 的 SingleCellExperiment2 进行分析;染色质可及性测量可以使用 muon150、ArchR140、snapATAC135 和 Signac143 进行分析;TCR 和 BCR 库分析可以使用 Scirpy164、Dandelion14 和 scRepertoire166 进行;表面蛋白表达可以使用 muon150、Seurat36 和 CiteFuse163 进行;空间分辨的单细胞数据集可以使用 Squidpy209、Seurat36、Giotto244 和基于 Bioconductor 的 SpatialExperiment211 等框架进行分析。这些框架还配备了众多用于特定后续分析任务的附加工具。
转录组¶
scRNA-seq 技术测量每个细胞中 mRNA 分子的丰度。提取的生物组织样本构成了单细胞实验的输入。组织在单细胞解离过程中被消化,随后进行单细胞分离,以分别分析每个细胞的 mRNA。基于板的协议将细胞隔离到板上的孔中,而基于液滴的方法则在微流体液滴中捕获细胞 18。在本文中,我们重点讨论由于其流行性而被广泛采用的基于液滴的检测方法。
获取的 mRNA 序列读数在使用细胞条形码或唯一分子标识符(UMIs)和参考基因组的原始数据处理管道中被映射到基因和原始细胞,以生成细胞与基因的计数矩阵(图 2a)。关于各种原始数据处理工具的详细比较,我们参考 Lafzi 等人的研究 19,并将计数矩阵视为我们单模态 scRNA-seq 数据分析工作流程的起点。
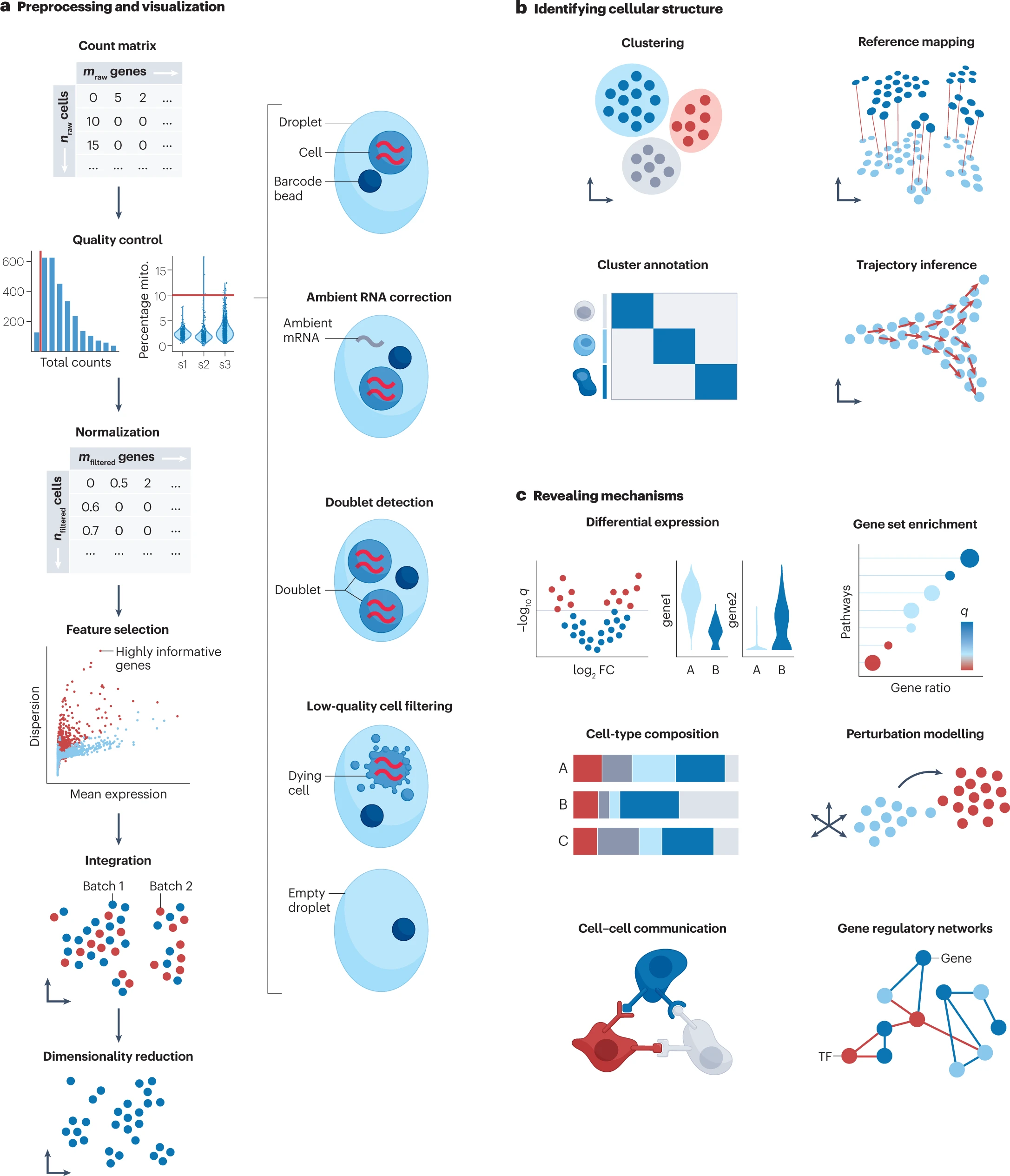
a. 通过原始数据处理管道获得细胞与基因的计数矩阵。为了确保只捕获高质量的细胞,计数矩阵需要校正去除无细胞环境 RNA,并过滤掉双细胞和低质量或正在死亡的细胞。后者通过移除质量控制指标(每个条形码的计数数量,称为计数深度或文库大小,每个条形码的基因数量和每个条形码中来自线粒体基因的计数比例(百分比线粒体))方面的异常值来完成。所有计数代表成功捕获、逆转录和测序的 mRNA 分子。这些步骤在细胞间有所不同,因此相同细胞的计数深度可能不同。因此,在比较细胞间的基因表达时,差异可能仅源于取样效应。为了解决这一问题,通过标准化获得细胞间正确的相对基因丰度。单细胞 RNA 测序(scRNA-seq)数据集可以包含多达 30,000 个人类基因的计数。然而,大多数基因并不提供信息,许多基因没有观测到表达。因此,选择最具变异表达的基因。将不同批次的数据整合以获得跨样本的校正数据矩阵。为了减轻计算负担并减少噪音,通常应用降维技术。这进一步允许低维嵌入转录组数据用于可视化目的。
b. 校正空间可以被组织成簇,代表具有相似基因表达谱的细胞群体,并用感兴趣的标签(如细胞类型)进行注释。注释可以使用先验知识手动进行,也可以使用自动注释方法进行。连续过程,如在分化或重编程过程中细胞身份的转变,可以推断出不符合离散类别的细胞多样性。
c. 根据感兴趣的问题和实验设置,数据集中的条件可以测试基因上调或下调(差异表达分析)、对通路的影响(基因集富集)和细胞类型组成的变化。扰动建模能够评估诱导扰动的效果和预测未测量的扰动。配体和受体的表达模式可以揭示细胞间通信的变化。转录组数据进一步使基因调控网络的恢复成为可能。
从原始计数矩阵到高质量的细胞数据¶
scRNA-seq 技术的进步带来了高通量的高质量运行。然而,scRNA-seq 数据集包含系统性和随机噪音(如低质量细胞产生的噪音),这些噪音会掩盖生物信号。scRNA-seq 数据的预处理尝试去除这些混杂的变异源。这包括质量控制、标准化、数据校正和特征选择(图 2a)。
过滤低质量细胞和噪音校正¶
大多数分析任务假设每个液滴包含一个完整的单细胞的 RNA。然而,这一假设通常因低质量细胞、无细胞 RNA 污染或捕获多个细胞而被违反(图 2a)。检测到的基因数量较少、计数深度较低和线粒体计数比例较高的细胞通常被称为低质量细胞,因为它们可能代表膜破裂的死亡细胞。低质量细胞通过手动设置阈值(如之前的指南 5 中建议的那样)或基于中位绝对偏差 20 的样本自动过滤来识别和过滤。这些指标被联合考虑,以防止误解细胞信号 5。质量控制在样本级别执行,因为阈值在样本之间可能有很大差异。
无细胞 RNA 可能存在于细胞溶液中,并在文库构建过程中被分配到细胞的本源 RNA 中。环境 RNA 污染可能导致细胞类型特异性标记基因转录本在其他细胞群体中也可检测到,从而混合不同的细胞群体 21。流行的方法如 SoupX 基于数据集中“空”液滴和细胞簇的表达谱估计细胞特异性的污染分数 21。CellBender 将去除环境 RNA 表述为一个无需事先了解细胞类型特异性基因表达谱的无监督贝叶斯模型 22。即使在没有系统性基准的情况下,也应考虑将去除环境 RNA 作为质量控制中的初始分析步骤,以改善许多组织的后续分析 21,22,23。
空液滴和双细胞(包含两个细胞的液滴)违反了每个液滴包含一个单细胞的假设。由不同细胞类型形成的双细胞(异型双细胞)难以注释,并可能导致错误的细胞类型标签。常见的双细胞检测方法通过结合两个随机抽样的细胞生成人工双细胞,并将其与测量的细胞进行比较。scDblFinder24 利用了这一理念,并可以与已知双细胞的先验知识结合。几个基准测试表明,scDblFinder 在双细胞检测准确性和计算效率方面优于其他方法 25,26,27。此外,应用多种双细胞检测方法并比较结果以提高双细胞检测的准确性也是有益的 27。
在下游分析过程中,当低质量细胞和双细胞聚集在一起时,选择的质量控制策略通常需要重新评估。因此,我们建议初始设置宽松的阈值,并在(重新)分析过程中根据需要移除更多细胞。
标准化和方差稳定化¶
细胞可能具有不同数量的基因计数,这可能是由于 mRNA 含量的体积(细胞大小)的差异或纯粹在测序过程中随机产生的。计数标准化使细胞轮廓可比。随后的方差稳定化确保异常轮廓对整体数据结构的影响有限【28】(图 2a)。最近的一项基准测试比较了基于 \(K\) 最近邻图(KNN 图)与真实情况重叠的 22 种单细胞数据转换方法【29】。移位对数转换 \(\log \left(\frac{y}{s}+1\right)\) 在这方面表现出色,常用于使数据结构更加稳定和一致。
移除混杂变异源¶
混杂变异源可分为技术性和生物性协变量,应分别处理它们,因为它们描述了不同的影响和挑战。
包含多个样本的数据集可能会受到反映技术变异的批次效应的影响。批次效应在聚类和可视化后可能会显现出来,应被移除以确保它们不被误认为是实际的生物学洞见 5。数据集成方法解决了相同实验设置中样本之间的批次效应。最近的一项基准测试基于 14 项指标比较了 16 种集成方法,评估了批次校正和生物变异保留的效果 35。线性嵌入模型,如典型相关分析 36 和 Harmony37,在具有明显批次结构的简单集成任务中的批次校正表现良好 38,39。scANVI40 可以结合细胞类型标签,这有利于保留生物变异 35。根据集成任务的复杂性,例如图谱集成,深度学习方法如 scANVI40、scVI41 和 scGen42 以及线性嵌入模型如 Scanorama43 表现最佳,而对于较简单的集成任务,Harmony37 是首选方法 35。可以使用 scIB 包来评估集成效果,使用上述基准测试的评估指标 35。
除了计数抽样效应,scRNA-seq 数据还可能包含生物混杂因素,如细胞周期效应,即细胞间的差异可能是由于不同的细胞周期状态而非细胞类型 44。移除这些效应有利于下游分析;然而,了解细胞是否在循环中可能为基础生物学提供宝贵的见解 5。最近的一项基准测试 44 建议使用 Scanpy4 或 Seurat45 中的内置细胞周期标记和校正功能作为基准,这些功能将平均表达值与参考特征进行比较。随后应应用更复杂的方法,如 Tricycle46,该方法将数据集映射到表示细胞周期的嵌入中 46。Tricycle 在具有高细胞类型异质性的数据集中表现良好 44。
选择信息性特征和降维¶
为了确保分析仅关注生物学上有意义的基因,并处理大型数据集,可以将计数矩阵减少到最具信息性的特征。特征选择方法应理想地选择解释数据集中生物变异的基因,优先选择在亚群体间变化而非在单一亚群体内变化的基因,而不影响小亚群体的可识别性 20。Deviance 通过拟合假设所有细胞中表达恒定的基因模型,并量化哪些基因违反这一假设,从而识别出高度信息性的基因 47。它在识别跨亚群体高变异基因方面表现良好,因此在选择信息性基因方面表现出色,如独立比较所示 20。此外,通过 deviance 排序基因是在原始计数上进行的,因此不敏感于标准化。在特征选择后,可以通过降维算法如主成分分析(PCA)进一步减少数据集的维度(图 2a)。降维技术可用于可视化或总结底层数据拓扑结构。基于其他研究,PCA 可用于数据总结,而 t-SNE、UMAP 和 PHATE 则用于更灵活地可视化 scRNA-seq 数据 5,48。值得注意的是,最近的一项研究表明,仅依赖于二维嵌入可能导致对细胞关系的误解,结果不应仅基于这些表示的视觉检查,而应结合定量评估 49。
从簇到细胞身份¶
在预处理后,数据集中的不需要的效应已被移除,信噪比得到改善。因此,现在可以开始提出生物学上相关的问题。作为下一个分析里程碑,可以识别不同的细胞群体,以进一步指导和结构化分析(图 2b)。
从单细胞到簇¶
识别细胞群体的第一步是将细胞聚类到具有相似表达谱的组中,以解释数据中的异质性。独立基准测试 5,50,51 表明,通过 Louvain 算法进行图模块优化的社区检测在聚类识别方面效果最佳。然而,Louvain 算法可能会导致连接不良的社区 52。Louvain 的继任者 Leiden 通过产生保证连接的社区来解决这一问题,并且在计算上更有效率 52。两种方法都应用于在低维表示上计算的 KNN 图,可以在不同分辨率下运行以控制识别的簇的数量。我们建议在不同分辨率下使用 Leiden 算法,以获得理想的细胞注释聚类 5。
将细胞簇映射到细胞身份¶
注释是赋予检测到的细胞簇生物学解释(如细胞类型)的过程(图 2b)。可以使用手动或自动方法进行。推荐采用三步法,即先进行自动注释,然后由专家进行手动注释,最后进行验证,以获得理想的注释结果 53。第一步,自动细胞类型注释,可以分为基于分类器的方法和参考映射。使用预训练分类器获得的注释结果受到分类器类型和用于创建分类器的训练数据质量的强烈影响 54,55。此外,不另外检查个体标记可能很难评估结果注释。基于先前注释数据集或图谱训练的分类器示例包括 CellTypist56 和 Clustifyr57。第二组自动注释方法是映射到现有的、注释好的单细胞参考,并在生成的联合嵌入上执行标签转移。参考可以是数据集的个体样本,理想情况下是经过精心策划的现有图谱。可以使用如 scArches58、Symphony59 或 Azimuth3 的方法进行查询到参考的映射。与基于分类器的方法类似,转移注释的质量取决于参考数据的质量、模型及其与数据集的适用性。第二步,手动注释,利用每个簇的基因特征来注释细胞簇。这些基因特征通常称为标记基因,可以使用简单的差异表达测试方法(如 t 检验或 Wilcoxon 秩和检验)来识别。统计测试应用于两组簇,以找到在感兴趣的簇中上调或下调的基因。为此,Wilcoxon 秩和检验表现最佳,但由于聚类的性质,P 值可能会膨胀,可能导致错误发现,因为使用相同的数据来定义我们测试差异的标签 60,61。然后将获得的标记与注释良好的参考中的标记基因进行比较,以注释细胞簇。最后一步,注释应由专家验证,特别是对于高复杂性数据集或涉及稀有细胞亚群体的研究,因为这些亚群体可能没有参考 53。
从离散状态到连续过程¶
在非静止的生物过程(如分化)中,细胞穿越一个连续的细胞状态空间。使用单细胞数据理解细胞命运及其在这一景观中的调控基因是具有挑战性的,因为测量仅是快照。底层轨迹可以是循环的、线性的、树状的,或最普遍的是图状的。根据表达模式的相似性对细胞进行排序的模型称为轨迹推断或伪时间分析方法。轨迹推断方法的性能取决于数据集中存在的轨迹类型。尽管 Slingshot62 在简单拓扑结构中表现较好,PAGA63 和 RaceID/StemID64 在复杂轨迹中得分更高 65。因此,我们建议使用 dynguidelines 选择适用的方法 65。当预期拓扑未知时,应通过使用不同基本假设的多种轨迹推断方法确认轨迹和下游假设。推断的轨迹不一定具有生物学意义 5。通过例如 RNA 速度测量等更复杂的方法和信息源可以恢复实际生物过程的更多证据。
为了推断动态、定向信息,velocyto66 和 scVelo67 使用未剪接和剪接的读数建模剪接动力学,以推断 RNA 速度:如果基因正在激活,未剪接 RNA 先于剪接 RNA,这可以在相图中可视化 67。获得的 RNA 速度场作为 CellRank68 的输入以估计细胞命运。RNA 速度推断假设基因独立性和转录、剪接和降解的恒定速率。在恒定速率假设下,相图形成一个杏仁形状,具有诱导(上半部分/弧)和抑制(下半部分/弧)阶段。因此,我们建议通过检查由 scVelo 的动态模型确定的高可能性基因的相图来验证模型假设是否成立。如果相图缺乏预期形状,RNA 速度可能被错误推断。此外,如果基因包含多个显著的动力学,系
谱特定模型更为合适 69。RNA 速度推断错误的情况包括转录突发的存在 70,71。此外,稳态群体提出了进一步的挑战,其中 RNA 速度在独立的终端细胞群体之间推断出错误的方向 70,71。
尽管基于伪时间的方法只要过程覆盖在足够细致的步骤中就没有时间尺度限制,但 RNA 速度无法覆盖所有时间尺度。因为是建模剪接动力学,观察到的过程也必须在这个时间范围内发生 70。
回顾性实验系谱追踪方法使用细胞中观察到的变异(如自然发生的遗传突变)来推断其系谱模型,总结克隆群体中的细胞分裂历史。系谱追踪数据的分析可以使用 Cassiopeia72,该工具实现了包括经典方法如 UPGMA73 或邻接 74 以及用于 CRISPR-Cas9 系谱追踪数据的较新方法的几种重建算法。算法的重建性能难以评估,因为它们可能很好地突出系谱的不同部分 75。因此,我们建议应用多种算法进行性能比较。此外,专门工具被引入用于包括时间过程信息的更复杂系谱追踪研究分析。其中包括 LineageOT76,一种适用于演变 CRISPR-Cas9 设置的最优传输框架 77,以及用于静态条形码系谱追踪的 CoSpar78。
揭示机制¶
在获得对高质量数据的可靠注释后,分析空间变得多样化,许多感兴趣的机制可以被调查。以下分析步骤的选择和顺序取决于研究问题和实验设计(图 2c)。
差异基因表达分析¶
负二项分布的 scRNA-seq 数据可以被测试以识别在特定条件下上调或下调的标记基因或基因。差异基因表达(DGE)分析目前有两种方法。样本级视角聚合每个样本 - 标签组合的计数以创建伪块,然后使用最初设计用于块表达分析的包(如 edgeR79、DEseq280 或 limma81)进行分析。另一种是细胞级视角,使用广义混合效应模型(如 MAST82)对单个细胞建模。DGE 工具之间的一致性和鲁棒性较低 83,84,但为块 RNA-seq 数据设计的方法表现较好 84,85,86。发现单细胞特定方法系统地低估了基因表达的变异,并容易错误标记高表达基因为差异表达 86。
当前的 DGE 分析方法仍然在真正阳性率(TPR)和精度之间存在权衡。高 TPR 导致低精度,因为假阳性较多,而高精度由于缺乏识别差异表达基因导致低 TPR83。伪复制导致 FDR 膨胀,因为 DGE 方法没有考虑重复样本(同一个体细胞)的内在相关性 86,87,88。在 DGE 分析之前,应通过在个体内聚合细胞类型特异性计数来考虑样本内相关性 87。通常,带有求和聚合的伪块方法和混合模型(如带随机效应设置的 MAST)被认为优于不考虑样本内相关性的简单方法,如流行的 Wilcoxon 秩和检验 88。
DGE 结果的有效性强烈依赖于统计模型中主要变异轴的捕获。中间数据探索步骤(如伪块样本上的 PCA)有助于识别变异来源,从而可以指导构建相应的设计和对比矩阵以建模数据 89。如果未考虑多个生物变异来源,实验会导致 FDR 膨胀 90,91。因此,我们推荐灵活的方法,如 limma、edgeR 或 DESeq2,这些方法允许复杂的实验设计。基于条件的 DGE 测试获得的 P 值必须进行多重检验校正 5,92 以获得 q 值。
基因集富集分析¶
scRNA-seq 数据的高通量性质使其难以解释。基因集富集分析允许将许多分子洞见总结为可解释的术语,如通过先前研究已知涉及的基因集定义的通路。常见数据库包括 MSigDB93、Gene Ontology94、KEGG95 或 Reactome96。这一概念的扩展是加权基因集,包括用于信号通路的 PROGENy97 和用于转录因子的 DoRothEA98。常见的富集方法包括超几何测试、GSEA99,100 或 GSVA101,这些方法可以在 DGE 分析之后或在单个细胞级别应用。基因集富集分析被发现对基因集的选择而不是统计方法更为敏感 102;因此,我们建议仔细选择数据库以确保覆盖潜在的基因集。为此,富集框架如 decoupleR103 提供了在单一工具中访问不同数据库和方法的功能。开发用于块转录组学的富集方法可以应用于 scRNA-seq102,但一些单细胞基于的方法(即 Pagoda2)可能表现优于它们 105。
解密细胞组成的变化¶
组成分析关注的不是细胞的基因表达谱变化,而是不同细胞类型相对丰度的条件变化。组成变化在发育 106 和疾病 107 中经常观察到,但组成分析方法缺乏独立的基准。单变量统计模型,如泊松回归或 Wilcoxon 秩和测试,分析每种细胞类型的丰度变化,可能将某些细胞类型群体的变化视为统计上显著的效应,尽管它们纯粹是数据组成性引起的统计假象 108,导致 FDR 升高。专门为单细胞数据设计的测试,使用细胞类型计数,包括 scDC109、scCODA108 和 tascCODA,可以纳入层次细胞类型信息 110。
对于发育数据,锐利的聚类边界可能具有欺骗性,基于已知注释确定组成变化可能不合适。DA-seq111 和 MILO112 使用 KNN 图定义亚群体,测试实验条件间的差异丰度。KNN 基于的方法对感兴趣的条件与混杂变异来源强相关时信息损失敏感。减少 KNN 图的 K 值或在特定系谱上构建图可以缓解此问题 112。如果在可视化中显著差异在大群体中显现,KNN 图基于的方法可能不适合,更直接地使用已知细胞类型计数的工具分析可能更适当。
推断扰动效应¶
单细胞实验方案的进展使得在数千种独特条件下测量细胞的大规模多重实验成为可能,通常称为“扰动”113。最近的技术如 perturb-seq114 或 CROP-seq115 允许多模态读出的 CRISPR-Cas9 筛选 116、全基因组扰动 117 和组合扰动 118 的分析。这些复杂条件的分析称为扰动建模 119,目前还没有独立的基准测试。
扰动建模的一个领域尝试区分成功和不成功靶向的细胞,以评估扰动效应。Mixscape116 和 MUSIC120 首先去除混杂变异源,然后区分成功和不成功扰动的细胞,最后可视化和评分扰动效应。Augur121,122 和 MELD123 仅覆盖第三步,按扰动响应程度对细胞类型进行排序,以识别最受扰动影响的细胞群体。
扰动建模的第二个领域涉及未被实验测量的扰动。潜在空间学习模型如 scGen42、CPA124 和 CellBox125 旨在预测未见扰动、组合或药物剂量的响应。这些模型通常对高表达基因效果良好,但由于缺乏变异,对低表达基因可能表现不佳。
跨细胞的通信事件¶
细胞在生物体发育和稳态中不断相互作用。如果这种相互作用受损,疾病就会发生。细胞 - 细胞通信推断方法通常使用配体、受体及其相互作用的库来预测注释簇之间的相互作用。发现这些数据库偏向特定通路、功能类别和组织富集蛋白 126。方法和相互作用数据库的选择对预测的相互作用有很大影响 126。CellChat127 和 CellPhoneDB128 也考虑异源复合物,被发现对数据和资源噪音具有鲁棒性 126。由于工具之间缺乏共识,我们建议使用 LIANA,其为多种方法和数据库组合提供整体排名 126。此外,Nichenet130 或 Cytotalk131 等工具提供的细胞内活动(如诱导基因表达变化或空间信息)的补充估计,可以增加预测相互作用的可信度。
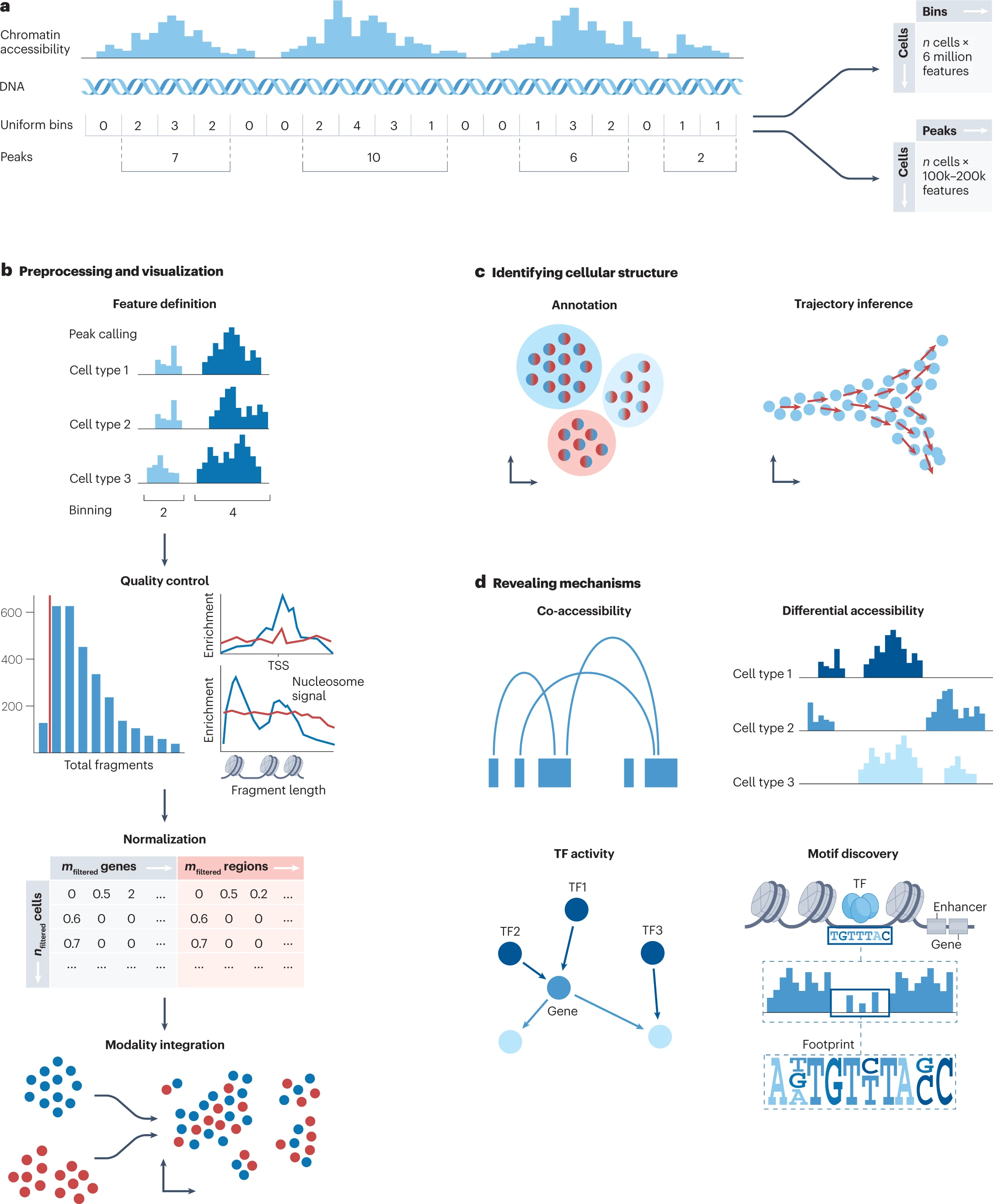
单细胞转座酶可及染色质测序(scATAC-seq)
a. 单细胞转座酶可及染色质测序(scATAC-seq)测量单细胞染色质可及性。数据可以以几种不同的方式表示。两种最常见的选择是细胞 - 峰值矩阵和细胞 - 箱体矩阵。峰值调用算法找到相对于背景噪音的高可及性区域,而箱体算法在等大小的箱体中捕获 Tn5 转座事件。
b. 为了确保后续分析专注于生物学上有意义的特征而不是噪音,对特征矩阵进行质量控制。数据需要控制每个细胞的总片段数(代表细胞测序深度)以及其他相关信号的测试:每个细胞中非零计数的峰值数、转录起始位点(TSS)富集得分、反映单核小体与无核小体片段比例的核小体信号,最后是与人为信号相关的基因组区域中的读数比例。然后,通过标准化校正稀疏分布的 scATAC-seq 特征。随后的预处理和可视化工作流程与典型的 RNA 分析步骤密切相关。
c. 可以根据已知的差异可及区域对 scATAC-seq 数据进行细胞类型注释,通常通过耦合到附近或注释的编码 DNA 区域。可以利用注释的细胞通过轨迹推断分析连续过程。
d. 根据研究问题,现在可以调查数据的共可及性以识别顺式调控相互作用、差异可及区域以理解条件之间的变化、转录因子(TF)活性以识别关键调控因子和基序发现以识别作为 TF 结合位点的 DNA 序列模式等。
染色质可及性¶
分析调控元件对于解码细胞多样性和理解细胞决策至关重要。基因表达由包括表观遗传学和染色质可及性在内的复杂调控机制控制 132。为了深入了解单细胞水平的染色质状态动态,单细胞转座酶可及染色质测序(scATAC-seq)测量单细胞中全基因组染色质可及性 133,134(图 3)。
特征定义和质量控制¶
与 scRNA-seq 数据中明确定义的基因特征相比,scATAC-seq 数据由于其全基因组特性缺乏标准化的特征集。大多数工作流程使用细胞 - 峰值或细胞 - 箱体矩阵作为分析的基础,这比基因或 TF 基序特征矩阵表现更好 135(图 3a)。箱体是跨基因组均匀大小的窗口,捕获所有 Tn5 转座事件,而峰值则指的是相对于背景噪音的开放染色质区域,Tn5 转座事件在这些区域富集。值得注意的是,细胞 - 峰值矩阵比 scRNA-seq 数据更稀疏,因为在二倍体生物的细胞中只有两个可测染色质拷贝,每个细胞中仅调用 1-10%的峰值 135。识别峰值需要足够数量的细胞,因此在稀有细胞类型中可能会失败 136。通过在聚类中调用峰值可以提高峰值检测的灵敏度,这减少了由于其他高度丰富的细胞类型的噪音而掩盖稀有细胞类型峰值的风险。对于这种方法,未排除基因组区域的细胞 - 箱体矩阵作为聚类的基础 136。
scATAC-seq 质量控制的最常见入口点是包含所有测序 DNA 片段的片段文件,这些片段由两个相邻的 Tn5 转座事件生成。使用这些文件计算一组 scATAC-seq 特定的质量指标,以确定低质量细胞(图 3b)。类似于 scRNA-seq 数据中的测序深度,检查每个细胞的总测序片段数、片段总数对数以及转录起始位点(TSS)富集得分(基于一般更开放的启动子区域与非启动子区域相比,每个细胞的信噪比)。低质量细胞通常形成一个结合低计数和低 TSS 富集得分的簇,应被移除 137。此外,核小体信号用于评估片段长度分布 137。还建议验证映射到与人为信号相关的基因组区域的读数比例 138。峰值调用后,通过数据集依赖的最小阈值控制每个细胞检测到的特征数。此外,峰值与非峰值区域中的低读数数是低信噪比的指示,类似于 TSS 得分 9。
为了评分双细胞,我们建议按照 Germain 等人 24 的推荐,使用专为 scATAC-seq 数据设计的两种正交方法,并在下游分析中考虑这两种评分。第一种方法是调整 scDblFinder,通过减少相关特征到一个小集合来使用完整信息,同时使计数数据更连续 24。第二种方法,AMULET139,利用染色体的二倍体性,通过评分具有意外高数目位置的细胞(超过两个计数)来捕捉同型双细胞 139。
学习低维表示¶
稀疏的 scATAC-seq 数据需要标准化,类似于 scRNA-seq。在 scATAC-seq 数据中,最常见的标准化策略是峰值二值化 136,140,141。然而,这也可能移除生物学信息,因此建议直接对 scATAC 计数建模 142。基于潜在语义索引(ArchR140 和 Signac143)、潜在 Dirichlet 分配(cisTopic141)和谱嵌入(snapATAC136)的降维方法被证明在下游聚类和细胞注释方面表现最佳 135。在批次校正方面,LIGER 在 scATAC-seq 数据中表现最佳 35。最近,深度学习模型如 PeakVI144 或 MultiVI145 被提议作为结合降维和批次校正的方法。获得校正的低维表示后,我们建议基于其在 scRNA-seq 衍生表示中的良好表现使用 Leiden 聚类。
基于可及区域注释细胞身份¶
可以根据差异可及区域(DARs)和基因活性得分对细胞簇进行注释(图 3c)。DARs 可以通过类似于 scRNA-seq 的差异测试方法获得。类似的测序深度差异需要通过将总计数视为混杂因素来处理 143,或选择总计数和其他质量控制指标(如 TSS 得分)匹配的偏差匹配细胞组进行比较 140。尽管尚未对 scATAC-seq 数据进行基准测试,但现有的块 ATAC-seq 数据基准测试推荐在样本量有限的情况下使用 edgeR,在样本量大的情况下使用 DESeq2146。DARs 可能包含信息性序列模式,如已知的顺式调控元件(CREs),或可与近端基因关联,这在功能富集分析工具如 GREAT147、LOLA148 或 GIGGLE149 中得以利用。与基因相关的 CREs 的染色质可及性可以汇总成基因表达的估计值(基因活性得分)。这可以通过汇总基因内和 TSS 上游一定距离内的计数来实现 136,143,150。更复杂的模型还集成了远端区域的信号,或者通过按距离加权方案 140,或者通过集成共可及性网络 151(图 3d)。为了指导细胞类型注释,简单模型通常就足够了,并且通过在邻近细胞中平滑基因活性得分可以增强可视化,这通常使用 MAGIC152 进行。
使用 TF 基序和足迹解析身份¶
TF 基序富集有助于表征细胞身份,可以在簇级别上通过对簇特异性 DARs 进行超几何测试进行。为了获得每个细胞的富集得分,可以使用 chromVAR 计算每个细胞中包含基序的所有峰值的可及性偏差,同时校正 Tn5 转座酶的插入偏好引起的偏差 153。TF 标记有助于簇注释,并代表决定细胞状态的调控蛋白的顶级候选者。一旦确定了感兴趣的 TF,scATAC-seq 数据允许通过足迹进一步验证 TF 的影响,足迹表明 TF 是否在给定的细胞簇中结合。为了执行此分析,生成簇级伪块以减少稀疏性,并绘制感兴趣基序周围的 Tn5 插入数 140。在给定细胞簇中 TF 的活性结合的情况下,结合位点本身受到 Tn5 转座事件的保护,而近邻核小体被置换,形成峰 - 谷 - 峰可及性谱。由于 Tn5 插入偏差也影响这一谱,当前的足迹工具通常使用 k-mer 模型校正这一偏差,该模型通过基于每个 k-mer 内切割位点数相对于全基因组出现次数的比率估计偏差 140,143,154。
链接单细胞染色质可及性和转录组学¶
诸如专有的 10x Multiome、sci-CAR155 或 scCAT-seq156 等检测方法允许基因表达和染色质可及性的联合分析。当前工作流程使用建立的单模态质量控制方法,并在所有模态的高质量细胞交集中进行综合分析 136,140,143。一旦选择了高质量细胞,可以学习捕获两种模态变异的细胞联合表示,从而去除混杂变异源(见框 1)。由于尚未确定这一整合的最佳方法,我们建议首先进行单模态分析,包括细胞类型注释。这使得可以通过将更新的聚类结果与单模态分析的细胞类型标签进行比较来评估联合表示。高质量的多模态表示然后可以作为大多数单模态分析方法(包括细胞类型注释、差异测试和轨迹分析)的输入。
配对的 scRNA-seq 和 scATAC-seq 数据还可以使用新的联合方法识别基因表达和细胞状态的调控因子。为了识别潜在的 CREs,基于相关性的方法用于在细胞簇内将峰值链接到基因 140,143,156。通过使用 SCENIC 推断活性 TF,然后将相应的基序与峰值区域匹配,可以增加解释性 156。为了深入了解局部或全局染色质景观是否影响特定细胞状态下的基因表达,可以比较基于局部邻域和全基因组染色质状态的表达可预测性 157。利用两种模态推断基因调控网络的方法(如 FigR154 或 Pando158)目前正在开发(图 3d)。
跨模态数据整合¶
跨模态分析可获得细胞的整体表示 245,其中联合检查相同细胞的几种模态。尽管实验测定的进步允许许多模态组合的配对测量 246,但不同模
态仍通常独立测量,导致非配对数据 247。需要正确整合这些数据集以获得可用于可视化感兴趣属性的信息低维嵌入。
联合测量模态的整合:配对整合¶
对于配对测量,细胞作为整合锚点(见图,部分 a)。可以使用 MOFA+248 实现的因子分析等线性方法进行配对整合,以获得联合、可解释的潜在空间。这种方法需要大小因子标准化以确保第一个因子不被每个样本的总表达差异所主导。或者,带权最近邻(WNN)3 分析学习细胞特定的模态权重,这些权重反映模态信息内容,以确定下游分析中模态的重要性,形式为邻居图。此图可以重用于计算嵌入或距离指标。
非配对测量的整合:非配对整合¶
整合非配对多组学数据(对角线整合;见图,部分 b)的主要困难在于不同的特征空间。基于先验知识将多模态数据映射到公共特征空间的初始方法,如转座酶可及染色质(ATAC)区域到附近转录本,然后应用单细胞数据整合方法已被证明会导致信息丢失 135。基于非线性流形对齐的方法(如基于最优传输的方法,如 SCOT249 或 UnionCom250)不需要先验知识,因此可以减少跨模态信息丢失。GLUE 通过结合先验知识的指导图,将细胞状态建模为通过模态特异性变分自编码器学习的低维嵌入,使用概率生成建模 251。已证明其在整合多于两种模态时效果良好,并在 NeurIPS 2021 多模态单细胞数据整合挑战赛中获胜 252。
联合和非配对测量的整合:拼接整合¶
尽管实验测定的进步,仍难以同时捕获同一细胞的几种模态。在不同的细胞群体上分析个体模态更为常见,导致完全缺失的数据矩阵 245。此类设置中的数据整合称为“拼接整合”,最近开始出现相关工具(见图,部分 c)。尽管 totalVI 和 MultiVI 也可用于拼接整合,但它们分别仅适用于 CITE-seq 和 Multiome 数据。所有模态组合的替代方法是 Stabmap253,它通过将所有细胞投影到参考坐标来遍历拼接拓扑的最短路径,以及 Multigrate254,它利用迁移学习来推测缺失模态。
多模态场景中的查询到参考映射¶
该领域的最新发展是多组学参考数据集的出现,因此可以针对多模态参考进行单模态和多模态查询(见图,部分 d)。通过将监督主成分分析(PCA)255 应用于使用 WNN 构建的参考,可以将单细胞 RNA 测序(scRNA-seq)查询细胞映射到多模态参考,进行可视化和注释 3。或者,Multigrate 学习配对和非配对测量的联合潜在空间。结合迁移学习,Multigrate 可以将单模态和多模态查询数据集映射到多组学参考,同时推测缺失模态 254。推测的模态可能是进一步重要的信息来源。桥接整合提出了第三种选择,使用多组学数据集作为分子桥,创建用于重建单模态数据集的细胞字典,这些数据集被转化为共享嵌入 256。尽管灵活,桥接整合的缺点是需要桥接数据集,这可能并不总是可用。
表面蛋白表达¶
转录和染色质可及性是细胞状态、活动和调控的代理。实际生成的产物,蛋白质,执行细胞内或细胞外任务,其中一部分蛋白质呈现在细胞表面。表面蛋白表达有助于识别细胞类型,例如免疫系统的造血细胞,其注释基于通常在流式细胞术或质谱细胞术实验中使用的标记。它们还可以用于验证特定基因的敲除,例如使用前述的 Mixscape 管道。最广泛使用的结合 scRNA-seq 和表面蛋白谱的协议是 CITE-seq10 和 REAP-seq159,主要区别在于用于量化表面蛋白表达水平的抗体衍生标签(ADTs)(图 4a)。

表面蛋白表达
a. 抗体衍生标签(ADTs)是带有独特条形码的抗体克隆,这些条形码附加到 poly(A) 序列和 PCR 柄上,在后续文库处理步骤中被特异性扩增。抗体与表面蛋白结合,测序得到的 ADT 计数代表这些蛋白的表达水平。
b. 虽然 ADT 数据可以单模态分析,但很少单独测量,通常与匹配的基因表达数据一起测量。这种基因表达和 ADT 的配对计数矩阵需要单独进行质量控制和标准化,然后进行单独或联合可视化嵌入。
c. CITE-seq 数据的注释可以在转录组数据、ADT 数据或通过将簇匹配到标记基因和标记 ADT 上联合进行。
d. 为了了解生物机制,可以对 ADT 数据进行差异丰度测试,推断细胞 - 细胞通信,并构建 RNA 和 ADT 信息的相关网络。q 值用于表示这些分析的统计显著性。
校正 ADT 计数¶
与基因计数的负二项分布相反,ADT 数据不那么稀疏。对于基于液滴的检测,ADT 通常会观察到非零计数,这是由于环境污染和非特异性抗体结合。大多数标记物表现出双峰分布,其中“负”峰(低计数)表示非特异性抗体结合,“正”峰表示特定细胞类型中细胞表面蛋白的富集 160。对于所有或大多数抗体面板计数为零的文库应被移除;然而,移除总 ADT 计数低的细胞可能会移除那些不表达特定蛋白集或仅表达少量蛋白的细胞类型 2。CITE-seq 实验还可以包含同型对照,这些是用于测量每个细胞非特异性结合的非目标特异性抗体(如抗体聚集体)。在异常细胞中可以检测到大量同型对照计数,这些细胞应被移除。鉴于这些考虑,应仔细评估 ADT 模态中的各个质量控制指标,RNA 和 ADT 的联合测量应分别进行质量控制。由于抗体效力是可变的,跨多个研究整合 ADT 数据可能会导致强烈的批次效应,需要校正 160。
考虑 ADT 组成偏差¶
细胞特征可能导致异质的捕获效率,从而导致细胞组成偏差。只有表达目标蛋白的细胞才会增加标签计数,这可能仅限于特定的细胞类型 2。可以通过使用中心对数比(CLR)变换 10 或去噪和按背景缩放(DSB)161 来标准化来解决这一问题。DSB 使用代表蛋白背景噪音的背景液滴来校正细胞中的值,同时结合同型对照水平和相应细胞的特定背景水平,消除细胞间变异。DSB 的作者发现,由于原始计数中背景分布的可用性,这种方法去除了更多噪音 161。
联合分析转录组和 ADT 数据¶
ADT 数据的单模态下游分析遵循类似于单模态 RNA 分析的管道,其中注释的簇可以进行差异丰度测试(图 2b 和图 4b)。然而,当与其他模态(如转录组测量)联合分析时,ADT 数据提供的见解最多。在各自预处理后,可以通过一般适用的多模态整合工具(见框 1)或基于深度学习的 CITE-seq 特定的 totalVI162 获得联合嵌入,totalVI 学习配对测量的联合概率表示,同时考虑噪音和技术偏差,包括每种模态的批次效应。另一种方法是使用 CiteFuse163,使用 CLR 标准化 ADTs,并结合相似网络融合算法合并两种模态矩阵。然后可以使用 Leiden 聚类对联合嵌入进行聚类,并基于差异表达的 RNA 和 ADT 使用 Wilcoxon 秩和检验对簇进行注释,通过将簇与所有其他簇进行比较 163(图 4c)。这两种模态可以用于下游任务,例如研究细胞 - 细胞通信,其中考虑配体簇的 RNA 表达和受体簇的蛋白质表达,或使用 CiteFuse 进行 RNA 和 ADT 相关性分析(图 4d)。将结果可视化在联合嵌入上。
适应性免疫受体库¶
TCR 和 BCR 是构成适应性免疫受体库(AIRR)的跨膜表面蛋白复合物(图 5a)。两种受体均检测病原体和肿瘤特异性抗原,但以不同方式相互作用。BCR 直接识别可溶性或膜结合的表位,而 TCR 与结合在细胞表面主要组织相容性复合物(MHC)分子的线性肽相互作用。活化的 B 细胞和 T 细胞执行各种功能,如效应免疫,形成通过增殖的记忆或调节进一步的免疫反应。个体 B 细胞和 T 细胞的特异性由 AIR 序列定义。为了捕获广泛的抗原,体细胞 V(D)J 重组在个体的 B 细胞和 T 细胞群体中生成高度多样的 AIR 序列(图 5a)。商业化的 10x Chromium 单细胞免疫谱分析和 BD Rhapsody TCR/BCR 多组学检测可生成配对的转录组和 AIRR 数据。免疫受体分析可以使用 scirpy164、Dandelion165 或 scRepertoire166 等框架进行。
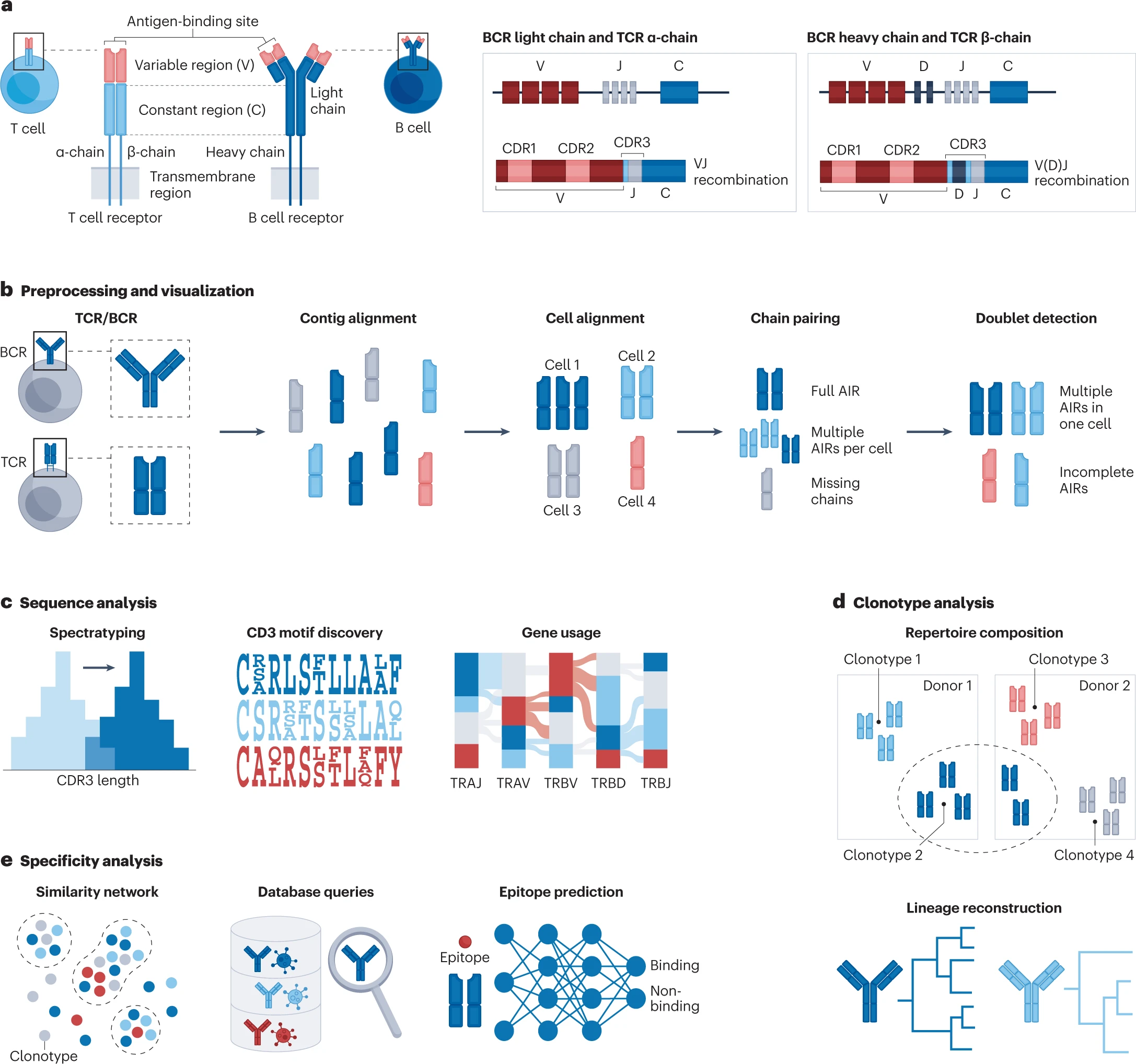
适应性免疫受体库
a. T 细胞受体(TCR)和 B 细胞受体(BCR)的结构。通过 V(D)J 重组生成多样的适应性免疫受体(AIR)库,其中 TCR α链和 BCR 轻链通过随机重排可变(V)和连接(J)基因片段生成,进一步的多样性(D)区域则整合到 TCR β链和 BCR 重链中。
b. 生成的 TCR/BCR 原始测序数据首先映射到 TCR/BCR 参考序列上,以获得由映射读数组装的连续序列(contigs)。在称为 contig 对齐的过程中,这些 contigs 通过 V(D)J 基因使用和互补决定区(CDR)1、2 和 3 氨基酸序列进行注释。细胞对齐后,获得的测量值需要匹配到理想的唯一完整 AIR 链。具有多个匹配 AIR、缺失链或双细胞的细胞会影响下游处理,应予以标记。
c. 通过频谱分析、基序发现和基因使用的过表达 V(D)J 序列调查,可以深入了解优先序列选择。
d. 通过克隆型组成分析和谱系重建可以识别克隆型,以重建最近的免疫反应。
e. 克隆型相似性网络的构建、数据库查询和表位预测提供了对 B 细胞和 T 细胞识别目标的见解。
解码 AIRR 序列特征¶
AIRR 序列可以通过 V(D)J 测序,然后进行比对和链配对来解码(图 5b)。尽管没有用于 TCR 序列重建的基准,MiXCR167 和 TRUST4(参考文献 168)被频繁使用。BALDR169、BASIC170 和 BraCer171 被证明可以稳健地恢复 BCR 序列 172,但不再维护。因此,我们鼓励分析人员也考虑使用 MiXCR 和 TRUST4 来处理 BCR 序列。V、D 和 J 基因的过表达组合提供了关于如何组合各种基因以创建 VJ 和 VDJ 链的宝贵信息。V(D)J 基因片段的重组和 V 与 J 片段的非精确连接产生了 VJ 和 VDJ 链中的 CDR3 区域,主要负责 AIR- 抗原结合。生发中心的 B 细胞在体细胞超突变期间进一步生成免疫球蛋白变体,免疫球蛋白基因在生产性重排的 V、D 和 J 片段内快速突变。AIRR 序列分析(图 5b)强调了与生物功能相关的 AIR 排列中优先选择的基因片段。对于频谱分析,观察在多种条件下的 CDR3 长度谱,这可能表明 AIRR 组成中的抗原特异性变化。序列基序通过频率分析揭示了 AIR 簇中 CDR3 位置上的保守和不同氨基酸(图 5c)。这些分析捕捉蛋白质序列特征以推断特异性并实现 AIR 设计。这些方法在 Scirpy、Dandelion 和 scRepertoire 中可用。
过滤功能适应性免疫受体¶
并非所有在等位基因重排期间生成的 AIR 链都形成功能性 AIR。在常规检测中会检测到仅分配给 VJ 或 VDJ 链的不完全 AIR,这些代表有效的细胞,但不能用于所有期望完整 AIR 的下游过程。淋巴细胞可以表达双重 AIR173,大约 10% 表达多个 VJ 链与单个 VDJ 链配对。表达双重 VDJ 链的淋巴细胞更为罕见(1%),应谨慎处理。然而,具有超过两个 VJ 或 VDJ 链分配的细胞总是表示双细胞。在下游分析中,将 AIR 状态与链配对信息和受体类型关联,可以实现特定任务的 AIR 选择,以确保尽可能多的数据被使用(图 5b)。例如,孤立的 VDJ 链仍可用于基于 CDR3-VDJ 链的数据库查询,但不能用于基于完整 AIR 的查询。可以在样本或条件等组中可视化链配对和受体类型的分布,应移除具有过多质量问题的异常簇。
识别和分类克隆型¶
由同一祖细胞衍生的 T 或 B 细胞群体形成克隆型,通常处于休眠状态,直到受到外部信号或自分泌因子的刺激。因此,特定细胞在克隆扩展期间显著增殖以履行各自预定的防御响应 174。克隆扩展的 T 或 B 细胞的持久性作为最近免疫响应的生物标志物。通过相同的 V 基因和 TCR 的 VJ 和 VDJ CDR3 的相同核酸序列或基于 BCR 的体细胞超突变系谱重建分析框架中实现的距离,可以识别克隆型(图 5d)。
在分析过程中,可以省略匹配 V 基因的要求,并可以将具有孤立链的细胞分配给相关克隆型。由于体细胞超突变,克隆系谱中的 B 细胞通常按其 CDR3 氨基酸序列中基于汉明距离的同源性分组,超过 80%175。公共克隆型出现在多个供体中,可能代表共享的免疫响应。相比之下,私人克隆型代表患者特异性克隆响应,可能对个性化医学有价值。样本级克隆型的丰度可以进一步用于通过 Jaccard 距离、多样性测量或层次聚类来比较 AIRR(图 5d)。
确定细胞特异性¶
影响 AIR- 抗原相互作用的最重要位置反映在 VDJ 链的 CDR3 中,较小程度上在 VJ 链的 CDR3176。T 细胞的抗原特异性由表位序列和整个 AIR- 表位复合物驱动。虽然可以通过条形码抗原实验确定 AIR 特异性 177,178,但有几种方法尝试计算推断(图 5e)。首先,可以直接或通过 Scirpy 或 immunarch179 查询包含现有研究中的 AIR- 表位对的数据库。常用的数据库有 IEDB180、PIRD181、vdjDB182(仅限 TCR)或 SAbDab(仅限 BCR)。与克隆型分配类似,数据库查询可以通过仅考虑 VDJ CDR3 序列或另外考虑 VJ CDR3 序列来进行,这可以减少 FDR。第二种方法使用距离度量直接应用于 CDR3 序列或序列嵌入来比较 AIR,因为具有相似序列的 AIR 可能具有共同特异性 183。虽然汉明距离常用于 BCR,因为它模拟了体细胞超突变,但 TCR 更常使用专门方法,如 TCRdist,通过变换成本和缺口惩罚比较两个 TCR 的所有 CDR3 序列 184,或 TCRmatch,使用 k-mer 基于其 CDR3β序列比较基序的重叠 185。第三种策略是最近的方法,使用机器学习工具如 ERGO-II176 直接预测 AIR 与表位之间的结合。所有三种方法都依赖于公共数据库,这些数据库主要包含常见研究疾病的数据,并缺乏关于 MHC 的信息以解码 T 细胞抗原特异性。
将适应性免疫受体与转录组测量结合¶
AIRR 测序通常与其他组学层结合,如表面蛋白和转录组测量,使得在感染或接种后对细胞命运进行详细观察 165。AIR 的存在可以通过分离免疫细胞簇并促进详细的 T 细胞注释来指导细胞类型注释。对于配对数据(见框 1),可以在特异性或克隆型网络等 AIR 条件下使用细胞类型簇进行表型 AIRR 分析,使用 Scirpy 和 scRepertoire。由于模态的固有结构差异,新方法如 TESSA186、mvTCR187 或针对 TCR 数据的 Conga188 以及针对 BCR 数据的 Benisse189 旨在整合两种模态,以便于联合注释和可视化。
空间中解析的单细胞数据¶
到目前为止,讨论的所有模态都是基于解离的单细胞组学技术,表征细胞身份和组织状态。然而,在多细胞生物中,细胞相互作用并形成空间结构的微环境,这些微环境在样本和条件之间可能有所不同。细胞组织弥合了组织生物学和病理学之间的差距,能够发现新的细胞功能,并为其需要不同分析方法的新计算挑战创造条件 190,191,192。空间组学通过向单细胞基因组学添加两个额外的模态:组织学成像和空间分析测量,来解析特征和细胞身份。个体细胞的空间定位有助于解开组织微环境及其功能依赖性。除了利用细胞的空间坐标生成更好理解组织结构之外,还可以使用组织学图像的非分子特征。添加从成像数据中提取的信息可以增强细胞识别 193,194 或分子特征的分辨率 195,或有助于识别空间变异模式 196。用于空间中基因表达谱的技术在空间分辨率(亚细胞与条形码区域,在区域间聚合特征)、检测效率、通量 192,197 和空间中解析的模态 198,199,200 上有所不同。目前开发的大多数分析方法都是针对空间转录组学的,因此我们重点推荐这些测量方法。两种主要的空间分子分析技术是基于阵列的 201,202(图 6a)和基于图像的方法 203,204,205(图 6b)。各种评论提供了对不同实验技术的详细概述 192,206,207,208。分析空间数据集需要专门针对这种模态的分析工具,可以使用 Squidpy209、Giotto210、Seurat45 或 SpatialExperiment211 等框架进行。
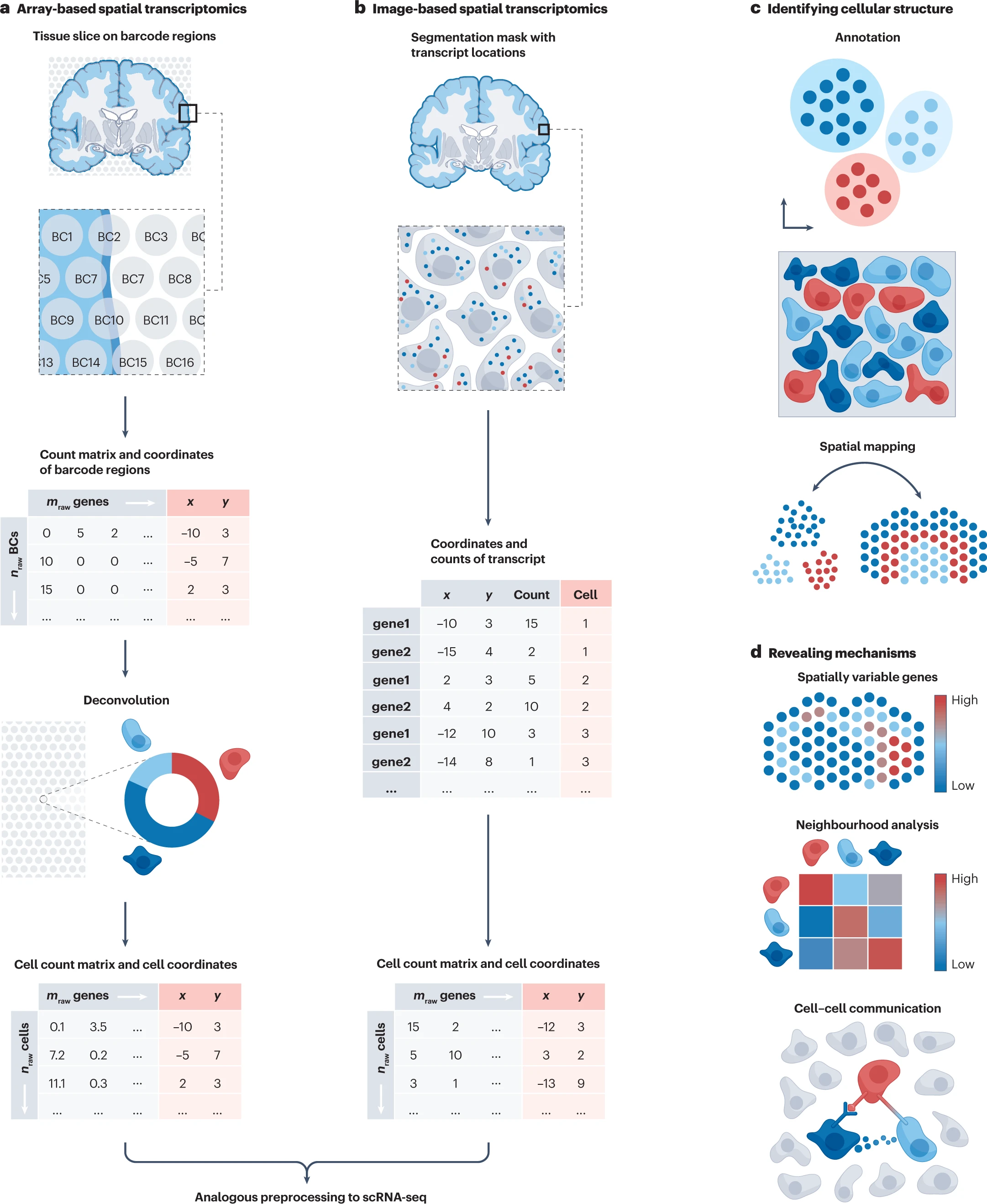
空间转录组学技术
a. 基于阵列的空间转录组学技术在预定义的条形码(BC)区域内量化基因表达,这些区域的跨度在 10μm 到 200μm 之间。BC 区域包含来自多个细胞的测量值,生成计数矩阵和空间坐标,其中每个观测值是一个 BC 区域。细胞类型解卷积方法将个体 BC 区域的细胞组成分解,获得计数矩阵和空间坐标,其中每个观测值是一个单细胞。进一步的预处理可以类似于单细胞 RNA 测序(scRNA-seq)数据集的分析进行。
b. 基于图像的空间转录组学,如荧光原位杂交(FISH)和原位测序(ISS)技术,通过多轮顺序杂交捕获转录本的个体位置。转录本位置可以聚合,获得单细胞水平的计数矩阵和空间坐标。后续处理同样类似于 scRNA-seq 的处理方式进行。
c. 在空间转录组学中,可以在单细胞或 BC 区域分辨率上识别细胞结构。基于图像的空间转录组学由于仅测量目标转录本子集而导致的小特征空间限制,可以通过空间映射解决,该方法将未测量的转录本推测到空间坐标上。
d. 空间转录组学中的机制可以通过识别在空间上变化的基因、分析细胞的邻域和基于受体和配体、紧密连接、机械效应或间接机制推断通信事件来相对于细胞的空间位置进行分析。
获得细胞的计数矩阵和空间坐标¶
无论是基于阵列还是基于图像的空间转录组学,都需要特定工具将测量的分子分配给单个细胞。由于基于阵列的检测不捕捉单细胞分辨率,因此斑点的基因表达谱反映的是细胞类型组成而非独立细胞类型。各种方法被提出用于分解基于阵列的基因表达谱中的基因表达谱。Cell2location212、SpatialDWLS213 和 RCTD214 基于单细胞分辨参考中细胞群体的基因表达谱估计每个斑点的细胞类型组成。对于模拟数据集,cell2location 在细胞类型解卷积方面表现优于其他方法,但需要更多计算资源,而对于真实数据集,SpatialDWLS 和 RCTD 在基于四种不同准确性指标的总体准确性评分上表现最佳 215,216。
对于基于图像的检测,如荧光原位杂交(FISH)和原位测序(ISS),通过细胞分割获得细胞计数矩阵和空间坐标 217,218,219,220。由于空间转录组学数据的复杂性(在检测方法、分辨率和组织变化方面),这些工具通常需要手动微调以获得有价值的分割结果。Giotto 和 Squidpy 等处理管道允许将定制的分割方法添加到分析管道中,这简化了所选方法的比较、选择和评估。此外,转录本的定位可以用于无分割方法,如 SSAM221 或 Baysor222,直接将细胞标签分配给空间邻近像素。Baysor222 还结合了通过组织学图像获得的细胞形状信息,以增强分割结果。这些工具可以作为基于分割方法的有用替代方案。
通过基于阵列的空间转录组学获得的基因表达矩阵,随后进行细胞类型解卷积,或通过基于图像的空间转录组学获得的基因表达矩阵,随后进行分割,可以类似于 scRNA-seq 数据进行过滤、标准化和可视化。
细胞身份和细胞微环境的表征¶
对于单细胞分辨率的基于成像的空间转录组学数据,可以类似于 scRNA-seq 数据对细胞进行注释(图 6c)。这些技术通常仅读出预定义的转录本集。基因通常基于从 scRNA-seq 获得的先前生物学知识选择(探针选择),可能不适合识别稀有细胞亚群体,这导致对已知细胞类型的偏倚。将标准的空间无关 scRNA-seq 数据与有针对性地空间解析的数据对齐,可以实现整个转录组(在标准 scRNA-seq 中测量)的空间解析方式推测,并尝试解决目标特征空间的限制。这种方法生成转录组范围内的单细胞分辨率空间转录组学数据。Tangram224 通过优化空间数据与 scRNA-seq 数据之间的基因相似性,推测空间样本中未检测到的转录本。证明其在各种准确性指标和可扩展性方面优于其他推测方法,如 gimVI225 和 SpaGE226。
除了仅基于基因表达谱注释细胞外,还可以利用空间位置识别细胞身份。BayesSpace227、stLearn228 和 spaGCN229 等工具通过考虑基因表达的共同性和空间邻域结构识别所谓的空间域。获得的标签可以用于识别组织中具有相似表达谱的区域,这些区域可能对应于数据集的整体形态。
跨不同样本识别细胞微环境可能因图像方向的差异而受到阻碍。图像在整个数据集中可能并不总是完美对齐,因此跨不同视野比较发现可能具有挑战性。Tangram224、GridNet230 和 eggplant231 生成跨样本的共同坐标框架,以减轻这一问题 232。
识别与细胞组织和组织结构相关的空间模式¶
细胞微环境为驱动组织状态的机制提供新的见解,可以通过多种方式进行分析(图 6d)。基因表达差异分析在 scRNA-seq 中广泛探索,用于识别高度可变基因和 DGE 分析。对于空间转录组学数据,这补充了空间可变基因(SVGs)的识别。为此目的的方法在其假设和 SVGs 的定义方面有很大差异,目前尚无关于如何最好地识别 SVGs 的共识。例如,SPARK233 和 SpatialDE234 利用空间相关性测试,BayesSpace227 使用马尔可夫随机场,spaGCN229 使用图神经网络整合基因表达数据、空间信息和组织学图像,sepal235 则利用基于扩散的建模识别具有空间模式的基因。
跨细胞的空间依赖通信事件¶
在组织中,细胞有直接接触,可以通过表面结合的配体和受体、长程旁分泌效应、生物力学力和代谢物交换等间接机制相互作用。这些事件通常被称为对基因表达变异的外在影响,应在描述细胞组织和组织生态位时考虑 236。可以如上所述在解离的 scRNA-seq 数据中识别细胞通信事件。然而,这些方法通常忽略了底层组织的空间组织,可能导致假阳性发现。空间细胞 - 细胞通信方法通常基于周围邻近细胞的基因表达模式进行比较。GCNG237、Misty238 和 NCEM236 将此任务表述为细胞的空间图和图神经网络,SpaOTsc239 使用最优传输,SVCA240 利用空间方差成分分析量化细胞 - 细胞通信事件对基因表达谱的影响。
结论和未来展望¶
我们在此回顾了典型的单模态和多模态转录组学、染色质可及性、表面蛋白、AIRR 和空间解析单细胞数据的分析步骤。我们的工作为该领域的新手提供了一个入门点,同时更新了经验丰富的分析人员关于最近分析最佳实践的知识。所有推荐基于独立基准测试,这些基准测试不可避免地落后于最新方法的开发。随着进一步的基准测试发布,个别工具推荐可能会发生变化,需要定期更新以确保最佳实践单细胞分析。因此,我们推荐我们的单细胞最佳实践在线书籍,该书提供详细的方法描述,展示如何将我们的推荐付诸实践,并作为分析模板。我们的在线书籍将包含定期更新,作为多组学单细胞分析领域的新手和专家的灵活且最新的指南。然而,我们预计本文中概述的分析工作流程在很大程度上仍将有效,并对应于最广泛使用的分析工作流程。
除了不断增加的方法数量,生成的单细胞数据集数量也在增加,我们预计从大型数据集中学习(如集成图谱)将变得更加重要。大型数据集使得开发描述细胞和个体异质性的模型成为可能,例如,通过潜在空间嵌入。单细胞变分推断 41 等框架学习的潜在表示可以用于批次校正、聚类、可视化和 DGE 分析。通过跳过手动质量控制步骤,这些模型简化了单细胞数据的分析。建立在这些潜在空间上的模型,通过查询到参考映射方法变得具有预测性,这将使单细胞分析从无监督的探索性分析方法转变为由监督预测补充。构建多模态参考图谱将进一步使同时在多个层次上表征细胞状态成为可能,即使对于单模态查询也能提供多模态见解。
理解这些多组学细胞状态上的扰动效应将变得越来越重要。高度平行的扰动筛选,如基因组规模的 Perturb-seq117,已经测量了全基因组的扰动效应。将基因组规模的 Perturb-seq 与进一步的模态结合,使得系统性探索基因组景观以揭示特定环境的基因调控网络成为可能。这进一步扩展了单细胞基因组学在药理学应用(如药物靶点筛选)中的应用。我们预计将引入更多分析方法,分解成功和失败的扰动,并从多模态数据中推断基因调控网络,如 CellOracle241 或 SCENIC+242(图 2c)。此外,新的分子测量方法如单细胞蛋白质组学这一快速发展的领域正在出现。这些测量的分析方法稀缺,选择性地进行了基准测试,尚未制定最佳实践。