Large scale foundation model on single cell transcriptomics
摘要¶
大型预训练模型已经成为基础模型,在自然语言处理及相关领域取得了突破性的进展。开发用于解码细胞“语言”并促进生物医学研究的基础模型具有广阔前景,但也面临挑战。在此,我们开发了一个大型预训练模型 scFoundation,也称为“xTrimoscFoundationα”,具有 1 亿个参数,涵盖约 20,000 个基因,并在超过 5,000 万个单细胞转录组数据上进行预训练。scFoundation 在可训练参数的规模、基因的维度和训练数据的体量方面是一个大规模模型。其不对称的类似 Transformer 的架构和预训练任务设计能够有效捕捉各种细胞类型和状态下基因之间复杂的上下文关系。实验显示,scFoundation 作为一个基础模型,在各种单细胞分析任务中表现出色,达到了最先进的水平,这些任务包括基因表达增强、组织药物反应预测、单细胞药物反应分类、单细胞扰动预测、细胞类型注释和基因模块推断。
主要内容 大规模预训练模型正在革新自然语言处理及相关领域的研究,并逐渐成为通用人工智能的新范式。这些在庞大语料库上训练的模型因其在许多下游任务中引领突破的基本作用以及在识别语言中的模式和实体关系方面的能力而成为基础模型。在生命科学中,生物体也有其内在的“语言”。细胞作为人体的基本结构和功能单位,构成了由 DNA、RNA、蛋白质和基因表达值等无数“词汇”组成的“句子”。一个引人入胜的问题是:我们能否基于大量细胞“句子”开发细胞的基础模型?
单细胞 RNA 测序(scRNA-seq)数据,也称为单细胞转录组学,提供了对细胞系统的高通量观察 2,为开发基础模型提供了所有类型细胞的转录组“句子”的庞大档案。在转录组数据中,基因表达谱描绘了细胞内基因共表达和相互作用的复杂系统。随着人类细胞图谱(HCA)3 及许多其他研究 4,5,6,7,8 的努力,数据规模正在指数级增长 9。大约 20,000 个编码蛋白的基因跨越数百万个细胞,其观察到的基因表达值达到了万亿“标记”(补充表 1)的规模,这与用于训练大型语言模型(LLM)如生成预训练变换器的自然语言文本的量相当。这为我们预训练一个大规模模型以类似 LLM 从大量自然语言文本档案中学习人类知识的方式,提取细胞内部复杂、多方面的模式提供了基础。
在 LLM 的预训练中 10,11,模型和数据规模的增长对于构建能够有效挖掘复杂多层次内部关系的基础模型至关重要。最近,在单细胞数据上的预训练模型取得了一些进展 12,13,14,15,但创建大规模基础模型仍然面临独特的挑战。首先,基因表达预训练数据需要涵盖不同状态和类型的细胞全景。目前,大多数 scRNA-seq 数据组织松散,仍缺乏一个综合和完整的数据库。其次,将每个细胞建模为一个句子,将每个基因表达值建模为一个词时,近 20,000 个编码蛋白的基因使得“句子”异常冗长,这是传统变换器难以处理的场景 16,17。现有工作通常不得不将模型限制在一小部分选定的基因上。第三,不同技术和实验室的 scRNA-seq 数据在测序深度上表现出高度差异。与由于污染等技术效应引起的随机噪声不同,测序深度的差异不是随机的,这种差异阻碍了模型学习统一和有意义的细胞和基因表示。
在本文中,我们解决了这些挑战,设计了一个处理大约 20,000 个基因、拥有 1 亿个参数的大规模基础模型 scFoundation。我们收集了超过 5,000 万个基因表达谱的 scRNA-seq 数据进行预训练。我们为 scRNA-seq 数据开发了一种不对称架构,以加速训练过程并提高模型的可扩展性。我们设计了一种感知读深度(RDA)的建模预训练任务,使 scFoundation 不仅能够建模细胞内的基因共表达模式,还能连接不同读深度的细胞。
为了验证 scFoundation 的能力,我们在多个下游任务上进行了实验,包括细胞聚类、大数据药物反应预测、单细胞药物反应分类、单细胞扰动预测和细胞类型注释。考虑到用户微调大规模模型的计算负担,我们通过将非微调或轻微微调的 scFoundation 的上下文 embedding 适配到相应的下游模型中,实现了先进的性能。我们还展示了使用 scFoundation 的基因 embedding 来推断基因模块和基因调控网络。所有结果都展示了 scFoundation 在转录组数据分析中的强大功能和价值,并作为基础功能促进生物学和医学任务学习。该工作探索并推动了单细胞领域基础模型的边界。
结果¶
scFoundation 预训练框架¶
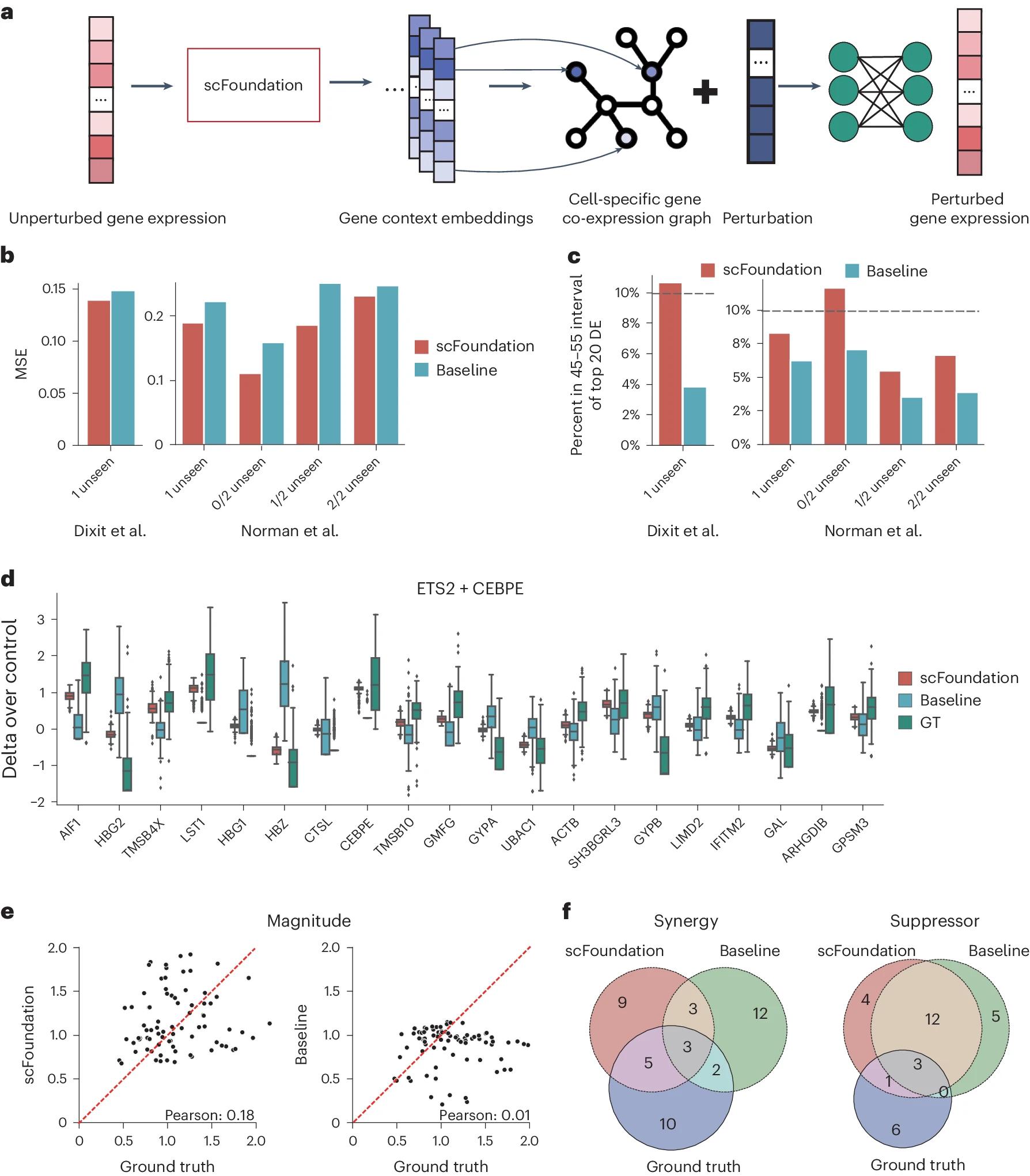
我们开发了 scFoundation 来建模 19,264 个基因,拥有大约 1 亿个参数,在超过 5,000 万个 scRNA-seq 数据上进行预训练。这是单细胞领域中具有大参数规模、基因覆盖范围和数据规模的大型模型。能够高效训练这样一个模型的能力由我们的预训练框架中的三个关键部分提供:模型设计、预训练任务和数据收集(图 1a)。
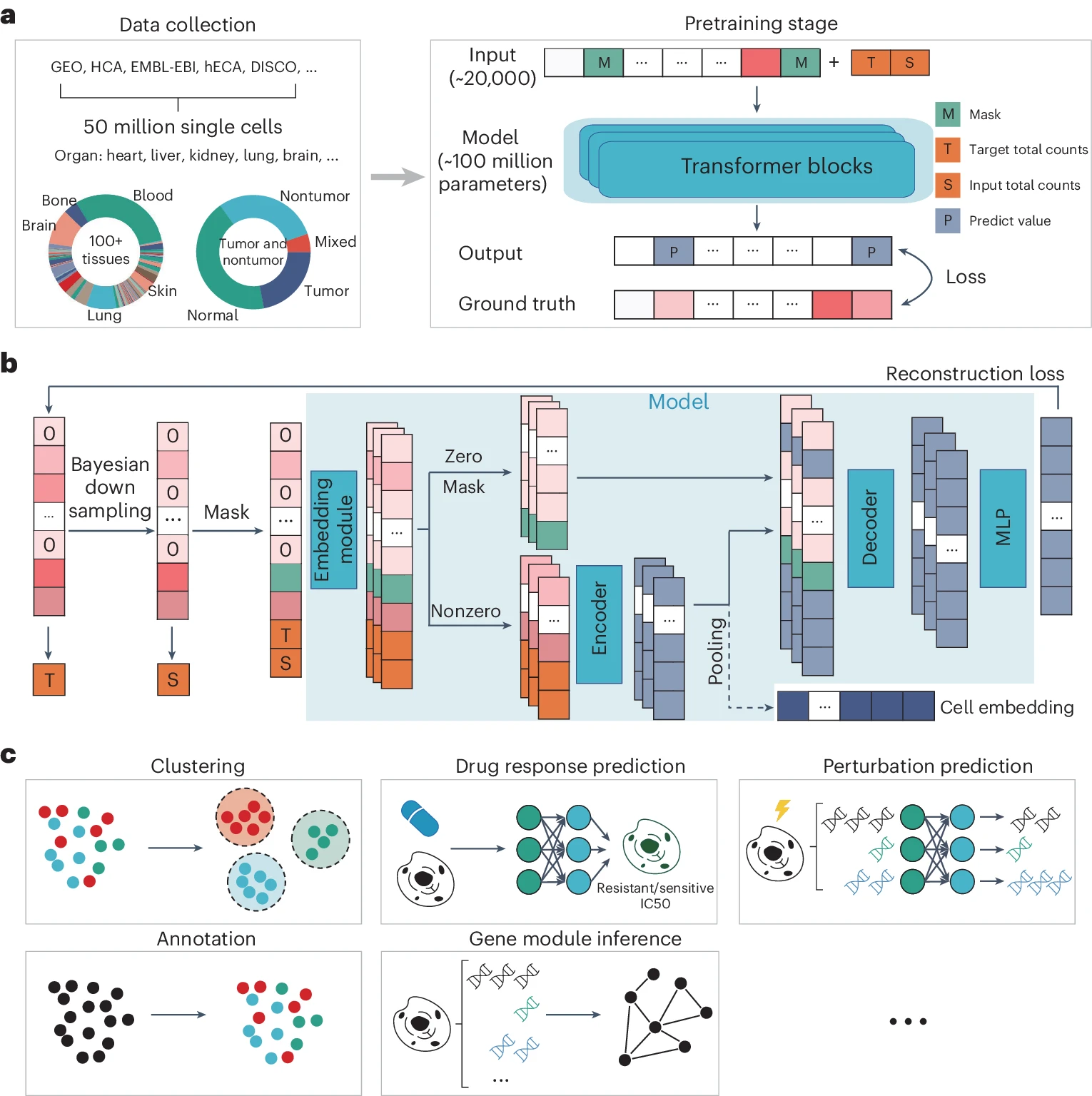
a. 收集了五千万单细胞基因表达谱,覆盖了来自各种组织的肿瘤和非肿瘤细胞。这些数据用于 RDA 建模任务来预训练模型。在 RDA 任务中,输入由被掩盖的基因表达向量和两个总计数指示器(T 和 S)组成。输出是所有基因的预测表达值,损失在掩盖位置处计算。
b. 预训练过程的概述。一个原始基因表达向量作为训练样本。层次贝叶斯下采样策略生成输入样本。计算原始和输入样本的基因表达总计数(T 和 S)。随机掩盖输入样本中的值。将标量表达值转换为 embedding。仅将对应于非零和非掩盖值(包括 T 和 S)的 embedding 输入到模型编码器中。然后将编码器的输出 embedding 与掩盖和零 embedding 结合并输入解码器。此外,编码器输出还可以被汇聚以生成用于下游使用的细胞 embedding。解码器输出 embedding 通过共享的 MLP 层投影到基因表达值上。计算预测值与原始样本的基因表达值之间的回归损失。
c. 预训练 embedding 可以作为基因表达谱的替代品,促进下游任务,如细胞聚类、药物反应预测、单细胞水平扰动预测、细胞类型注释、基因模块推断等。
我们开发了 xTrimoGene,这是一种具有算法效率和工程加速策略的可扩展 Transformer 模型 18。该模型包括一个 embedding 模块和一个不对称的编码器 - 解码器结构。embedding 模块将连续的基因表达标量转换为可学习的高维向量,确保完全保留原始表达值,这是对先前模型中使用的离散化值的显著改进 13,19。不对称编码器 - 解码器结构类似于计算机视觉中的掩码自动编码器 20 模型,但设计上适应了 scRNA-seq 数据的高稀疏性,实现了对所有基因关系的高效学习,无需任何选择。此外,我们在模型部署中结合了多种大规模模型训练优化技术,以确保高效训练(方法部分)。
我们设计了一种名为 RDA 建模的预训练任务,这是掩码语言建模 21 的扩展,考虑了大规模数据中读取深度的高度差异。在 RDA 建模中,模型根据其上下文基因预测细胞的掩码基因表达。上下文来自该细胞基因表达谱的复制或低读取深度变体(方法部分)。我们将总计数视为一个细胞的读取深度,并定义了两个总计数指示器:T(“目标”)和 S(“源”),分别用于原始和输入样本的总计数。我们随机掩盖输入样本中的零和非零表达基因,并记录其索引。然后模型使用掩码输入样本和两个指示器来预测掩码索引处的原始样本的表达值(图 1b)。这使得预训练模型不仅能够捕捉细胞内的基因 - 基因关系,还能协调不同读取深度的细胞。在推理过程中,我们将细胞的原始基因表达输入预训练模型,并将 T 设置为高于其总计数 S,以生成增强读取深度的基因表达值。我们通过细胞聚类性能评估进行了一些消融实验,以展示我们模型架构和预训练任务设计的优势(方法部分和补充说明 1)。
我们通过收集所有公开可用的单细胞资源的数据构建了一个全面的单细胞数据集,包括基因表达综合数据库(GEO)22、单细胞门户、HCA3、人类集成细胞图谱(hECA)4、深度集成的人类单细胞组学数据(DISCO)7、欧洲分子生物学实验室 - 欧洲生物信息学研究所数据库(EMBL-EBI)8 等。我们将所有数据对齐到一个由 HUGO 基因命名委员会 23 确定的包含 19,264 个编码蛋白质和常见线粒体基因的基因列表上。经过数据质量控制(方法部分),我们获得了超过 5,000 万个用于预训练的人类 scRNA-seq 数据。丰富的数据来源使预训练数据集中充满了生物学模式。在解剖学上,它跨越了 100 多种不同疾病、肿瘤和正常状态下的组织类型(图 1a),几乎涵盖了所有已知的人类细胞类型和状态。
在预训练之后,我们将 scFoundation 模型应用于多个下游任务(图 1c)。scFoundation 编码器的输出被汇聚成细胞级 embedding,用于细胞级任务,包括细胞聚类(跨数据集和单个数据集)、大数据和单细胞级药物反应预测和细胞类型注释。scFoundation 解码器的输出是基因级上下文 embedding,用于基因级任务,如扰动预测和基因模块推断。
无需微调的可扩展读取深度增强模型¶
在我们的研究中,我们发现验证损失与模型规模和计算量的增加呈幂律下降,这在 LLM 中被称为“缩放定律”10,24。我们分别训练了参数规模为 300 万、1000 万和 1 亿的三种模型,并记录了它们在验证数据集上的损失。随着模型参数和浮点运算总数(FLOPs)的增加,验证数据集上的损失呈幂律下降。然后我们估计了各种规模的 xTrimoGene 架构模型的性能,其参数规模与之前的基于 Transformer 的模型相当 13,14,15,并与 scVI25 进行了比较(补充说明 2)。具有 1 亿参数的 scFoundation 模型超越了所有其他模型(图 2a)。我们进一步在细胞类型注释任务上评估了我们的三种模型,并观察到随着模型规模的增加,性能得到了提升(补充表 2)。
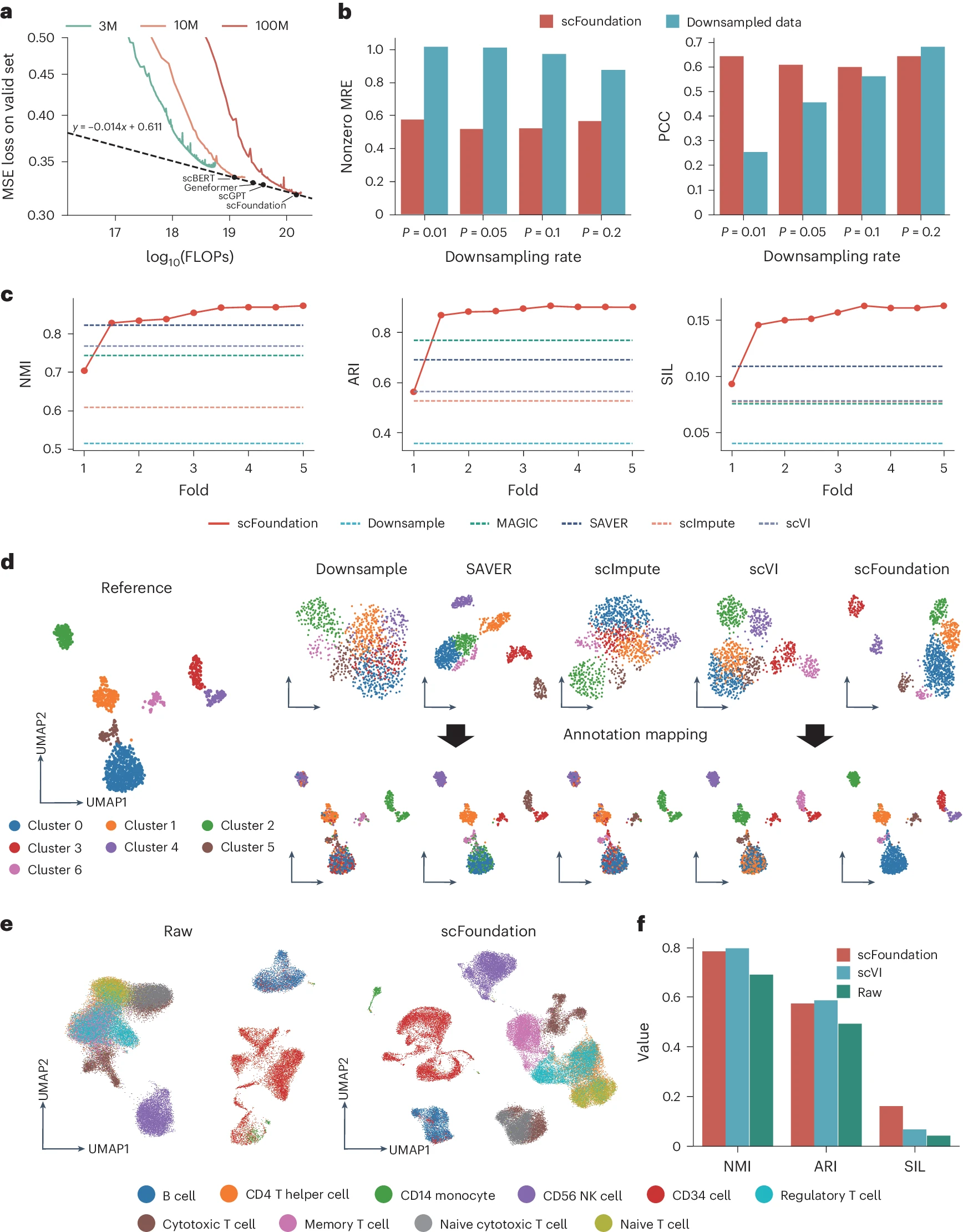
a. 在不同参数规模和 FLOPs 下的训练损失。图中标注其他模型名称的点表示参数规模相当于其他模型的不同规模 xTrimoGene 架构模型的性能。scVI 模型的均方误差(MSE)为 0.98。由于它不是基于 Transformer 的模型,因此不适合在图中绘制。
b. 在未见过的数据集上评估读取深度增强性能。使用非零基因的相对均方误差(MRE)和所有基因的皮尔逊相关系数(PCC)来评估恢复的基因表达性能。较低的 MRE 和较高的 PCC 表明性能更好。
c. 基于细胞聚类指标比较 scFoundation 模型与其他插补方法。x 轴表示期望总计数与输入总计数之间的倍数变化,y 轴表示得分。
d. 不同方法生成的细胞 embedding 的 UMAP 图。左图显示了使用原始基因表达获得的参考 UMAP 图,颜色表示细胞簇。右上图显示了不同方法获得的聚类结果:下采样(无插补)、SAVER、scImpute、scVI 和 scFoundation。簇的数量是对齐的。右下图显示了映射到参考 UMAP 图上的每种方法的聚类结果。
e. 比较原始基因表达和 scFoundation 插补的细胞 embedding 在 Zheng68K 数据集上的 UMAP 图。
f. 比较 scFoundation、scVI 和原始数据在 Zheng68K 数据集上的聚类性能。
RDA 建模通过设置 T 值高于 S 值,使 scFoundation 能够增强输入细胞的读取深度。我们在独立测试数据集上评估了这一能力,从验证数据集中随机抽取了 10,000 个细胞。我们将总计数下采样至原始谱的 1%、5%、10% 和 20%,生成四个具有不同总计数倍数变化的数据集。对于每个数据集,我们使用未微调的 scFoundation,通过将期望总计数 T 设置为采样率的倒数来增强低总计数的细胞。我们测量了预测和实际非零基因表达之间的平均绝对误差(MAE)、平均相对误差(MRE)和皮尔逊相关系数(PCC)。如图 2b 和补充图 1 所示,即使下采样率低于 10%,scFoundation 也显著降低了 MAE 和 MRE 至原始数据的一半。这些观察结果表明 scFoundation 在极低总计数情况下也能增强基因表达。
接下来,我们将 scFoundation 与包括 MAGIC26、SAVER27、scImpute28 和 scVI25 在内的插补方法进行了比较,使用的是由 SAVER 处理的人类胰岛数据集。该数据集包含手动生成的下采样基因表达谱及其对应的参考数据。对于 scFoundation,我们通过设置 T 为 S 的不同倍数(从 1 到 5),从未微调的编码器中获得了五组细胞 embedding。对于其他方法,我们首先使用下采样数据训练这些方法,然后分别从 scVI 和其他方法中获得插补的细胞 embedding 和基因表达。参考数据中的簇标签用作地面真相(方法部分)。为了评估聚类准确性,我们使用了归一化互信息(NMI)、调整后的兰德指数(ARI)和轮廓系数(SIL)等指标(补充说明 3)。使用下采样数据获得的聚类性能作为基线。
当 T 设置为等于 S 时(倍数变化为 1;图 2c),scFoundation 在所有指标上均优于基线和 scImpute,但与 SAVER 等较小模型相比表现较低。最近的研究 29 中也报告了这种读取深度未改变的现象。随着 T/S 倍数的增加,我们观察到 scFoundation 的性能迅速提高,超过了所有其他方法。在更高的 T/S 倍数下,其性能达到平台期,表明细胞 embedding 对高于 3.5S 的 T 值不敏感。我们在倍数变化为 5 时可视化了 scFoundation 的 embedding 结果及其他方法的结果(图 2d)。值得注意的是,scFoundation 的细胞 embedding 显示出比基线和其他方法更明显的簇边界。此外,我们将所有方法的结果进行聚类,并将簇标签应用到参考统一流形近似与投影(UMAP)上。其他方法显示出混合标签,尤其是地面真相中的簇 0。scFoundation 是唯一将所有细胞簇分配与参考结果一致的方法。
然后,我们将 scFoundation 应用于 Zheng68K 数据集 30,该数据集包含约 60,000 个人类外周血单个核细胞,在早期 10x Chromium 平台上测序。每个细胞表达约 500 个基因,总读取数少于 2,000,使得细胞类型区分具有挑战性 13,31(补充图 2)。我们在未微调情况下使用 scFoundation,通过设置 T 值为 10,000 来增强细胞 embedding。生成的 UMAP 图显示,scFoundation 有效地将记忆 T 细胞与其他 T 细胞分离,并更好地区分了 CD14 单核细胞和 CD34 细胞(图 2e)。我们将结果与在相同数据集上训练的 scVI 进行了比较。两种方法在聚类方面均优于原始数据。尽管它们的 NMI 和 ARI 指标相似,scFoundation 的 SIL 得分更高,显示了其在未微调模式下的泛化能力(图 2f)。
scFoundation 还展示了其在不同批次间促进读取深度增强聚类的能力。需要注意的是,单纯对齐读取深度无法完全消除批次效应,因为批次效应可能涉及其他变化,如供体性别、实验处理、细胞周期等 32。我们通过将读取深度增强的细胞 embedding 输入到非可训练的下游头部 BBKNN33 中,将来自不同批次的单细胞数据进行映射。模拟数据以及来自类器官和体内实验的数据的结果表明,scFoundation 可以更好地实现细胞映射,同时略微减少不同细胞类型的离散性(补充表 3 和补充图 3 和 4;详情见补充说明 4)。
这些结果表明,scFoundation 具有增强细胞读取深度的能力。值得注意的是,scFoundation 与其他插补方法之间的一个重要区别在于,scFoundation 无需进行特定数据集的微调即可达到最佳性能。
改进癌症药物反应预测¶
癌症药物反应(CDR)研究肿瘤细胞在药物干预下的反应。计算预测 CDR 对于指导抗癌药物设计和理解癌症生物学至关重要 34。我们将 scFoundation 与 CDR 预测方法 DeepCDR35 结合,预测多个细胞系数据的半数抑制浓度(IC50)值。这个实验验证了 scFoundation 是否可以为大规模基因表达数据提供有用的 embedding,尽管它是在单细胞上训练的。
原始 DeepCDR 模型使用药物结构信息和多组学数据作为输入,输出预测的 IC50 值。在这里,我们专注于基因表达数据,并用 scFoundation 替代 DeepCDR 中的转录组多层感知器(MLP)子网络(图 3a)。我们使用癌症细胞系百科全书 36 和癌症药物敏感性基因组 37 数据集获取输入的细胞系基因表达数据、输入药物和 IC50 标签(方法部分)。
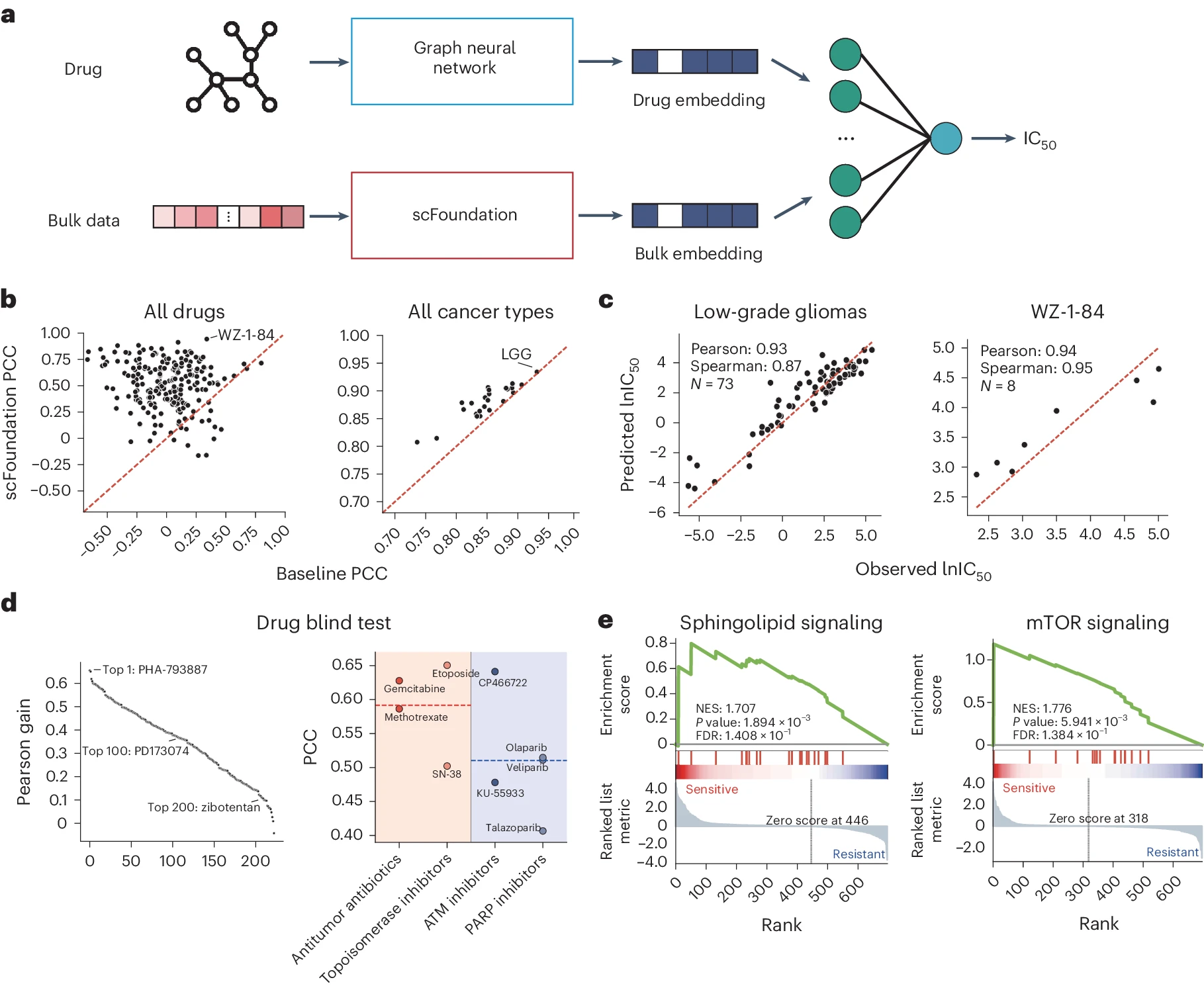
a. 基于 scFoundation 的 CDR 预测模型的示意图。
b. 测试集中所有药物和癌症类型之间的 PCC。每个点代表一种药物或癌症类型,x 轴和 y 轴分别显示基线 CDR 模型和基于 scFoundation 模型获得的 PCC。
c. 对药物 WZ-1-84 在癌症类型低级别胶质瘤上的预测和观察到的 IC50 值的比较。每个点代表一种药物和细胞系组合。
d. 留一药物盲测性能。皮尔逊增益图显示了用 embedding 替换基因表达获得的 PCC 增益。每个点代表一种药物,y 轴表示获得的 PCC 值,x 轴表示排名。排名较高的药物具有更高的 PCC 增益。在 PCC 图中,每个点代表一种药物,y 轴表示预测和真实 IC50 之间的 PCC,x 轴代表四种药物类型。前两种属于化疗,后两种属于靶向治疗。虚线显示了每种治疗类型内的平均 PCC。
e. GSEA 在预测 IC50 值较低的细胞系数据上的结果。鞘脂信号通路在阿霉素敏感的细胞系中富集,而 mTOR 信号通路在伏立诺他敏感的细胞系中富集。P 值是单侧的,并从标准 GSEA 置换测试中计算得出。对于假发现率(FDR)值,进行了多重比较的调整。NES,归一化富集分数。
我们评估了基于 scFoundation 的结果与基因表达结果在多种药物和癌症细胞系上的性能(图 3b)。使用 scFoundation embedding,大多数药物和所有癌症类型都获得了更高的 PCC。我们可视化了药物和癌症类型的最佳预测案例(图 3c)。无论 IC50 高低,基于 scFoundation 的 DeepCDR 模型都能预测出准确的值,PCC 超过 0.93。在药物盲测中,我们一次从数据集中留出一种药物,基于 scFoundation 的模型始终优于原始模型(图 3d)。排名第 1 的 PCC 增益药物 PHA-793887 是一种强效的 ATP 竞争性 CDK 抑制剂,其 PCC 从 0.07 提高到 0.73。即使是排名第 200 的用于阻断内皮素 A 受体活性的药物 zobotentan,其 PCC 也从 0.49 提高到 0.64。
我们进一步将药物分为不同的治疗类型,以检查 IC50 预测性能是否与其内在机制有关。基于 scFoundation 预测结果,属于化疗的药物(如抗肿瘤抗生素和拓扑异构酶抑制剂)比属于靶向治疗的药物(如失调性毛细血管扩张突变(ATM)和聚(ADP- 核糖)聚合酶(PARP)抑制剂)具有更高的 PCC(图 3d)。这可能是因为特定基因突变往往对靶向治疗有重要影响 34,但这些信息在基因表达数据中难以揭示,而化疗药物被广泛报道与基因表达相关 38,39,因此其 IC50 更容易预测。至于基因表达结果,整体 PCC 较低,我们没有观察到治疗类型之间的性能差异。
然后我们使用我们的模型预测数据中未知的 CDR。为了验证这些预测,我们对新预测的相对低 IC50(表明细胞系对药物敏感)进行了基因集富集分析(GSEA)40(图 3e)。例如,鞘脂信号通路在阿霉素敏感的细胞系中富集。根据京都基因和基因组百科全书(KEGG)数据库 41,该通路与鞘磷脂及其代谢有关。鞘磷脂被报道与阿霉素协同作用,通过改变细胞膜通透性,使这些细胞系的药物 IC50 降低 42。mTOR 信号通路在伏立诺他敏感的细胞系中富集。先前的研究表明,伏立诺他通过抑制 mTOR 信号通路抑制癌症生长 43。其他临床研究也表明,mTOR 抑制剂常与伏立诺他联合使用 44,45,这表明伏立诺他与 mTOR 通路之间存在关系。这些例子支持了我们预测的有效性。
尽管 scFoundation 在单细胞转录组数据上进行预训练,但学到的基因关系可以转移到大规模表达数据上,以生成简洁的 embedding,从而促进更准确的 IC50 预测。这些发现展示了 scFoundation 在扩展对癌症生物学中药物反应的理解以及可能指导更有效抗癌治疗设计方面的潜力。
转移大规模药物反应到单细胞¶
推断单细胞水平的药物敏感性可以帮助识别表现出不同药物耐药特性的特定细胞亚型,提供关于潜在机制和潜在新疗法的宝贵见解 46。我们将 scFoundation 应用于基于名为 SCAD47 的下游模型的单细胞水平药物反应分类这一关键任务。由于单细胞药物反应数据有限,SCAD 使用领域适应来消除单细胞和大规模数据的差异,并转移在大规模数据上学习的知识以推断单细胞的药物敏感性。该过程同时将大规模和单细胞数据作为输入,并输出每个细胞的预测敏感性。在我们的设置中,我们使用未微调的 scFoundation 获取大规模和单细胞数据的统一 embedding,并使用这些 embedding 来训练 SCAD 模型(图 4a)。
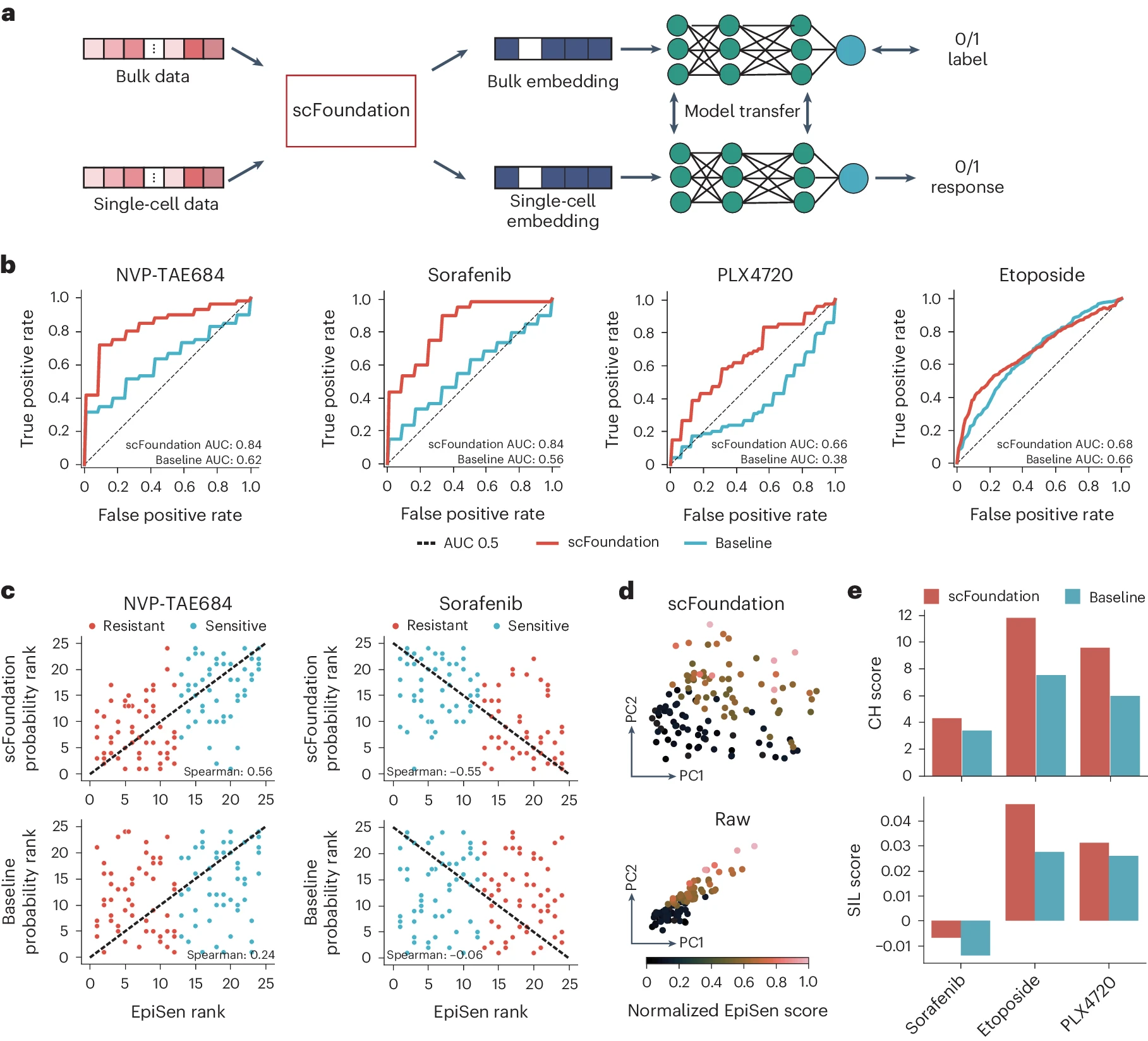
a. 基于 scFoundation 的单细胞响应分类模型的示意图。
b. 四种药物的受试者工作特征(ROC)曲线。红线和蓝线分别代表基于 scFoundation 模型和基线 SCAD 模型的性能。AUC,受试者工作特征曲线下面积。
c. 药物敏感性概率与标准化 EpiSen 评分之间的相关性。每行对应一个模型,每列代表一种药物。
d. 用 scFoundation embedding 和原始数据绘制的 SSC47 单细胞数据集的主成分分析(PCA)图。颜色表示参考 EpiSen 评分。具有不同 EpiSen 评分的细胞对药物的反应不同。
e. 在所有三个与药物相关的大规模数据集上的聚类性能。每个大规模数据集都有两种标签:敏感和耐药。
我们专注于四种在原始研究中表现较低的受试者工作特征曲线下面积(AUC)值的药物(索拉非尼、NVP-TAE684、PLX4720 和依托泊苷)。这些药物在癌症药物敏感性基因组数据库中有大规模数据的药物敏感标签,而真实的细胞水平药物敏感标签通过不同方式获得。对于药物 PLX4720 和依托泊苷,未处理细胞系中的细胞被认为是敏感的,而药物暴露后存活的细胞被认为是耐药的 48。对于索拉非尼和 NVP-TAE684,细胞的敏感标签通过与药物反应相关的衰老相关(EpiSen)程序分数的值来确定 49(方法部分)。
我们将基于 scFoundation 的模型与以所有基因表达值为输入的基线 SCAD 模型进行了比较。基于 scFoundation 的模型在所有药物上的 AUC 值均有所提高,尤其是在 NVP-TAE684 和索拉非尼上,AUC 增加超过 0.2。所有四种药物的基线结果至多为 0.66,有一个结果甚至低于随机,这突显了任务的难度(图 4b)。我们使用 Spearman 相关性评估了预测的药物敏感性与 EpiSen 分数之间的关系。对于 NVP-TAE684 和索拉非尼,分别应与 EpiSen 分数呈正相关和负相关。scFoundation 模型在这些药物上的 Spearman 相关性分别为 0.56 和 -0.55,而基线模型仅为 0.24 和 -0.06(图 4c),表明使用 scFoundation embedding 能够捕捉药物敏感性生物标志物的信号。这些结果进一步激励我们调查 embedding 是否比基因表达更具信息量而无需提取信号。我们对单细胞数据集 SSC47 的 embedding 进行了主成分分析(PCA),并可视化了前两个主成分。结果显示,与原始数据 PCA 相比,embedding 捕捉到的信息更为丰富,线性相关性较低(图 4d)。此外,我们基于 embedding 和基因表达计算了大规模和单细胞数据的聚类性能,使用药物敏感性作为标签。较高的 Calinski-Harabasz(CH)和 SIL 得分(图 4e 和补充图 5)的结果表明,scFoundation 比基因表达基线更好地将具有相同药物反应的细胞或大规模细胞系分组。
这些发现强调,从 scFoundation 获得的统一 embedding 将大规模和单细胞数据对齐到统一的表示空间。这种简化表示在敏感和耐药状态的数据之间产生了清晰的区分,促进了下游模型更好地将药物基因组学信息从大规模细胞系转移到单细胞数据。
促进扰动反应预测¶
了解细胞对扰动的反应对于生物医学应用和药物设计至关重要,因为它有助于识别不同细胞类型间的基因 - 基因相互作用和潜在药物靶点 50。使用 Perturb-seq51,52 数据资源训练模型来建模细胞对扰动的反应是计算生物学的关键任务 53,54,55。我们将 scFoundation 与一个高级模型 GEARS53 结合,用于预测单细胞分辨率的扰动。原始 GEARS 模型使用基因本体知识图通过学习先前观察到的基因扰动节点的组合来表示未见的基因扰动,并结合基因共表达图和扰动信息来预测扰动后的基因表达。共表达图中的每个节点代表一个基因,最初 embedding 是随机的,边连接到共表达的基因。这个图在所有细胞间共享。在我们的方法中,我们从 scFoundation 解码器中获取每个细胞的基因上下文 embedding,并将这些 embedding 设置为图中的节点(方法部分),从而生成细胞特异性的基因共表达图来预测扰动(图 5a)。
a. 基于 scFoundation 细胞特异性基因 embedding 的扰动预测模型示意图。
b. 预测和真实基因表达之间的均方误差(MSE)。红色和蓝色分别显示了基于 scFoundation 的 GEARS 模型和基线 GEARS 模型的结果。
c. 预测值在前 20 个差异表达基因(DE 基因)中的比例,落在对应的真实表达分布区间的 45-55% 分位区间内的平均比例。黑色虚线表示预期百分比(10%)。
d. 在组合扰动(ETS2 + CEBPE)后,前 20 个最差异表达基因的预测基因表达相对于对照的变化。红色和蓝色框分别表示基于 scFoundation 的 GEARS 模型和基线 GEARS 模型的基因预测结果。绿色框代表真实的基因表达分布。对于每个基因,n=313 个细胞进行了检查。一个框的两个边缘和框内的水平条表示所有值的四分位间距和中位数。须的长度从四分位间距向外延伸 1.5 倍。
e. 在 Norman 数据集上对所有测试扰动组合计算的幅度分数。每个点代表一个特定的扰动组合。y 轴显示从预测结果计算的幅度分数,而 x 轴表示使用真实后基因表达计算的真实幅度分数。
f. 使用 scFoundation 和基线方法识别出的具有协同和抑制基因相互作用类型的前 20 个扰动。Venn 图展示了识别出的扰动集与验证的扰动集之间的关系。
我们在三个扰动数据集上训练和测试模型,遵循原始研究(补充说明 5)。由于扰动数据中没有单细胞级别的真实数据,我们计算了前 20 个差异表达(DE)基因的平均均方误差(MSE)在基因表达前后的差异表达谱之间进行评估。基于 scFoundation 的模型在所有数据集中均取得了较低的 MSE 值,相比原始的 GEARS 基线模型表现更好。在更具挑战性的两基因扰动预测中,模型在未见的 0/2 情况下实现了最低的平均 MSE,并在所有情况下均优于 GEARS 和另一个基线模型 CPA56(图 5b 和补充图 6)。对于测试集中的每个两基因扰动,我们进一步检查了前 20 个 DE 基因的平均预测值落在真实表达分布区间 45-55% 分位区间的比例。基于 scFoundation 的模型表现出更高的比例(图 5c),表明它预测的基因表达值分布更合理。图 5d 展示了两基因扰动 ETS2+CEBPE 的前 20 个基因的表达变化。
预测两基因扰动的一个应用是将两基因扰动分类为不同的基因交互(GI)类型。我们使用幅度评分(方法部分)识别协同和抑制 GI 类型。我们首先计算了测试集中所有两基因扰动的预测和真实幅度评分之间的 PCC,发现基于 scFoundation 的模型相比基线模型取得了更高的 PCC(图 5e)。然后,我们根据预测的幅度评分对两基因扰动进行排序,将前 20 个视为潜在的协同 GI,后 20 个视为抑制 GI。图 5f 中的 Venn 图显示,基于 scFoundation 的模型识别的真实协同和抑制 GI 的数量更多。
这些结果突显了从 scFoundation 获得的细胞特异性基因上下文 embedding 作为扰动预测的宝贵基础表示。两基因扰动的分析强调了模型准确分类不同类型 GI 的能力。
细胞类型注释¶
细胞类型注释在单细胞研究中至关重要,已有多种方法用于此目的。为了评估 scFoundation 的性能,我们使用 Zheng68K 数据集 30 和 Segerstolpe 数据集 57 进行了实验,这些数据集在之前的研究中被认为具有挑战性 13。我们仅微调了 scFoundation 编码器的一层,并添加了一个 MLP 头来预测标签。我们将 scFoundation 与 CellTypist58、scBERT13、scANVI59、ACTINN60、Scanpy61 和 SingleCellNet62 等方法进行了基准测试(方法部分)。补充表 4 显示,scFoundation 在两个数据集上都获得了最高的宏 F1 得分。与排名第二的方法 CellTypist 相比,scFoundation 在稀有细胞类型(如 CD4+ 辅助 T 细胞 2 型和 CD34+ 细胞)上表现更好(补充表 5)。我们在 UMAP 上可视化了 scFoundation 和 CellTypist 的预测,分别从潜在 embedding 和 PCA 成分获得。补充图 7 和 8 显示,scFoundation 在不同细胞类型之间有明显的分离。
这些结果表明,scFoundation 利用整个基因集作为输入的能力,能够比其他使用基因子集或离散化基因表达的方法更准确地进行注释。
推断基因模块和基因调控网络¶
scFoundation 的一个优势是将基因表达值扩展为上下文 embedding,相比其他架构如普通的 MLP 模型(补充说明 6)。这些 embedding 不仅可以促进基于图的下游方法如 GEARS,还可以用于推断基因 - 基因网络。在此,我们使用三种免疫细胞类型(单核细胞、细胞毒性 CD8+ T 细胞和 B 细胞)的基因 embedding 进行验证和探索这种用法(方法部分)。
我们根据 embedding 的相似性对基因进行聚类。结果显示,scFoundation 可以识别每种细胞类型的差异表达基因模块(补充图 9 和 10)。基因富集分析验证了识别出的基因模块在各自的细胞类型中富集(补充图 11),表明基因 embedding 已经学习了基因之间的功能关系。此外,我们还探索了在 T 细胞中前 1 个 DE 基因模块内构建的基因网络(补充图 12)。编码 CD8 分子的基因 CD8A 和 CD8B 表现出强烈的相似性,而 S100A8 基因如预期的那样显示出与其他 T 细胞标记物的有限相关性。这表明 embedding 可以提供模块内基因关系的见解。此外,我们还进行了基因调控网络(GRN)推断实验,使用下游模型 SCENIC63(方法部分)。我们识别了单核细胞、B 细胞和 CD8+ T 细胞的特异性调控因子,如 KLF6、SPIB 和 MXD4,这些调控因子在先前的研究中分别被证实为单核细胞 64、B 细胞 65 和 CD8+ T 细胞 66 的调控因子(补充图 13)。这些示例强调了 scFoundation 基因 embedding 在推断 GRN 中的潜力。
讨论¶
最近的 LLM 突破激励我们探索大规模模型是否也能有效地从单细胞转录组数据中学习生物学的细胞和分子“语言”,这些数据具有大规模数据、复杂的生物模式、多样性和技术噪声。结合 xTrimoGene 架构和 RDA 预训练任务,我们开发了 scFoundation,一个在超过 5000 万个单细胞数据上预训练的拥有 1 亿参数的大规模基础模型。消融实验和下游任务应用显示了预训练任务和模型设计的优势。补充表 1 提供了与已发布的类似模型的主要特征比较。
scFoundation 作为一个通用基础模型进行了预训练,可用于许多下游任务:它在读取深度增强、药物反应预测、单细胞药物敏感性预测、扰动预测和细胞类型注释任务中表现优越。它还在基因模块推断和通过与下游批次去除头部如 BBKNN33 合作更好地促进细胞映射方面显示出很高的潜力。
scFoundation 模型在大多数任务中不需要进一步微调。这种设计减少了用户的计算和时间成本,并提供了下游模型设计的灵活性,使 scFoundation 能够更好地作为单细胞生物学领域各种下游任务的基础模型。
我们建议在没有显著批次效应或模态差异的数据集中使用 scFoundation 提取 embedding。考虑到批次效应或模态差异可能包含一系列变化,我们在 scFoundation 中采取了只考虑读取深度的策略,并将其他可能的差异留给下游任务中的协作方法,如 BBKNN 和 SCAD。此外,我们建议使用细胞和基因 embedding 而不是预测的基因表达值,因为当前用于预训练标签的数据存在高丢失率,且模型预训练损失未优化为零。
scFoundation 仍面临一些限制。尽管预训练数据包含了我们收集时几乎所有公开可用的人类 scRNA-seq 数据,但它们可能仍不足以完全反映人类器官发育和健康状态的复杂性。预训练需要大量的计算资源,需进一步优化以提高效率。目前的模型仅关注转录组数据,未包含基因组或表观基因组数据。此外,其无监督预训练过程有不依赖于大量数据的人类注释的优势,但忽略了元数据中的丰富信息。将细胞的元数据与转录组数据结合到模型中可能有潜力将细胞的分子特征与表型联系起来。
未来,我们将使用我们有效的预训练框架,预训练更多参数和更大数据集的模型,我们相信可以基于 scFoundation 的见解开发多项工作。例如,设计更有效的预训练任务可能会进一步提高模型的性能 29。还需探索各种数据集特征对训练性能的影响 29。此外,新兴的单细胞多组学数据 67,68 为开发能够描述细胞多层复杂规律的模型提供了机会。一种可行的情况是基于 ATAC-seq 上下文预测基因表达值,反之亦然(补充说明 7)。
scFoundation 在各种任务中的普遍适用性表明,它在不同类型细胞中的基因表达关系学习方面取得了成功。我们期望预训练架构和预训练 scFoundation 模型能够作为支持大型生物模型研究和各种下游研究的基础贡献。这项工作以及其他最近的报告表明,在高通量单细胞数据上预训练的大型生物模型正在开辟解码和建模复杂分子系统的新途径。
方法¶
预训练数据收集和预处理¶
数据收集¶
许多人体 scRNA-seq 数据被存储在基因表达综合数据库(GEO)、HCA、单细胞门户、EMBL-EBI 等中。还有几个研究整合了来自多个资源的人类单细胞,如 hECA4、DISCO7 等。每个数据库中的数据集都链接到已发布的研究,因此有对应的 DOI ID。我们手动从这些数据库中收集了 scRNA-seq 数据,并删除了具有重复 ID 的数据集。大多数数据集提供了原始计数矩阵。对于具有归一化表达谱的数据集,我们将其转换回原始计数形式:我们将原始矩阵中最小的非零值视为原始计数值 1,所有其余非零值除以此最小值并取整数部分。对于无法转换回原始计数的每百万转录本(TPM)或每百万映射片段的转录本每千碱基(FKPM)表达谱,我们保持不变。
我们的数据收集包括来自健康供体和各种疾病及癌症类型样本的 50 多万个单细胞,代表了人类单细胞转录组的全谱。我们将所有数据分为训练和验证数据集。验证数据集是随机抽样的,包含 100,000 个单细胞,并对所有测试模型保持一致。
基因符号统一¶
我们使用 HUGO 基因命名委员会提供的基因符号映射参考,统一了所有原始计数基因表达矩阵的基因符号。我们包括了人类蛋白编码基因和常见的线粒体基因,总共 19,264 个基因。如果某些符号缺失,我们用零值填充。
质量控制¶
为了过滤受污染的空滴、极低质量细胞和损坏细胞,我们保留了超过 200 个基因表达的细胞(即表达向量中非零值计数>200)进行预训练,使用 Seurat69 和 Scanpy61 包。
scFoundation 模型架构¶
我们开发了 xTrimoGene 模型作为 scFoundation 的骨干模型。它有三个模块:embedding 模块将标量值转换为 embedding,这是 Transformer 块所需的;编码器将非零和未掩盖的表达基因作为输入,使用普通的 Transformer 块并具有较大的参数规模;解码器将所有基因作为输入,使用 Performer 块并具有相对较小的参数规模。消融实验表明,与其他架构相比,这种不对称设计减少了计算和内存挑战(补充表 6)。
Embedding Module¶
对于一个细胞的基因表达值向量 \(\mathbf{X}^{\text{input }} \in \mathbb{R}^{n=19,264}\),基因 \(i\) 的表达值 \(x_i^{\text{input }}\) 是一个大于或等于零的连续标量。与之前的语言模型或最近开发的基于单细胞 Transformer 的模型不同,对于每个基因 \(i\),embedding 模块直接将表达标量转换为可学习的值 embedding \(\mathbf{E}_i\),而无需任何离散化。然后,将值 embedding 与基因名称 embedding \(\mathbf{T}_i^G\) 相加以形成最终的输入 embedding \(\mathbf{E}_i^{\text{input }}\)。值 embedding 是 embedding 集的加权总结,其中权重由基因表达标量值学习。基因名称 embedding 从查找表中检索,该表中的 embedding 随机初始化并可在预训练期间学习(补充说明 8)。连续 embedding 方案的消融实验显示了我们设计相比其他值离散化方法的优势(补充图 14)。
Encoder¶
编码器仅处理非零和未掩盖值的 embedding(即表达的基因和两个总计数值),因此编码器的输入长度约为全基因长度的 \(10 \%\)。设 \(S^E=\left\{S_0^E, S_1^E, \ldots, S_K^E\right\}\) 为具有 \(K\) 个元素的非零和未掩盖值的索引集,编码器的输入定义为:
编码器的设计大大减少了所需的计算资源,使得编码器可以使用一系列的普通 Transformer 块
来捕捉基因依赖关系,而无需任何核或低秩近似。编码器的输出是中间 embedding \(X^{\text{inter }}\):
这种设计不仅优化了计算资源,还保留了对基因表达的详细依赖关系的捕捉能力,从而提高了模型的表达能力和预测准确性。在编码器中使用的普通 Transformer 块能够有效地处理和整合基因表达数据的复杂结构,为后续的解码和预测任务提供了高质量的中间表示。
在这些块中的核心函数是注意力机制,其公式如下:
其中 \(Q=X W_q\),\(K=X W_k\) 和 \(V=X W_v\) 是输入 \(X\) 的线性变换,\(W\) 是训练参数。\(\mathbf{1}_{\mathbf{K}}\) 是长度为 \(K\) 的全 1 向量,\(\operatorname{diag}(\cdot)\) 是以输入向量为对角线的对角矩阵。
中间 embedding \(X^{\text{inter }}\) 有两个用途:(1) 它们与零和掩码 embedding 一起送入解码器,(2) 它们被汇聚为细胞 embedding,用于下游应用。
Decoder¶
为了建立转录组范围的基因调控关系,应考虑零表达的基因,以在掩码位置恢复表达值。编码器的中间 embedding 与零和掩码 embedding 连接,形成长度为全基因的解码器输入张量 \(X^{\text{Dec-input }}\):
其中 \(K_0\) 和 \(K_m\) 分别是零和掩码 embedding 的数量。我们使用基于内核近似的 Transformer 变体 Performer 作为解码器的主干,因为对于长序列的注意力计算具有挑战性。在 Performer 中,使用了可内核化的注意力机制:
其中 \(\varnothing(\cdot)\) 是一种内核函数,用于将输入映射到高维空间。
解码器的输出为 \(X^{\mathrm{Out}}\),其中
为了预测表达值,将 \(T\) 和 \(S\) 的 embedding 丢弃,并通过一个 MLP 将 \(X^{\text{Out }}\) 投影为标量。这些标量形成一个预测向量 \(\mathbf{P}\),其中
所有参数 \(\Theta=\left\{\mathbf{E}_{\mathrm{i}}, \mathbf{T}_i^G, \Theta_{\text{Encoder}}, \Theta_{\text{Decoder}}, \Theta_{\text{MLP}}\right\}\) 在预训练过程中被优化。不同模型的详细超参数设置可以在补充表 7 中找到。
RDA 预训练任务¶
我们使用 RDA 基因表达预测任务来训练模型。对于每个原始预训练的单细胞基因表达向量,我们使用分层贝叶斯下采样策略生成其低总计数变体或不变的谱作为输入向量。我们对原始和输入基因表达进行归一化和对数变换,并将原始和输入向量的总计数分别设为两个总计数指示器 T 和 S。在归一化基因表达后,细胞的原始总计数值被移除。通过通过标记重新引入此信息,我们相信它可以增强模型的预训练性能,因为细胞中的掉落通常与总计数值相关。有关采样策略和计数指示器计算的详细信息,请参见补充说明 9 和 10。
然后我们随机掩盖输入向量的基因表达。在本研究中,我们对零值和非零值都使用 30% 的掩盖比例。然后掩盖的输入向量与两个总计数指示器 T 和 S 连接,并输入到模型中。在获得模型预测的原始基因表达后,我们在预测值和原始值之间的掩盖基因上进行回归损失(补充说明 11)。如果输入向量保持不变,模型学习捕捉单个细胞内基因之间的关系。如果输入向量是低总计数变体,模型学习不同读取深度的细胞之间的关系。采用下采样策略(补充表 8)和回归损失(补充图 15)的消融研究表明,当前设置可以促进细胞特征的学习(补充说明 1)。
scFoundation 的整体模型架构如补充图 16 所示。有关模型和预训练的实现,请参见补充说明 12。
读取深度增强分析¶
为了评估基因表达预测,我们从 5000 万个单细胞数据中采样了 10000 个总计数较高(高于 1000)的细胞作为验证数据集。这些 10000 个细胞在训练阶段被排除。然后,我们使用二项分布生成低总计数的基因表达向量并将其输入到我们的模型中。我们只评估非零基因表达值,因为下采样后 0 表达值不会改变值。除了使用 MSE 作为评估指标外,我们还使用了 MRE,以反映相对误差。
聚类分析
我们使用 scFoundation 和 scVI 编码器获取细胞嵌入。对于其他方法,我们获取了填补的基因表达谱。所有方法均使用默认参数设置。然后,我们按照 SCANPY pbmc3k 教程,通过 sc.tl.leiden 函数获得细胞簇。
在评估聚类结果时,我们首先使用 ARI 和 NMI(scikit-learn71 包)作为指标,评估不同方法得到的聚类结果与实际细胞类型标签之间的一致性程度。考虑到聚类标签的获取也会受到聚类算法选择的影响,我们使用 SIL 作为另一个评估指标,SIL 测量不同方法给定的细胞邻域图上真实细胞类型标签的聚集程度,因此独立于聚类算法的选择,反映了细胞表示的内在特性。
下游方法¶
所有基准模型都使用默认参数进行训练。我们导出了用于 DeepCDR 和 SCAD 任务的细胞嵌入,以及用于 GEARS 任务的基因嵌入。对于细胞嵌入,我们发现将通过对所有基因嵌入进行最大池化和平均池化获得的嵌入,以及 S 标记和 T 标记的嵌入进行连接,能够实现最佳性能(补充说明 13 和补充表 9 和 10)。四个嵌入的连接构建了具有 3,072 维度的新细胞嵌入,并基于这些细胞嵌入训练了下游模型。
DeepCDR¶
我们使用 DeepCDR 预处理的细胞系和药物配对数据。细胞系数据包含 697 个基因表达谱,我们将这些基因与我们的统一基因符号列表对齐。药物表示为具有一致特征矩阵和相邻矩阵大小的图。总共考虑了 31 种癌症类型的 223 种药物和 561 种细胞系数据。我们遵循原始研究随机划分 5%的数据作为测试集,得到 89,585 个训练样本和 4,729 个测试样本。对于每个细胞系,我们将指标 S 和 T 均设置为所有基因表达值的总和。我们将非零基因表达值和两个指标输入模型编码器,得到每个基因的上下文嵌入。通过对所有基因的每个嵌入维度进行最大池化操作获得了总体水平的细胞系嵌入。
我们通过将参数 -use_gexp 设置为 True,将 -use_mut 和 -use_methy 设置为 False 来训练基准 DeepCDR 模型。然后,对于基于 scFoundation 的模型,我们直接用细胞系嵌入替换基因表达,并使用相同设置训练 DeepCDR。对于每个基因,我们计算了所有细胞系上预测的 IC50 与真实 IC50 之间的皮尔逊相关系数(PCC)。对于每个细胞系,我们计算了在该细胞系上进行的所有药物的 PCC。
SCAD¶
我们按照原始 SCAD 研究的实验设置,进行了五折交叉验证。对于每次拆分,使用四折的总体和单细胞数据来训练模型,另一折用于预测,并合并所有拆分结果以获得所有细胞的预测。我们使用了所有基因,并在模型训练过程中进行了加权采样。对于训练基准模型,将基因表达值转换为 z 分数。
对于训练基于 scFoundation 的模型,我们使用了归一化的基因表达数据。对于总体数据,我们将 S 和 T 均设置为所有基因表达值的总和,以保持原始细胞系特征。对于单细胞数据,我们将 S 标记设置为基因表达值的总和,T 标记设置为 10,000,这是每个细胞的经验最大测序深度。然后,将每个样本的非零值和两个指标输入预训练模型的编码器。输出是每个样本基因的上下文嵌入,然后凝聚成细胞嵌入。
扰动预测¶
我们将基因符号列表统一为 19,264,并在每个数据集上生成基因共表达网络。根据原始 GEARS 研究,对于单基因扰动,我们随机分配 75% 的扰动作为训练数据。对于双基因扰动,我们将 75% 的两基因均在已知集合中的扰动(0/2 未知)指定为训练集,而其他所有组合(1/2 和 2/2 未知)则保留用于测试。然后,我们通过设置 epoch 为 15 和批次大小为 30 来训练 GEARS 基准模型。CPA 模型没有基因嵌入,它将药物或基因扰动嵌入作为输入模型。我们使用 GEARS 研究中的相同参数设置训练 CPA 模型。基因扰动被编码为 one-hot 向量,双基因扰动表示为两个单基因扰动向量的相加。在基于嵌入的模型中,每个细胞的 T 和 S 值等于其总计数,基因表达和指标输入模型。scFoundation 的最后一层 MLP 被删除,以从解码器中提取基因上下文嵌入,作为共表达图的节点特征。我们冻结 scFoundation,仅训练下游 GEARS 模型,使用梯度累积保持与基准训练期间一致的批次大小。
我们遵循 GEARS 中的定义和指标。我们重点关注了协同和抑制基因交集类型,因为它们是最基本的类型。这两种类型的识别是基于量级分数,该分数测量了双基因扰动和两个单基因扰动组合之间的相似性。具体来说,令 A 扰动前后细胞的平均变化为 \(\delta g_a\)。使用线性模型拟合 \(\delta g_a, \delta g_b\) 和 \(\delta g_{a+b}\) 的效果:
其中 \(\epsilon\) 表示模型拟合中的误差。我们使用 Theil-Sen 估计器的鲁棒回归,按照之前研究中使用的相同程序。利用系数的值,量级定义为:
所有测试的双基因扰动根据量级分数进行排序,排名最前和最后的分别被认为是协同和抑制类型。
细胞类型注释¶
我们将每个数据集随机拆分为训练集、验证集和测试集,比例为 8:1:1。对于 scFoundation,我们在编码器之后添加了一个具有 ReLU 激活函数的两层 MLP。MLP 的输出是预测标签。考虑到不同细胞类型的不平衡细胞数量,我们使用加权交叉熵损失。给定总共有 \(C\) 种细胞类型,细胞类型 \(i\) 有 \(A_i\) 个细胞,每种细胞类型在损失中的权重 \(w_i\) 定义为:
其中 \(B_i\) 是缩放后的数量。我们将学习率设置为 0.001,梯度累积步数设置为 5,每步的批次大小为 64。我们选择验证集上 F1 分数最高的模型作为测试的最佳模型。
对于 scBERT,我们将基因表达矩阵转换为其所需的基因符号列表,并微调其预训练模型。我们使用验证集选择最佳模型。对于 CellTypist、scANVI、ACTINN 和 SingleCellNet 方法,我们将训练和验证数据集输入模型,并使用默认参数设置进行训练。我们通过如 CellTypist 的‘celltypist.annotate’等相应函数获取测试数据的预测结果。对于 Scanpy,我们使用‘sc.tl.ingest’函数基于 PCA 组件将细胞类型标签转移到测试数据,并将转移的标签视为预测结果。对于测试集上的每种方法,我们计算了表现最好的三个模型复制的平均宏 F1 分数。
基因模块和基因调控推断¶
我们从 Zheng68K 数据中的三种细胞类型(单核细胞、CD8+ 细胞毒性 T 细胞和 B 细胞)中随机选择了 100 个细胞,总计 300 个细胞。这些数据通过 scFoundation 处理,获得所有基因的上下文嵌入,形成一个 300 × 19,264 × 512 的矩阵。在选择了高变异基因并对基因嵌入在细胞间进行平均后,我们得到了 495 个基因嵌入,每个嵌入有 512 个维度,并使用 Leiden 聚类方法基于嵌入获得了 34 个基因模块。然后,我们使用‘scanpy.tl.score_genes’函数计算了 300 个细胞中每个基因模块的平均表达,生成了一个 300 × 34 的评分矩阵。我们对该评分矩阵进行差异分析,以识别每种细胞类型的标记基因模块。接着,我们通过在线 EnrichR73 工具对差异表达基因模块进行富集分析。我们使用了‘PanglaoDB Augmented 2021’数据集,并选择调整后的 P 值最低的术语来解释基因模块。对于基因网络,我们计算了基因嵌入在模块中的相似性,并标记了具有最高值的前 5 条边。
在基因调控推断方面,我们从 SCENIC 获取了所有已知的转录因子(TF)–目标基因对,并根据它们的基因嵌入相似性量化了它们的关系。对于每个 TF,我们选择相似性高的前 1000 对作为候选对。由于转录组数据不能在序列水平上直接提供 TF- 基因结合的洞察,我们使用 SCENIC 的 RcisTarget 模块来优化我们选择的对。使用 SCENIC 的 auc_cell 模块,我们在细胞类型中得出了 TF 富集评分,并确定了排名最高的细胞特异性 TF。然而,我们要指出的是,直接从嵌入中计算相似性是一种简单的方法,可能无法充分利用向量中的丰富信息。未来的工作可以探索利用上下文嵌入进行更复杂的 GRN 推断的算法,例如使用图神经网络的算法。
报告总结¶
有关研究设计的更多信息,请参阅链接到本文的 Nature Portfolio Reporting Summary。
数据可用性¶
本研究使用的所有数据均可公开获取,具体用法在方法部分说明。预训练数据集主要从 GEO(https://www.ncbi.nlm.nih.gov/geo/)、Single Cell Portal(https://singlecell.broadinstitute.org/single_cell)、HCA(https://data.humancellatlas.org/)和 EMBL-EBI(https://www.ebi.ac.uk/)下载,详细的数据集列表在补充数据 1 和 2 中。下游任务使用的数据集可以从以下链接下载:Baron 数据集(https://github.com/mohuangx/SAVER-paper);Zheng68K 数据集(https://www.dropbox.com/sh/w3yg2nucnng5v1u/AAAM8Ym_KU9XF4z51RT81eNEa?dl=0);Segerstolpe 数据集(https://zenodo.org/records/3357167);CDR 数据集(https://github.com/kimmo1019/DeepCDR);单细胞药物反应分类数据集(https://github.com/CompBioT/SCAD);扰动数据集(https://github.com/snap-stanford/GEARS);用于细胞映射的模拟参考和查询数据集(https://doi.org/10.6084/m9.figshare.21456645.v4);和用于细胞映射的类器官和体内数据(https://doi.org/10.17632/sm67hr5bpm.1)。通过 scFoundation 生成的基因表达数据和嵌入可以在我们的 GitHub 仓库(https://github.com/biomap-research/scFoundation)和 figshare(https://doi.org/10.6084/m9.figshare.24049200)(参考 74)中找到。
代码可用性¶
使用在线 API 的代码、模型代码和权重、推理嵌入的示范、生成下游任务结果的代码在 GitHub 仓库(https://github.com/biomap-research/scFoundation)或 Zenodo75 中可以找到。所有代码和数据信息的总结在补充数据 3 中。