Museum of spatial transcriptomics
摘要¶
许多生物系统(如胚胎、肝小叶、肠绒毛和肿瘤)的功能依赖于其细胞的空间组织。在过去的十年中,高通量技术已被开发出来以量化空间中的基因表达,同时也开发了利用空间基因表达数据来识别具有空间模式的基因和划定组织内邻域的计算方法。为了全面记录空间基因表达技术和数据分析方法,我们呈现了一篇精心整理的关于自 1987 年以来空间转录组学文献的回顾,以及对该领域趋势的详尽分析,例如实验技术的使用情况、研究的物种、组织以及所采用的计算方法。我们的回顾将当前的方法置于历史背景中,并从中得出关于该领域的见解,以指导当前的研究策略。随附的补充材料提供了对所分析技术和方法的更详细介绍:https://pachterlab.github.io/LP_2021/。
主体部分¶
早在果蝇胚胎到肝小叶等生物系统中,就已认识到许多基因需要在空间上正确调控才能使系统正常运作。为了研究基因表达的空间模式,已经开发了许多不同的空间转录组学方法,这些方法通过对信使 RNA (mRNA) 转录本的空间定位量化来作为基因表达的代理。由于对这一领域的兴趣日益浓厚,过去五年里已有几篇综述提供了数据收集的实验技术概述 1,2,并描述了这些技术如何应用于特定的生物系统,例如肿瘤 3、大脑 4 和肝脏 5。这些综述通常从 20 世纪 90 年代末的激光捕获显微切割 (LCM) 或单分子荧光原位杂交 (smFISH) 开始,尽管在空间中描绘转录组的探索历史更为悠久。
与以往的综述不同,本论文呈现了一个可以追溯到 1987 年的文献数据库,全面记录了空间转录组学数据收集和分析的历史演变和当前发展情况。此外,我们还对该数据库中的文献元数据进行了分析,以展示该领域的趋势。本文展示了数据库和分析中的主要亮点,更多详细内容请参见我们的书长补充材料:https://pachterlab.github.io/LP_2021/。本文中的章节和图表编号对应的是 DOI PDF 版本,而在线 HTML 版本的编号可能会有所变动,因为它会不断更新以反映该领域的变化。该数据库通过在 PubMed 和 bioRxiv 上搜索“spatial transcriptomics”和“Visium”等关键词,并手动筛选引用该领域重要论文的文献来进行整理。收集的文献元数据包括发表或发布的日期和第一作者的机构。此外,对于涉及新数据集的出版物,还包括数据收集的物种和组织、用于收集数据的实验技术以及用于分析数据的编程语言。对于涉及新数据分析方法的出版物,元数据包括实现过程中使用的编程语言、实现的代码库,以及代码是否被打包和记录。该数据库通过手动筛选 PubMed 和 bioRxiv 的 RSS 源中相关关键词,或通过 Google 表单提交,持续更新。
前传时代¶
我们所说的“空间转录组学”是指在组织和细胞的空间背景中量化大量基因的 mRNA 表达的尝试。某些重要技术追溯到 20 世纪 70 年代(补充信息第 2 章)。各种形式的原位杂交 (ISH) 已经被长期使用以可视化空间中的基因表达。放射性 ISH 首次引入于 1969 年,用于可视化非洲爪蟾卵母细胞中的核糖体 RNA6 和 DNA7,并在 1973 年首次用于可视化特定基因(珠蛋白)的转录本 8(图 1a)。非放射性荧光或显色 ISH 在 1970 年代和 1980 年代早期被开发出来,提高了空间分辨率,允许三维(3D)染色,并缩短了所需的曝光时间 9,10(图 1a)。早期的 ISH 是在组织切片中进行的,这使得它很难应用于囊胚和重建 3D 组织结构;整体装载 ISH(WM ISH)于 1989 年首次在果蝇中引入 11,并在 1990 年代早期被改编到其他物种,如小鼠 12。
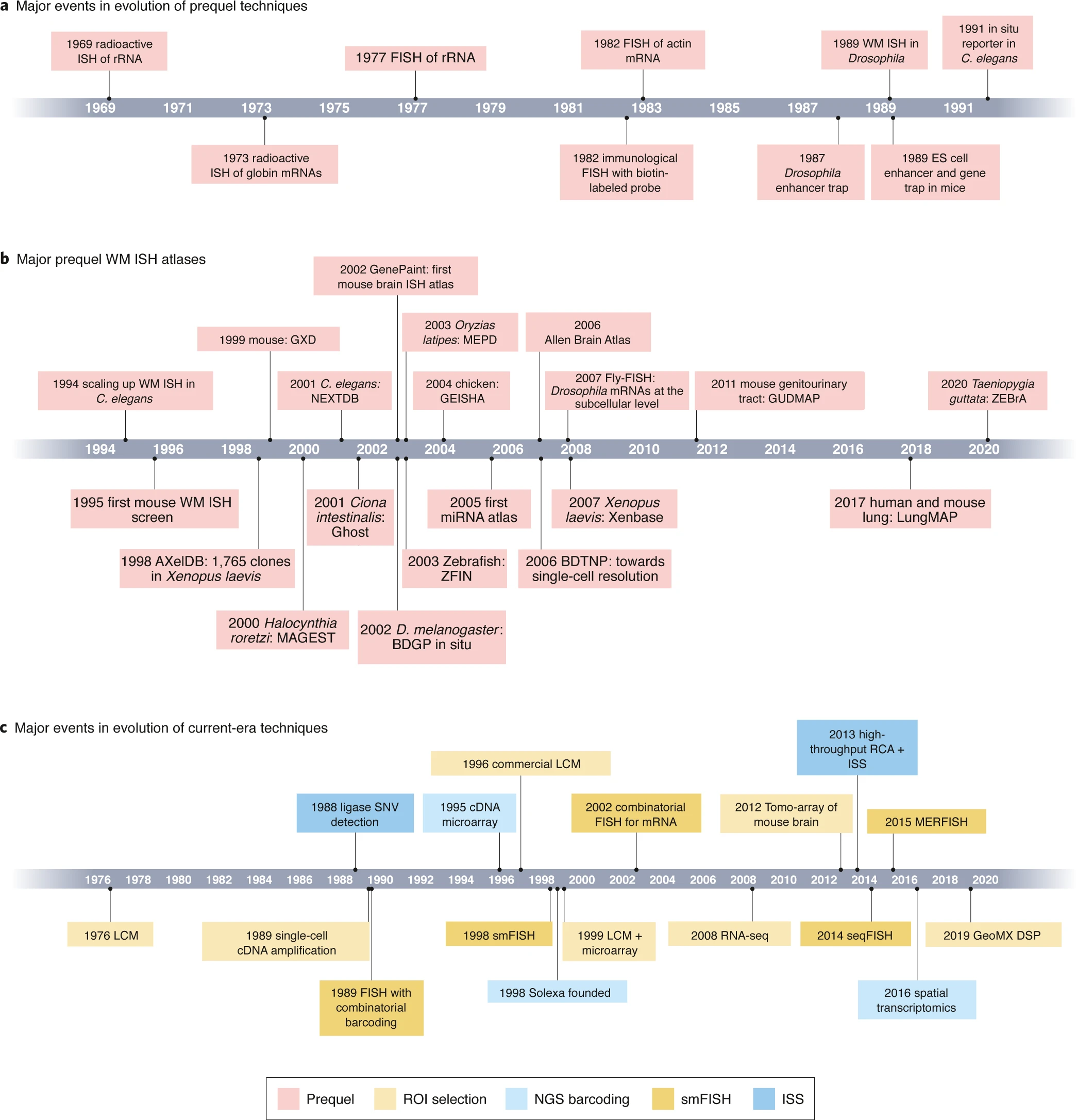
a. 前传时代技术的发展。参考文献:1969 年放射性核糖体 RNA(rRNA)原位杂交(ISH)6,7,1973 年珠蛋白 mRNA 的放射性原位杂交 8,1977 年 rRNA 的荧光原位杂交(FISH)10,1982 年带有生物素标记探针的免疫 FISH9,1982 年肌动蛋白 mRNA 的 FISH135,1987 年果蝇增强子捕获 14,1989 年果蝇整体装载 ISH11,1989 年小鼠胚胎干细胞增强子和基因捕获 15,1991 年秀丽隐杆线虫中的原位报告基因 136。
b. 主要的整体装载 ISH 图谱和基因表达模式数据库。参考文献:1994 年扩展秀丽隐杆线虫的整体装载 ISH137,1995 年首个小鼠整体装载 ISH138,1998 年 AXelDB139,1999 年小鼠:基因表达数据库(GXD)140,2000 年 Maboya 基因表达模式和表达序列标签(MAGEST)141,2001 年线虫表达模式数据库(NEXTDB)142,2001 年 Ghost143,2002 年 GenePaint144,2002 年果蝇:伯克利果蝇基因组项目(BDGP)24,2003 年青鳉表达模式数据库(MEPD)145,2003 年斑马鱼信息网络(ZFIN)31,2004 年鸡表达原位杂交分析(GEISHA)25,2005 年 miRNA 图谱 29,2006 年艾伦脑图谱 26,2006 年果蝇基因表达图谱(BDTNP)146,2007 年 Fly-FISH147,2007 年 Xenbase148,2011 年小鼠泌尿生殖系统发育分子解剖项目(GUDMAP)27,2017 年肺图谱项目(LungMAP)28,2020 年斑马鱼表达图谱(ZEBrA)149。
c. 当前时代技术及其显著前体的发展,按技术类型分类。参考文献:1976 年激光捕获显微切割(LCM)17,1988 年连接酶介导的单核苷酸变异(SNV)检测 150,1989 年单细胞 cDNA 扩增 151,152,1989 年组合条形码 FISH21,1995 年 cDNA 微阵列 153,1996 年商业 LCM18,19,1998 年单分子荧光原位杂交(smFISH)23,1999 年 LCM + 微阵列 154,2002 年组合 FISH22,2008 年 RNA-seq155,2012 年 Tomo-array156(Tomo-seq 的 cDNA 微阵列前体),2013 年高通量 RCA + ISS59,2014 年 seqFISH20,2015 年 MERFISH50,2016 年空间转录组学(ST)87,2019 年 GeoMX DSP44。
早期空间转录组学发展的另一条线是增强子和基因捕获筛选,这在 1980 年代 DNA 测序通量增加 13 以及后生动物基因组成为新前沿时被开发出来。第一个在果蝇 14 和小鼠 15 中进行的筛选是在 1980 年代末进行的,目的是可视化未定向且常常是未知基因的表达。随着通量的增加,增强子和基因捕获成为 1990 年代空间转录组学的首选技术,直到 1990 年代末的整体装载原位杂交(WM ISH)的兴起,这利用了自动化。WM ISH 还避免了转基因系的需求,并受益于 2000 年代初参考基因组的可用性,从而进行计算探针设计。尽管现在被新方法所取代,但增强子捕获、基因捕获和原位报告方法在整个 2000 年代和 2010 年代仍被用于构建转基因系中基因表达和增强子使用模式的参考数据库 16。
当前时代许多技术的基础是在 1970 年代到 2000 年代之间奠定的(图 1c)。例如,1976 年首次使用紫外线(UV)激光切割组织 17。流行的红外(IR)和紫外线 LCM 系统首次报道于 1996 年(参考文献 18,19)并很快商业化。一些高度多重的 smFISH 技术,例如序列 FISH(seqFISH)20,依赖于组合条形码,即使用颜色的组合对每个基因进行编码,从而可以同时量化更多基因的转录本,这些基因具有易于辨别的颜色(最多 5 种)。组合条形码首次在 1989 年免疫 DNA FISH 中报道(参考文献 21),并于 2002 年首次用于转录本(参考文献 22)。1998 年首次明确展示每个 mRNA 分子作为一个斑点的 smFISH(参考文献 23)。没有这些技术的发展,高度多重的 smFISH 将是不可能的。
在 1990 年代后期和 2000 年代,WM ISH 是首选技术,然后才是高度多重、高分辨率和更定量的技术的兴起。它已被用于创建多个物种胚胎的基因表达图谱,例如果蝇 24,小鼠和家鸡 25;在各种小鼠器官中,例如大脑 26、泌尿生殖道 27 和肺 28;以及特定类型的基因,例如微小 RNA(miRNA)29(图 1b)。对于 miRNA 和除小鼠和人类之外的许多物种,目前可用的空间转录组资源大多是 WM ISH 图谱。在此期间,还建立了从各种来源收集的基因表达模式的模型生物数据库,例如基因表达数据库(GXD)30 和斑马鱼信息网络(ZFIN)31(图 1b)。WM ISH 的黄金时代似乎在 2010 年代结束了(图 1b),可能是由于该技术的一些缺点,例如需要典型的组织结构,需要数千只动物来生成图谱,以及结果主要是定性的。
空间转录组学的早期应用动机包括识别具有受限模式的基因,这些模式表明其在发育中的功能,识别新的细胞类型标志物,以及识别从组织形态学上看不明显的新细胞类型 14,15。在 1980 年代和 1990 年代,分析通常是手动完成的,尽管最近已经开发了自动化方法(补充信息第 3 章)。技术的汇聚,包括更强大的计算基础设施、测序成本的降低以及更定量数据的生成,使得空间转录组学主流化并带来了新的可能性。然而,前传时代的遗产仍然存在,例如引用艾伦脑图谱(ABA)32 和艾伦小鼠共同坐标框架 33,以及像艾伦脑研究所和杰克逊实验室这样的机构,它们正在为当前时代的研究做出贡献 34,35。
数据收集¶
当前时代的技术在获取空间信息方面大致分为五类:感兴趣区域(ROI)选择(补充信息第 5.1 节)、smFISH(补充信息第 5.2 节)、原位测序(ISS)(补充信息第 5.3 节)、带有空间条形码的下一代测序(NGS)(补充信息第 5.4 节)和不需要先验空间位置的方法(补充信息第 5.6 节)。这些技术的开发者通常希望实现转录组范围的分析、单细胞分辨率和高基因检测效率的三位一体。尽管这一目标似乎越来越接近,但当前时代的技术在这些目标之间存在权衡。
ROI 选择¶
可以通过选择和隔离已知位置和形状的 ROI 来获取空间位置,这可以通过物理(补充信息第 5.1.3 节)和光学标记的 ROI 进行隔离(补充信息第 5.1.4 节)。然后,可以通过互补 DNA(cDNA)微阵列或 RNA 测序(RNA-seq)分析隔离的 ROI,或将其解离成单个细胞进行单细胞 RNA 测序(scRNA-seq)。
物理显微切割包括 LCM、2000 年代的体素化 36 和 Tomo-seq37,后者使用冷冻切片机沿感兴趣的轴对组织进行切片,然后对每个切片进行 RNA-seq。自 1999 年以来,最广泛使用的显微切割技术是 LCM,它已被用于各种生物学领域,如肿瘤学、神经科学、免疫学、发育生物学和植物学(见补充信息第 6 章 PubMed 和 bioRxiv LCM 文献的主题建模)。在 LCM 中,通过紫外线激光切割(由蔡司和徕卡制造的激光器)或红外线激光将组织与膜融合(由 Arcturus 制造)来切割组织切片中的 ROI;在 Arcturus 的最新版本中,红外线融合去除使用紫外线切割的 ROI。结合 LCM 和 Tomo-seq,可以在 3D 中绘制空间转录组,例如地理位置测序(Geo-seq)38,尽管空间分辨率有限。一个创新的物理显微切割方法是 STRP-seq39,它将相邻的组织切片以不同角度切成条状,并通过一种受射线计算机断层扫描启发的算法在 3D 中重建基因表达模式。手动切割常用于沿植物的一个空间轴轮廓化基因表达 40。
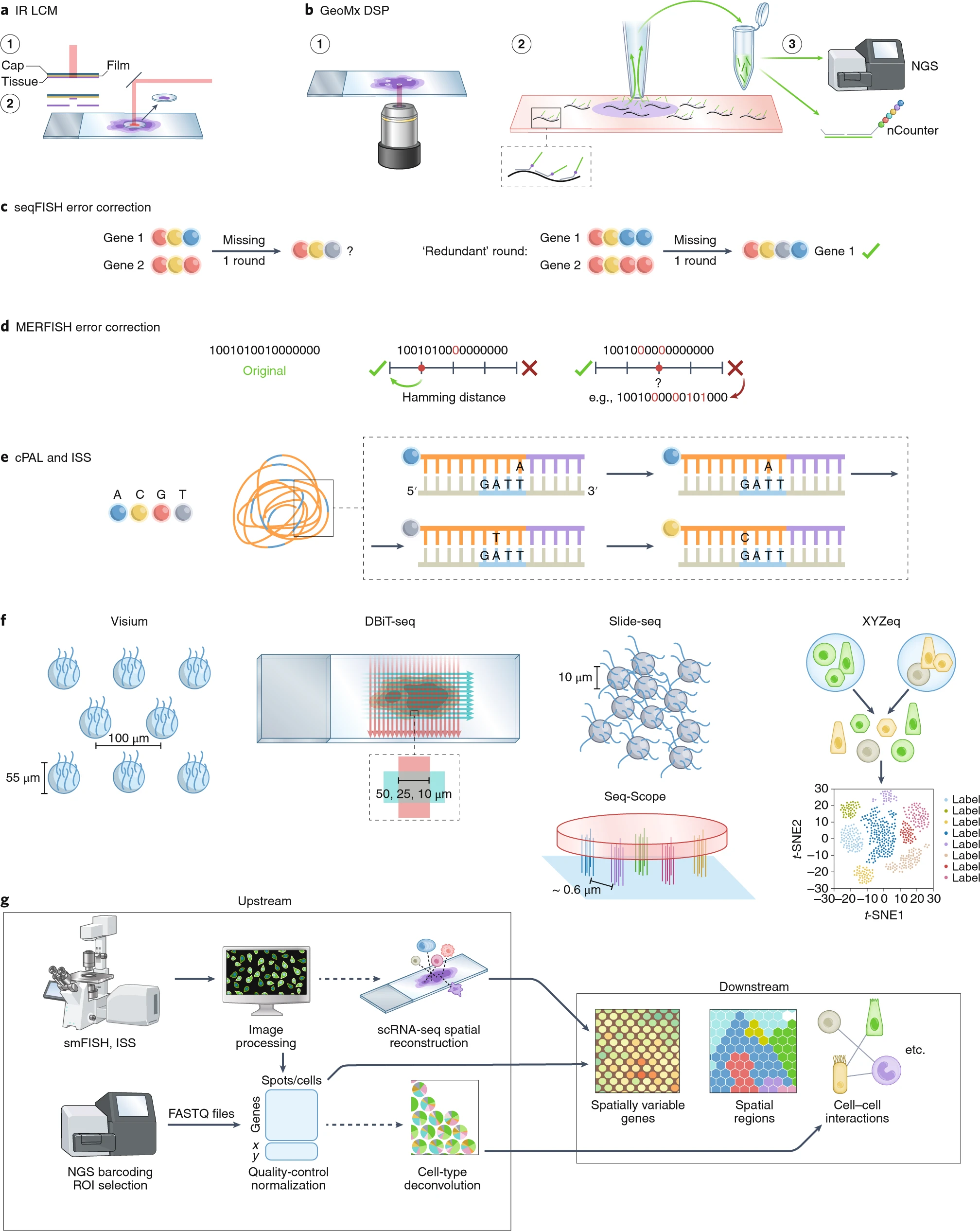
a. 红外线激光捕获显微切割(IR LCM)。
b. GeoMX DSP。在步骤 2 中,紫色圆圈是紫外线照射的感兴趣区域(ROI)。
c. seqFISH 条形码和错误校正方案:如果一个轮次的杂交信号缺失,其余轮次仍然可以唯一识别被条形码编码的基因。
d. MERFISH 汉明距离 4 的条形码和错误校正方案:从条形码的设计来看,如果一个轮次的杂交信号缺失,可以恢复正确的条形码。如果缺失两个轮次,剩余的信号与两个不同的条形码等距,因此无法恢复原始条形码。
e. 使用 cPAL 测序的 Cartana 原位测序(ISS):通过滚环扩增(RCA)对基因条形码进行信号放大,然后使用 cPAL 进行原位测序。橙色线代表 RCA 扩增产物。短蓝色线代表基因条形码。棕色代表探针;未标记的碱基是退化的。灰色代表匹配恒定区的引物。
f. 下一代测序(NGS)条形码技术。在 Visium 中,点排列在一个六边形网格中,中心间距为 100µm,直径为 55µm。在 DBiT-seq 中,位置条形码被沉积在微流体通道中,空间分辨率由通道的宽度(最小可达 10µm)和通道间距决定。在 Slide-seq 中,直径为 10µm 的条形码珠子被单层分布在载玻片上。在 XYZeq 中,空间条形码被赋予直径为 500µm 的孔中的多个细胞,然后这些细胞被解离用于单细胞 RNA 测序(scRNA-seq)。在 Seq-Scope 中,组织被安装在重新利用的 Illumina 流动细胞上,条形码聚合物点平均间距为 0.6µm。对于 Visium 和 Slide-seq,线条代表附着在载玻片或珠子上的寡核苷酸。对于 DBiT-seq,红色和绿色线条代表携带条形码寡核苷酸的微流体通道中的流动。对于 Seq-Scope,组织(粉色块)安装在重新利用的 Illumina 流动细胞上,每个条形码聚合物都有其自己的空间条形码,用不同颜色表示。对于 XYZeq,细胞的不同颜色代表微孔中的不同空间条形码,细胞被解离用于 scRNA-seq。t-SNE,t 分布随机邻域嵌入。
g. 数据分析工作流程:上游分析是技术特定的,包括 smFISH 和基于 ISS 技术的图像处理,以及基于 NGS 技术的 FASTQ 文件处理、基因计数矩阵的质量控制和数据标准化。可以通过将非空间的 scRNA-seq 数据与 smFISH 或 ISS 数据中的标志基因映射到位置上,或者通过 Visium 点中的细胞类型解卷积来整合。下游分析通常与技术无关,包括寻找空间变化的基因、转录定义的空间区域和细胞间相互作用。
光学标记的 ROI 选择包括 NICHE-seq41,它使用双光子照射标记来自表达光活化绿色荧光蛋白(PA-GFP)转基因小鼠的组织中的 ROI,然后使用荧光激活细胞分选(FACS)分离带有活化 PA-GFP 的细胞进行单细胞 RNA 测序(scRNA-seq)。类似于 NICHE-seq 但不需要转基因小鼠的是空间光活化颜色编码细胞地址标签(SPACECAT)42,它对培养的活细胞或类器官用光笼罩荧光染料进行染色,并对 ROI 进行光活化以进行 FACS 和 scRNA-seq。同样使用光笼罩,ZipSeq43 通过抗体或脂质插入将锚定寡核苷酸与光笼罩的延伸段连接到组织上,并向光活化的 ROI 添加与延伸段杂交的空间“邮政编码”。更受欢迎的商业光学 ROI 选择技术是 NanoString 的 GeoMX 数字空间分析仪(DSP)44 和全转录组图谱(WTA)45(图 2b),它通过在 ROI 上照射紫外线来释放光可切割的基因条形码,并使用 nCounter 或 NGS 进行定量。由于 GeoMX 使用预定义的基因面板而不是 poly-A 捕获,NanoString 提供了包含 1800 多个基因的癌症转录组图谱(CTA)基因面板,以及包含 18000 多个基因的人类和小鼠全转录组面板。
单分子 FISH¶
按时间顺序,当前时代开发的下一项技术是高度多重的单分子 FISH(smFISH),其始于 2012 年的原型(seqFISH),依赖于超分辨率显微镜(SRM)通过对酵母中的转录本杂交不同颜色的探针来同时分析 32 个基因的位置 46。SRM 不再需要;2014 年,seqFISH20 发布,每轮杂交每个基因可视化一种颜色,在下一轮进行下一种颜色的条形码之前,探针被剥离。相同基因的所有转录本具有相同的条形码。四种颜色和 8 轮杂交(48=65536)足以编码人类或小鼠基因组中的所有基因。实际上,会进行一轮错误校正的杂交,以便在一轮杂交信号缺失的情况下仍能区分基因 47(图 2c)。最近,在基于 RNA 连续探测的 seqFISH 版本中 48,“颜色”本身通过一系列杂交进行独热编码,将调色板扩展到每通道 20 种“颜色”,使得能够分析 10000 个基因 49。
另一种 smFISH 技术是多重错误鲁棒 FISH(MERFISH)50,它使用不同的条形码策略,其中每个基因都由二进制代码编码。每次实验的颜色代码必须以汉明距离 4 分离,以允许纠正一轮中的缺失信号,并以距离 2 分离,以识别错误而无需纠正功能(图 2d)。条形码的长度可以增加,以编码 10000 个基因 51。由于仅去除荧光团而不剥离探针,因此 MERFISH 中的多轮杂交比 seqFISH 中的耗时少。大多数其他基于 smFISH 的技术,如基于杂交的 ISS(HybISS)52 和 split-FISH53,使用类似 seqFISH 或 MERFISH 的条形码。
smFISH 面临许多挑战,这些挑战已通过各种方法得到解决。信噪比可以通过滚环扩增(RCA)52、分支 DNA(bDNA)54、杂交链反应(HCR)47、引物交换反应 55 和组织清除 56 来提高。随着分析基因数量的增加,转录点可能会重叠,导致光学拥挤。通过扩展显微镜(ExM)57,仅一次成像一部分探针并使用计算超分辨率 49,对高表达基因进行成像而无需组合条形码 50,以及计算解决重叠点 58,可以减轻这种情况。
原位测序¶
ISS 方法通过测序获得空间转录组信息,通常通过连接(SBL)、基因条形码(靶向)或 cDNA 的短片段(非靶向)进行原位测序。这种方法依赖于连接酶仅在引物与探针的序列匹配时连接两个 DNA 片段,并洗去不匹配的探针。使用的探针是退化的,除了一个或两个由颜色编码的查询碱基。RCA 常用于信号放大。2013 年的 ISS59,后来由 Cartana 商业化,以及条形码寡核苷酸连接到 RNA 上用于多重平行原位分析(BOLORAMIS)60,每个探针使用一个查询碱基,如组合探针锚连接(cPAL)61,用于测序基因条形码(图 2e)。在 cPAL 中,每个探针查询基因条形码中的一个碱基。荧光 ISS(FISSEQ)62 和一种后来的 ExM 改编版,称为 ExSeq63,使用 SOLiD,每个探针使用两个查询碱基来测序环化和 RCA 扩增的 cDNA。在空间分辨转录本扩增产物读出映射(STARmap)56 中,通过动态退火和连接的错误减少测序(SEDAL)测序基因条形码,类似 SOLiD 使用两个查询碱基来拒绝错误,但也可以使用一个碱基编码。条形码分析通过测序(BAR-seq)也通过 RCA 扩增带有基因条形码的探针,但使用合成测序(SBS)而不是 SBL 来测序条形码 64。
带有空间条形码的 NGS¶
转录本的空间位置可以通过在原位阵列上捕获组织切片上的转录本来保留。这种阵列可以通过在商业微阵列载玻片上打印点条形码、唯一分子标识符(UMI)和 poly-T 寡核苷酸来制造,以捕获 poly-A 化的转录本,如空间转录组学(ST)和 Visium 技术中(图 2f)。它们也可以是 Drop-seq 样珠子 65,带有分池条形码、UMI 和 poly-T 寡核苷酸,单层分布在载玻片上(例如 Slide-seq66)或限制在刻有孔的载玻片中(例如高分辨率空间转录组学(HDST)67),通过原位 SBL 随后定位珠子条形码。或者,在组织确定性条形码空间组学测序(DBiT-seq)68 中,通过微流体通道生成阵列,先在一个方向上沉积一种条形码,然后在垂直方向上沉积另一种条形码,通过正交条形码连接使每个点都能用独特的成对组合识别。虽然 NGS 条形码技术通常设计用于 3' 末端 Illumina 测序,Visium 已被改编为纳米孔长读测序 69。
NGS 条形码技术已应用于大面积组织 33,并且其使用量在增加(图 4b)。然而,它们没有单细胞空间分辨率。常用的 Visium 在中心距 100µm、直径为 55µm 的六边形阵列中具有点(图 2f)。在 Slide-seq 中,珠子直径为 10µm,在 HDST 中为 2µm(图 2f)。Slide-seq 和 HDST 使用的珠子尺寸小于单细胞,但并不总是提供单细胞分辨率,因为一个珠子可以跨越两个或多个细胞。DBiT-seq 的分辨率由通道宽度决定(50、25 或 10µm,图 2f)。最近,点大小可以减小到 1µm 以下,带有 RCA 扩增的 DNA 纳米球,跨越 0.22µm,带有点条形码的孔 0.5 或 0.715µm 在立体测序(Stereo-seq)70 中沉积,和在 Seq-Scope 聚合酶群落(polonies)上,空间条形码平均 0.6µm 中心到中心,捕获组织切片的转录本 71(图 2f)。另一种基于 polony 的方法,PIXEL-seq,点直径约为 1.22µm,但与流动细胞不同,polony(或 DNA 聚集体)索引库测序(PIXEL-seq)没有每个 polony 周围的间距 72。技术如 XYZeq73 和 sci-Space74 已经开发,用于解离空间条形码点中的单细胞或细胞核进行 scRNA-seq,因此数据具有单细胞转录组学,但没有空间分辨率(图 2f)。
空间信息的去 novo 重建¶
一些技术已经开发出来,可以在不知晓或收集空间位置的情况下,保留必要的信息,以便计算重建空间基因表达模式。这样的一种技术是 DNA 显微镜 75,76,它记录 cDNA 之间的接近度。该信息可用于重建转录本的相对位置。在细胞层面,可以通过故意检测多重测定并将它们映射到空间参考中的位置来重建稀有细胞类型的基因表达,这基于来自常见细胞类型的细胞附着在稀有细胞类型的细胞上的基因表达 77。术语“空间转录组学”的变体也被用于描述将转录本定位到细胞器的技术(例如 APEX-seq78),尽管没有记录空间坐标。
多组学¶
转录组只是细胞功能的一个方面。其他方面,如蛋白质组、神经连接组和 3D 染色质构象对于细胞功能也很重要,并且已经开发出一些方法来在相同的细胞中分析它们及转录组(补充信息第 5.8 节)。对于蛋白质组,使用带有寡核苷酸标签的抗体检测感兴趣的蛋白质,可以使用基于 smFISH 的方法检测表示蛋白质种类的寡核苷酸。这样的抗体面板已经与转录组学结合,例如在 DBiT-seq68、SM-Omics79、GeoMX DSP44 和 MERFISH80 中使用。使用寡核苷酸条形码,可以使用 100 多种抗体,例如在使用 GeoMX DSP 的所有可用抗体面板时。对于 3D 染色质构象,MERFISH 和 seqFISH+ 已经被改编为可视化染色质结构,通过靶向 DNA 基因组位点 81 或新生转录本的内含子 81,82。对于神经连接组,多重转录定量也可以与神经投射追踪相结合。例如,霍乱毒素 b 亚单位(CTb)逆行追踪已与 MERFISH 结合使用以可视化轴突 83。此外,BAR-seq 最初设计用于使用 ISS 通过测序注射到大脑中的病毒引入的神经元特异性条形码进行轴突追踪,但后来被改编为测序基因条形码 64。同样,尽管不是一个 -ome,电生理学已经在相同细胞的转录组分析之前记录,例如在移植的人类神经元中用补片钳,随后是 HCR–smFISH84,以及在培养的心肌细胞中用细胞外电极,随后是电映射中的 STARmap85。
跨类别比较¶
在本节中,我们讨论了不同类型技术在高检测效率、转录组范围分析、高空间分辨率以及有时较大组织面积之间的权衡,以及与选择技术相关的实用因素,如 FFPE 兼容性和成本/可用性。
检测效率¶
检测效率通常通过对同一细胞类型进行非条形码 smFISH(近 100% 灵敏度的选择性标记基因)进行比较,并比较每个技术在单细胞分辨率技术中检测到的每个基因的转录本平均数,或在没有单细胞分辨率的技术中每单位组织面积检测到的转录本平均数。对于带有 UMI 的 NGS 技术,有时会比较每个细胞或单位面积检测到的 UMI 和基因数量与其他带有 UMI 的技术。
高度多重的 smFISH 技术在这方面表现出色,例如汉明距离 4 的 MERFISH86 相对于非条形码 smFISH 的~95%;多轮杂交往往会降低效率,部分原因是具有不可纠正错误的条形码被丢弃。NGS 条形码技术往往效率较低。在相同组织类型的特定基因中,ST 检测到的 UMI 数量约为单位面积非多重 smFISH 检测到的转录点的 6.9%,相当于每个细胞分析的 scRNA-seq 的检测效率。Visium 的效率似乎略高于 ST,DBiT-seq 的效率更高,为~15.5% 单位面积相对于 smFISH68。子微米技术的效率,按相同组织中的单位面积 UMI 数量,可能与 Visium 相当 72。ISS 往往效率较低,部分原因是逆转录(RT)和 SBL 的效率较低。虽然 scRNA-seq 技术的检测效率在 3% 到 25% 之间(参考文献 65,88,89,90,91),Cartana ISS 和 FISSEQ92 的检测效率分别约为 5% 和~0.005%,而 STARmap 的检测效率仅略高于 scRNA-seq。然而,与 smFISH 相比,ExSeq 声称每个细胞的基因测试效率高达 62%63。较新的技术往往跳过 RT 并使 RNA 模板上的锁环探针连接更高效,如在 BOLORAMIS 和基于杂交的 RNA ISS(HybRISS)93 中,或用 seqFISH 样条形码替代 SBL,如在 HybISS 中,以提高检测效率。
转录组范围分析¶
不使用特定基因探针面板的技术是转录组范围的,例如 ROI 选择后跟 NGS,以及 NGS 条形码,其中 NGS 在 poly-A 捕获的转录本上进行,以及非靶向 ISS,如 FISSEQ 和非靶向 ExSeq。然而,这些转录组范围的技术往往检测效率较低。可以使用某些需要基因探针面板的技术定量超过 10000 个基因的转录本,如 seqFISH+、MERFISH 和 GeoMX WTA,尽管不像 NGS,未被探针靶向的新转录本无法检测到。虽然 GeoMX WTA 已在 Nanostring 以外的研究中使用,GeoMX 起源 94,但总体上基于 smFISH 的技术每个数据集分析的基因数量并未增加(图 3g)。相反,在使用基于 smFISH 和 ISS 的技术的研究中,分析的基因数量较少,并且 smFISH 或 ISS 数据集是转录组范围的 scRNA-seq 数据集的补充 95。
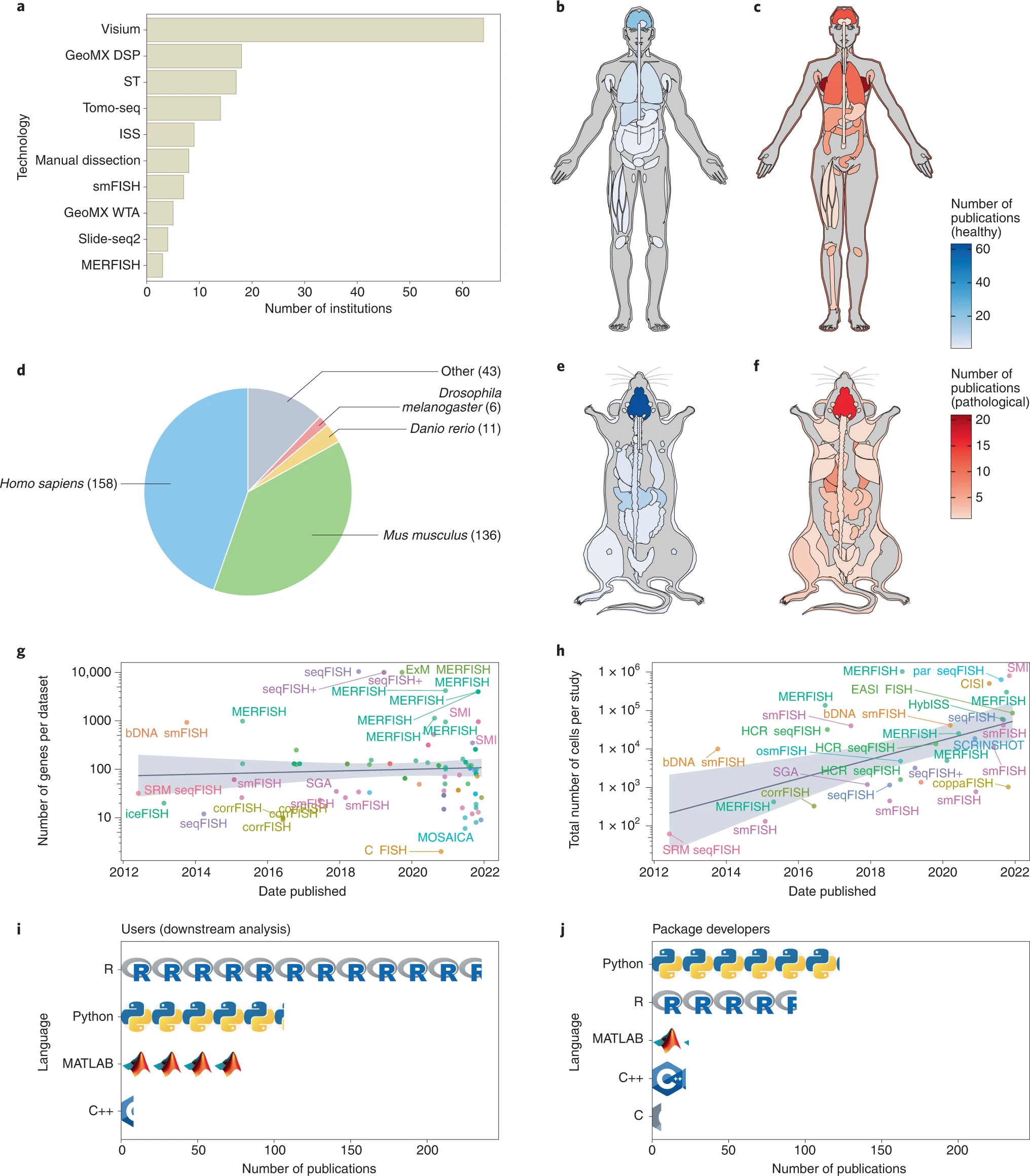
a. 使用每种技术发表论文或预印本的机构数量,除去 LCM 文献过于庞大无法手动整理。仅显示至少有三个机构使用的技术。
b. 人类健康器官(男性,如今没有针对人类女性特有健康器官的研究)的每个器官的出版物数量。
c. 人类病理器官(女性,但有两项关于前列腺癌的研究)的出版物数量。
d. 每个物种的出版物数量。
e. 小鼠健康器官的每个器官的出版物数量。
f. 小鼠病理器官的每个器官的出版物数量。
g. 随时间推移,每个数据集的基因数量。g 和 h 中的灰色带表示 95% 置信区间。在 g 中,斜率与 0 无显著差异(t 检验)。在 g 和 h 中,y 轴为对数转换。
h. 随时间推移,使用基于 smFISH 技术分析的研究中报告的每项研究的细胞总数。
i. 使用每种五种最流行的编程语言进行下游数据分析的数据收集出版物数量。在 i 和 j 中,每个图标代表 20 个出版物。请注意,一篇出版物中可以使用多种编程语言。
j. 使用每种五种最流行的编程语言进行软件包开发的数据分析出版物数量。
检测到的基因数量受限于光学拥挤问题,MERFISH 和 ExSeq 使用扩展显微镜(ExM)来解决这个问题。然而,扩展会减少每个视野覆盖的组织量,从而限制成像吞吐量。
空间分辨率¶
smFISH 和 ISS 技术具有单细胞和单分子分辨率,尽管细胞分割可能具有挑战性。此外,smFISH 和 ISS 技术可以应用于清除后的厚组织切片 80,尽管在这种情况下分析的基因数量远小于大多数二维(2D)高度多重的 smFISH 研究。所有其他类型的技术需要组织切片,因此仅限于 2D,或者 3D 的 z 分辨率受限于切片厚度,冷冻切片通常至少为 10μm。尽管存在亚微米分辨率的 NGS 条形码技术,并且 LCM 和 GeoMX 的 ROI 理论上可以是单细胞或更小,但这些技术在实际使用中往往具有较低的空间分辨率,例如 Visium 的 55μm 和 GeoMX 的几百微米(例如参考文献 94 中的 700×800μm),因为在单细胞或亚细胞分辨率下转录本检测的灵敏度不足 96。
组织面积¶
总体而言,检测效率较低的技术在分析较大组织面积时表现更好,对于 smFISH,细胞数量和基因数量之间似乎存在权衡。在当前时代的空间转录组学中,几个毫米见方的组织切片,例如可以放入 Visium 或 ST 组织捕获区域的小鼠脑冠状切片的相当部分,被认为是大的,增加组织面积和测序深度以提高灵敏度会增加测序成本。Cartana ISS 和 HybISS 也被用于分析几毫米见方的大片组织,但仅约 100 个基因 97。HybISS 在这里的优势是强 RCA 信号和较少的光学拥挤,因为较低的检测效率有助于较低的放大倍数(×20;MERFISH 使用 ×60)和更快的成像。虽然大多数高度多重的 smFISH 数据集仍停留在数百个基因(图 3g),但在报告细胞数量的研究中,每项研究的总细胞数量增加了(图 3h,P<0.001,双侧 t 检验)。ROI 选择技术通常用于少量 ROI,因为选择大量 ROI 并分别处理它们而不使用空间条形码是劳动密集型的。然而,当高空间分辨率不是至关重要或实用时,可以选择非常低分辨率的 ROI 来覆盖更多组织,例如在艾伦人脑图谱 98 中的 LCM 数据集。
可用性¶
虽然大多数技术最初是为冷冻切片开发的,但有些技术与福尔马林固定石蜡包埋(FFPE)兼容,因为这是一种常见的组织存档,有时可能是唯一可用的组织类型。在基于 smFISH 的技术中,ACD 的 RNAscope99 与 FFPE 兼容,但在 FFPE 中一次只能分析 12 个基因,而在冷冻切片中可以分析 48 个基因。在 NGS 条形码技术中,Visium100 和 DBiT-seq101 与 FFPE 兼容,但由于在存档存储中的交联和 RNA 片段化,在 FFPE 组织中每个点检测到的 UMI 和基因数量的检测效率比其冷冻对应物低 5 到 10 倍。LCM 长期以来一直应用于 FFPE 组织,甚至在单细胞分辨率下使用灵敏的 SMART-3Seq102。GeoMX 不仅与 FFPE 兼容,而且主要用于病理人类 FFPE 组织(补充信息第 5.8 图)。
虽然已经开发了许多新技术,但大多数从未超出其发源机构(补充信息图 4.9)。在那些传播范围广的技术中,最受欢迎的往往是商业平台,例如 LCM、10X Visium 及其前身 ST、Cartana ISS(被 10X 收购)和 Nanostring GeoMX(图 3a)。此外,许多主要机构都有 NGS 核心设施,如果没有 LCM、Visium 和 GeoMX(例如,加州大学洛杉矶分校的 TPCL 和密歇根大学安娜堡分校的高级基因组核心),这减少了在各个实验室购买新设备和培训人员的成本。Tomo-seq 也得到了传播,也许是因为它易于使用标准设备实现。相比之下,基于 smFISH 的技术传播得不那么广泛,可能是由于复杂的自制流体系统、长时间的成像时间、TB 级的图像和昂贵的探针。然而,一些 smFISH 技术正在商业化,具有自动成像和流体平台,例如由 Vizgen 商业化的 MERFISH 作为 MERSCOPE,以及另一种基于 smFISH 的技术 Resolve Biosciences 的分子制图平台。此外,Rebus Esper 可以编程自动化不同的 smFISH 技术,并且可以像 Illumina 测序一样在线处理图像,并已被用于自动化循环 -ouroboros smFISH(osmFISH)103。随着新的自动化商业平台的出现,基于 smFISH 的技术可能会流行起来,特别是如果这些平台被核心设施采用。
数据分析¶
高通量空间转录组学数据的处理和分析需要新方法和工具,特别是用于图像预处理、scRNA-seq 数据的空间重建、NGS 条形码数据的细胞类型解卷积、空间变化基因的识别和细胞 - 细胞相互作用推断的问题(图 2g)。
上游分析¶
上游数据分析将原始数据转换为更适合生物学解释的形式,依赖于数据收集技术。
对于基于 smFISH 和 ISS 的方法,原始数据包括荧光点的图像,必须处理这些图像以识别转录点,将点与基因匹配并将点分配给细胞(补充信息第 7.1 节)。smFISH 和 ISS 研究经常使用经典的图像处理工具,例如顶帽滤波器,以去除背景,对齐不同轮次杂交的图像,并使用分水岭进行细胞分割 47,56,86。还可以使用 Ilastik 中的机器学习、DeepCell104 等深度学习包以及结合 scRNA-seq 数据的替代工具 105 进行细胞分割。然而,在没有可视化质膜的情况下,细胞分割的准确性有限。某些分析,例如组织区域识别,可以在没有细胞分割的情况下进行 105。直到 2019 年,图像处理通常使用在专有语言 MATLAB 中编写的文档不全且技术特定的代码进行,但最近,这些代码越来越多地使用开源语言 Python 编写。软件包 starfish106 旨在提供一个统一且文档齐全的用户界面,用于处理来自不同技术的图像,例如 seqFISH、MERFISH 和 ISS,但尚未被广泛采用。
scRNA-seq 技术的改进激发了新方法,以利用高分辨率转录组定量与空间转录组数据的互补性质。对于不是转录组范围的 smFISH 和 ISS 数据,可以使用 scRNA-seq 数据填充空间数据中未分析的基因的表达模式,或者通过将解离的 scRNA-seq 细胞映射到空间参考中,或者通过直接使用 scRNA-seq 的表达谱在空间中填充基因表达(补充信息第 7.3 节)。可以通过在共享的基因上将解离的 scRNA-seq 细胞映射到现有空间数据集上的空间位置来映射细胞,使用倾向于细胞和位置相似性的临时分数 107 或通过最佳传输建模 108。虽然临时评分易于实现,但结果往往是定性的。还可以在没有明确地将 scRNA-seq 细胞映射到位置的情况下,从 scRNA-seq 中填充空间中的基因表达。一种常见的方法是将空间和 scRNA-seq 数据投影到共享的低维和无批次潜在空间中,并通过将空间细胞投影到潜在空间中来估计基因表达。这种方法的例子包括 Seurat3(参考文献 32)和 gimVI109。这些方法还可用于在空间技术不适用于某些多组学数据时,为单细胞多组学数据添加空间上下文。
在没有单细胞分辨率的空间数据中,例如从 ST 和 Visium 获得的数据,scRNA-seq 数据可以为点或体素的细胞类型组成提供信息(补充信息第 7.4 节)。负二项式模型和非负最小二乘法(NNLS)是细胞类型解卷积方法的常见原理。负二项式模型通常以速率和离散度参数化,速率建模为来自 scRNA-seq 的细胞类型特征的加权和,带有图书馆大小和技术灵敏度
的缩放因子;非负权重可以标准化为 1 作为每个点的细胞类型比例。基于负二项式的方法包括 stereoscope110 和 cell2location111。比负二项式更简单,基因表达在 RCTD112 中建模为泊松。细胞类型解卷积也可以通过将每个点的基因表达建模为细胞类型特征的加权和,而不在负二项式分布的速率参数之外,并且使用 NNLS 推断权重。例如,AdRoit113 使用拟合到点基因表达和 scRNA-seq 细胞类型特征的负二项式分布的均值。细胞类型特征可以是来自 scRNA-seq 的非负矩阵分解(NMF)细胞因子分配给细胞类型,例如在 NMFreg66 和 SPOTlight114 中。细胞类型权重可以常规化或阈值化,以限制分配给每个点的细胞类型数量。还可以在文本挖掘中将细胞类型解卷积与主题建模进行类比;细胞类型类似于主题,基因类似于词。主题建模中的潜在狄利克雷分配(LDA)已被适应于细胞类型解卷积,例如在通过主题建模的空间转录组学解卷积(STRIDE)115 和 STdeconvolve116 中;后者是无监督的,不需要 scRNA-seq 参考。
下游分析¶
下游分析通常应用于基因计数矩阵和细胞或点位置,因此在很大程度上独立于数据收集技术。
鉴于 scRNA-seq 与空间数据的相关性,以及空间数据在探索性数据分析(EDA)中经常像 scRNA-seq 数据一样进行分析,流行的 scRNA-seq EDA 生态系统,例如 Seurat32、SCANPY(其在 Python 中的单细胞分析扩展 Squidpy)117 和 SingleCellExperiment(由 SpatialExperiment 扩展)118,增加了对空间数据的功能,例如更新数据容器和功能,以方便可视化空间位置的基因表达和细胞或点元数据(补充信息第 7.2 节)。专门针对空间数据的 EDA 包,具有漂亮的图形和良好的文档,也已经编写,例如 Giotto119 和 STUtility120。Seurat 和 Giotto 还实现了识别空间变化基因的基本方法。此外,Giotto 还实现了在 ST 和 Visium 点中识别细胞类型富集、识别基因共表达和基因表达与细胞类型共定位的关联,以及识别空间区域的方法 121。
空间变化基因是其表达与空间位置相关的基因(补充信息第 7.5 节)。通常有三种方法用于这些基因:高斯过程回归(GPR)122 及其广义到泊松 123 和 NB124,拉普拉斯评分 125 和 Moran’s I。基于 GPR 的方法将标准化的基因表达或泊松或 NB 基因表达的速率参数建模为 GPR,并找出带有空间项的模型是否比没有更好地描述数据。基于拉普拉斯评分的方法识别其表达更能反映空间邻域图结构的基因。还可以将细胞的位置建模为带有基因表达标记的空间点过程;空间变化基因可以被识别为与位置相关的标记 126。拟合 GPR 模型到众多基因可能耗时,特别是在使用马尔可夫链蒙特卡罗的贝叶斯方法时。基于拉普拉斯评分的方法中使用的排列测试也可能耗时。由于 GPR 和拉普拉斯评分方法都寻求识别空间自相关,有时经典的空间自相关指标 Moran’s I 直接用于识别空间变化基因,例如在 Seurat v3 及以上版本中。MERINGUE127 使用 Moran’s I 的局部版本。Moran’s I 及其显著性测试在已建立的地理空间包中实现,运行速度快且简单,但其统计功效可能不如基于模型的方法 123。
空间信息还使识别潜在的细胞 - 细胞相互作用成为可能(补充信息第 7.8 节)。这通常通过了解配体 - 受体(L-R)对来完成,可以测试哪些 L-R 对在邻近细胞或点中更有可能表达 128,或者是否分别表达配体和受体的两种细胞类型更有可能共定位 127。空间点过程中的跨类型 L 函数可用于寻找共定位的细胞类型 129。还可以建模感兴趣基因的表达,包括细胞 - 细胞共定位项;如果模型带有该项比没有更好地描述数据,则该基因被认为与细胞 - 细胞共定位相关 130。
还有许多其他类型的下游分析对空间转录组学分析有用,包括识别典型基因模式(补充信息第 7.6 节)、由转录组定义的空间区域(补充信息第 7.7 节)、推断基因 - 基因相互作用(补充信息第 7.9 节)、亚细胞转录本定位(补充信息第 7.10 节)以及从 H&E 图像中推断基因表达(补充信息第 7.11 节)。
空间转录组学领域的趋势¶
现有技术固有的质量与数量的权衡意味着目前没有单一的“最佳”解决方案,实施方法的难度导致许多技术从未传播到其发源机构之外。LCM、Visium、ST、GeoMX DSP 和 Tomo-seq 被广泛采用(图 3a),几乎所有案例都在美国和西欧(补充信息图 4.12、5.27 和 5.33)。在分析的组织方面,多重的现代技术被广泛用于表征人类组织 131、肿瘤 87(特别是乳腺肿瘤)和不一定具有典型结构的病理组织 132(图 3b,c)。在 SARS-CoV-2 大流行期间,GeoMX DSP 被用于因 COVID-19 死亡的人的肺部尸检中的空间转录组学分析(参考文献 94)。
一些处理的数据和相关的空间变化基因可以从 SpatialDB133 下载和可视化。除去庞大到无法手动整理的 LCM 文献,绝大多数现代研究都是在人类或小鼠中进行的(图 3d),大脑是研究最多的健康器官,而肺(特别是由于 COVID-19)和乳腺肿瘤也是人类中常研究的器官(图 3b,e,f)。特别是国际项目 BRAIN Initiative-Cell Census Network(BICCN)正在构建人类、小鼠和非人灵长类动物大脑的多模式图谱,包括 MERFISH 和 seqFISH 等空间数据 34。
在“数据分析”部分提到的所有软件包都是开源的,使用 R、Python 和 Julia 等语言编写。主要涉及新数据和数据分析包的研究中的下游分析主要使用开源编程语言,如 R、Python 和 C++(图 3i,j)。虽然 MATLAB 仍然流行,但其使用没有像 R 和 Python 那样增加(补充信息图 7.12)。虽然 R 在下游分析和 EDA 中更受欢迎,Python 和 C++ 在软件包开发中更受欢迎(图 3i,j)。大多数软件包未托管在标准存储库上,例如综合 R 存档网络(CRAN)、Bioconductor、pip 或 conda(补充信息图 7.13)。虽然使用 R、Python 和 C++ 的大多数软件包都有良好的文档,但许多 MATLAB 软件包没有(补充信息图 7.12)。标准存储库和文档使软件包更易用;这在补充信息第 7.12 节中有更详细的讨论。
未来展望¶
虽然过去的技术迅速贬值,但它们所依赖的理念和方法是当前时代空间转录组学的基础。该领域在过去五年中迅速扩展(图 4a),大量新技术和 Visium 的普及推动了这一增长(图 4b 和补充信息图 4.9、5.38 和 8.1)。
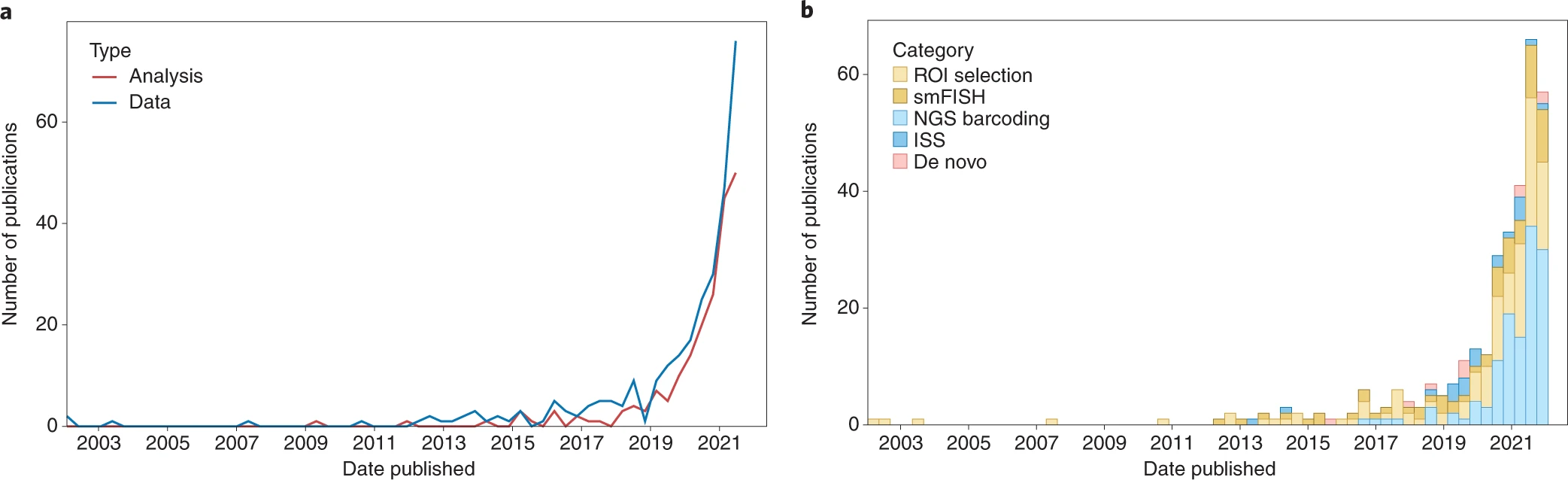
a, 当前时代的数据收集和数据分析的出版物数量随时间变化。柱宽为 120 天;曲线下降是因为图表是在新柱开始时制作的。非整理的 LCM 文献被排除。
b, 图 a 中的数据收集曲线,按技术类别细分。颜色堆叠并按使用该类别技术的出版物总数降序排列。
未来的趋势如何(图 4)?首先,可以在改进数据收集技术方面做更多工作。例如,目前大多数技术需要组织切片。应开发高度多重的整体装载 smFISH 和组织清除协议,以及更高效的计算工具,这些工具将对齐可能来自多个个体甚至不同发育阶段的多个切片,以将当前技术扩展到 3D 和时空分析。未来的技术还可能将当前时代从毫米级扩展到厘米级,并跨越其他模式,如表观基因组学和代谢组学,以全面了解细胞功能。此外,smFISH 和 ISS 技术可以通过信号放大来减少每个转录本所需的探针数量,进而适应于靶向特定异构体的外显子或非翻译区,而不是所有基因的转录本。
其次,目前时代的数据尚未整合到综合数据库中。前传数据库,如 GXD 和 e-Mouse Atlas and Gene Expression(EMAGE)134,包含来自多个来源的数据,可以通过基因符号和发育和空间本体进行查询。此外,ABA26 和 EMAGE 将 ISH 图像对齐到共同坐标,并可以通过表达模式进行查询。虽然一些当前时代的作者提供在线互动可视化其研究数据集的功能 33,但尚未开发出如前传时代那样的整合、查询和可视化多个来源数据的综合数据库。此外,虽然前传本体仍在当前时代的研究中使用,但这些本体可以通过当前时代的全转录组定量数据得到改进。
第三,除 LCM 外,当前时代高度集中于人类和小鼠,而对其他物种(如植物和无脊椎动物)的空间转录组学研究则相对滞后。对非人类和小鼠的前传联盟进行技术现代化改造,为开发有用的空间转录组学图谱提供了很大希望。
第四,一个开源的、文档齐全的、可互操作的、可扩展的工作流程,带有集成的、易于使用的界面,将大大简化空间转录组学数据的收集和分析。目前,对于 EDA 以外的任务,用户仍然需要学习新的语法、转换对象类型,甚至学习新语言来使用某些数据分析工具。最后,我们对方法的调查显示,空间转录组学方法需要更加开放和可访问,以便它们能够在世界范围内被采纳,而不仅限于西方精英机构。
数据可用性¶
空间转录组学文献数据库可以访问:https://docs.google.com/spreadsheets/d/1sJDb9B7AtYmfKv4-m8XR7uc3XXw_k4kGSout8cqZ8bY/edit#gid=1363594152。本文写作时使用的版本位于 GitHub 存储库的 metadata.xlsx 文件中,以重现本文中的图表并呈现补充网站:https://doi.org/10.5281/zenodo.5774128
代码可用性¶
本文中生成图表并呈现补充网站所使用的所有代码都在 GitHub 存储库中:https://github.com/pachterlab/LP_2021。本文最终提交时的存储库冻结 DOI 版本位于 Zenodo:https://doi.org/10.5281/zenodo.5774129。