Proteomic and interactomic insights into the molecular basis of cell functional diversity
摘要¶
生物系统适应变化条件的能力源于它们改变其分子构成的能力。这是通过多种机制来实现的,这些机制调节分子库存的定量组成和多样性。在蛋白质组水平上,分子多样性尤为明显,多个源自同一基因的蛋白质形式可以组合形成不同的蛋白质复合物,从而扩展细胞中功能模块的种类。通过新的“组学”筛选技术的发展,研究分子和模块多样性及其在应对变化条件中的作用才刚刚成为可能。本综述探讨了我们目前对沿基因表达轴调节功能多样性的机制的认识,重点关注蛋白质组和互作组。我们探讨了不同分子层次之间的相互依赖关系,以及这如何促进功能多样性。最后,我们重点介绍了几种研究分子多样性的最新技术,特别是基于质谱分析的蛋白质组及其组织成功能模块的分析,并审视了这一快速发展的领域的未来方向。
引言¶
分子生命科学研究的一个基本目标是理解基因型和表型之间的复杂关系。Beadle 和 Tatum 在 Neurospora spp.上的开创性工作揭示了生化反应的遗传根源,确立了“一个基因,一种蛋白质,一种功能”的范式。这一范式做了两个基本假设:基因与其对应的蛋白质产物的功能之间存在单一的联系(因此暗示了生物系统的功能多样性直接由其蛋白质编码基因编码),并且一个基因的表达对任何其他基因产物的表达或功能没有影响。
随着基因组时代的技术进步以及以越来越高的精度和分辨率解析细胞分子组成的能力的提升,显而易见,“一个基因,一种蛋白质,一种功能”的范式并不能完全解释有机体的复杂功能表型。在对人类基因组进行测序后,国际人类基因组测序联盟报告了约 20,000 个蛋白质编码基因,这个数字明显低于基因组前的估计。这一发现表明,有机体的功能多样性程度与蛋白质编码基因的数量并不直接相关。这一点在大规模筛选方法(如全基因组关联研究和 RNA 干扰筛查)有限的进展中更加明显,这些方法未能提供基因型和表型之间直接联系的见解。尽管使用这些技术发现了具有高度显性的一基因缺陷,许多研究表型的遗传基础比预期更为复杂,通常由基因组变化或涉及其他分子层(如转录组、蛋白质组和互作组)的机制网络支撑,并且这些层之间存在非线性交叉对话。
对生物系统功能复杂性的认识导致了基因型–表型关系的系统生物学视角的范式转变。系统生物学的基本假设是,细胞的功能多样性源于基因组之外的多层次,要全面理解复杂的生物过程,必须将分子及其关系作为整合系统的一部分进行研究。系统生物学方法依赖于在转录组、蛋白质组和互作组水平上检测和精确量化细胞的分子多样性。在本综述中,我们描述了在基因组之外的转录组、蛋白质组和互作组水平上确定分子多样性的已知机制,重点关注蛋白质组和互作组。此外,我们讨论了不同分子层之间的非线性信息流动,包括使不同“组”的精细调节成为可能的交叉对话和反馈回路。最后,我们重点介绍了蛋白质识别和量化、蛋白形式表征以及通过质谱进行互作组分析的最新技术,并指出了在研究分子和功能多样性方面的未来方向。
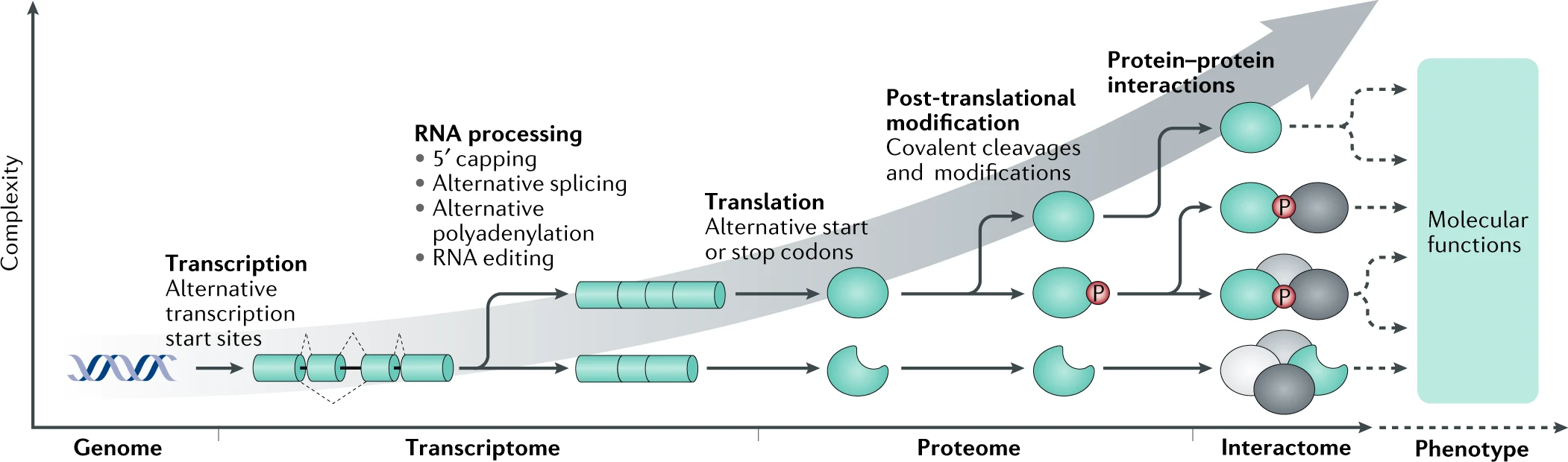
与“一个基因,一种蛋白质,一种功能”的范式相反,细胞复杂性来自许多扩展分子多样性的机制,这些机制超越了蛋白质编码基因组所编码的范围。这些机制包括通过使用替代转录起始位点以及 5′ 端加帽、可变剪接、可变聚腺苷酸化和在共转录或转录后水平进行 RNA 编辑来增加编码潜力。通过在翻译过程中使用替代起始和终止密码子,蛋白质的多样性进一步增加。后翻译修饰(包括共价切割和共价修饰,如磷酸化)引入了高度的多样性。最后,蛋白质可以相互作用,形成多个独特的功能单元,这些单元可能执行各种下游功能。尽管最近的技术进步在全面表征任何给定状态下的转录组、蛋白质组和互作组及其关系方面取得了进展,但评估它们的功能影响和表型仍然是一个尚未完全探索的挑战(虚线部分)。
转录组指导蛋白质组¶
一种生物的基因组中内在嵌入的功能多样性在很大程度上反映了其所包含的蛋白质编码基因的数量。除了表观遗传修饰和三维组织的构象变化外,单个细胞,甚至整个生物体的基因组,基本上是稳定的。因此,细胞具有不同表型并动态调整以应对环境扰动的能力主要源于转录组及其之外的变化。生物系统的转录组包括蛋白质编码的 mRNA 以及非编码的 tRNA、microRNA、核糖体 RNA 和长非编码 RNA。与基因组不同,同一生物体中不同细胞的转录组可能存在显著差异,并且它们可以动态地响应内部或外部刺激。最近的研究估计,在稳态下细胞中蛋白质的变异有 56-84% 可以通过 mRNA 变异来解释。此外,研究表明,表达水平特别高的 mRNA 转录本更有效地被翻译。这些结果支持 mRNA 表达在大多数情况下可以作为稳态下蛋白质表达的有用替代指标的观点。
调节 mRNA 表达水平¶
细胞通过多种调控机制仔细调整 mRNA 表达水平。顺式调控元件与反式作用因子的组合相互作用,创建了一个复杂的网络,该网络调节 RNA 聚合酶介导的基因转录以响应变化的条件,最终影响转录组的组成和数量。除了转录本身,几种转录后机制调节真核细胞的转录组组成。首先,异常处理的 mRNA 转录本可以被 RNA 外切体靶向降解。这种降解是转录水平质量控制的关键组成部分,由于外切体存在于细胞核和细胞质中,它可以靶向新生和成熟的 mRNA 转录本。其次,异常处理的 mRNA 也可以通过无义介导的降解(NMD)被降解。尽管主要与异常处理的 mRNA 降解有关,已知细胞利用外切体和 NMD 机制来微调基因表达,从而直接影响蛋白质组水平的功能多样性。例如,在粒细胞分化过程中,特定剪接异构体的选择性靶向 NMD。第三,RNA 可以在转录后进行修饰。已有超过 100 种不同的 RNA 化学修饰不会改变核苷酸序列,但会改变表转录组。这些修饰中最显著的是腺苷的甲基化生成 N6- 甲基腺苷(m6A)。这种修饰可以影响效应蛋白的募集和 RNA 二级结构的形成,进而调节 mRNA 代谢,包括成熟、翻译和降解。据当前估计,哺乳动物 mRNA 中约 0.1-0.4% 的所有腺苷核苷酸受到 m6A 表转录组修饰,从而影响约 25% 的人类转录本。尽管对 mRNA 修饰的调节及其对蛋白质表达的总体影响知之甚少,但一些疾病表型与 m6A 及其调节因子相关。
调节转录组的编码多样性¶
除了定量变化,真核细胞还进化出了扩展转录序列多样性的机制。多样性的第一级别是通过选择替代转录起始位点引入的。通过在转录起始过程中选择不同的顺式调控启动子区域,可以转录出长度不同的蛋白质编码前体 mRNAs(pre-mRNAs),这些 mRNAs 可能包含不同的开放阅读框和替代的第一个外显子,从而增加翻译蛋白质的数量。例如,由 PKLR 编码的丙酮酸激酶异构体 PKL 和 PKR,是通过组织特异性启动子使用产生的,这些启动子从同一个基因中产生具有不同第一个外显子的替代 mRNA 转录本。人类基因平均有超过四个转录起始位点,这表明这一过程可以显著增加蛋白质组的多样性。
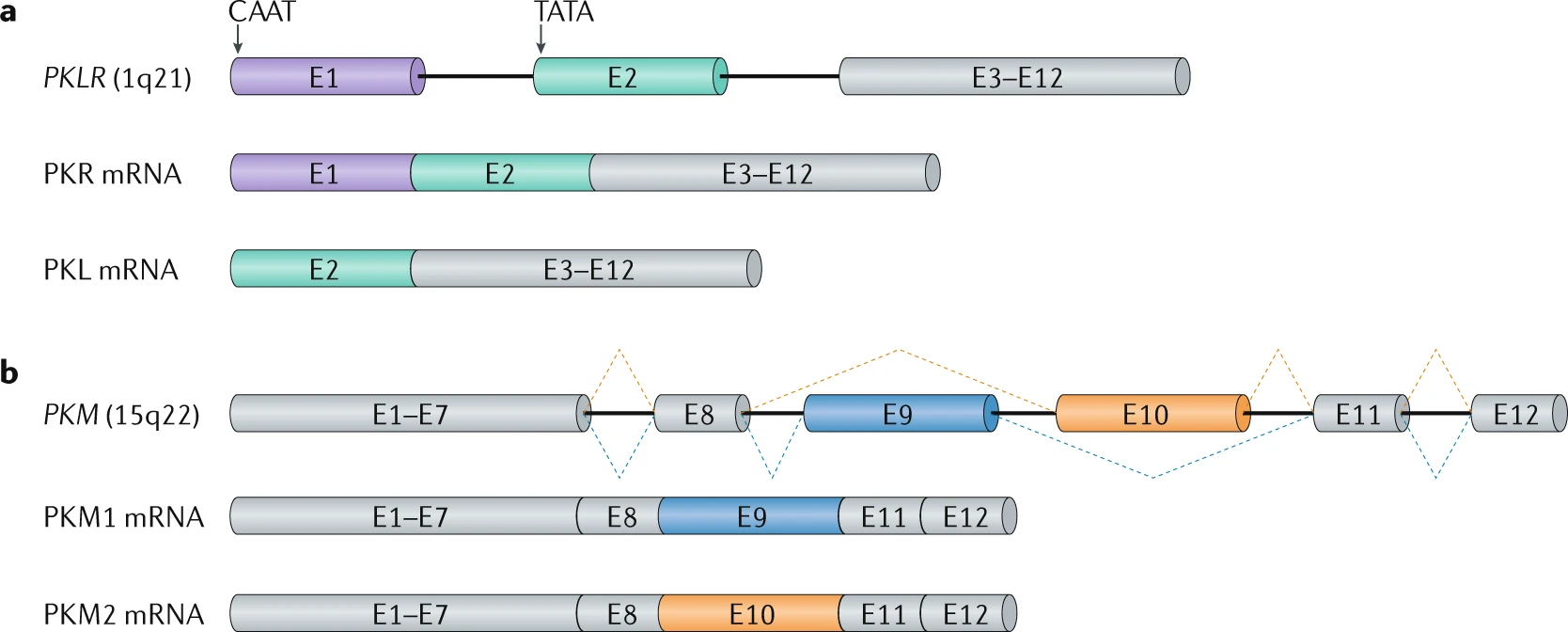
丙酮酸激酶(PK)是一种糖酵解酶,催化糖酵解的最后一步。在哺乳动物中,PK 有四种同工酶(PKR、PKL、PKM1 和 PKM2),由 PKLR 和 PKM 基因编码。
a | PKLR 基因可以根据组织特异性启动子使用情况生成 PKR 和 PKL 同工酶。PKR 同工酶从经典启动子的 CAAT 盒转录,包含所有的外显子(E),即 E1–E12;而 PKL 同工酶从基因内部的 TATA 启动子转录,因此缺少 E1(紫色),从 E2(绿色)开始。PKL 在激素和营养可用性变化时表达,特异于肝脏和肾脏组织。
b | PKM 基因的两个同工酶通过可变剪接生成,导致 E9(蓝色)在 PKM1 中或 E10(橙色)在 PKM2 中互斥性地包含。E9 和 E10 都对应于蛋白质的调控区域:E9 的包含使 PKM1 酶能持续形成四聚体并保持活性,而 E10 的包含允许通过营养可用性调节 PKM2 的活性。PKM1 主要在脑和肌肉等高能量需求的组织中表达,而 PKM2 则在胚胎细胞和癌细胞中表达。
前 mRNA 的多样性通过多种改变转录本核酸序列的过程进一步增加。在前 mRNA 转录本能够从细胞核转运并翻译之前,必须经历三种(共转录)加工事件,这些事件可能扩展其编码潜力。这些过程包括在 5′末端获得 7- 甲基鸟苷帽,剪接以去除基因内区域(内含子)并连接表达区域(外显子),以及在 3′末端切割并添加聚腺苷酸尾巴。这些加工步骤确保转录本免受降解,辅助翻译起始,并能引入显著的分子多样性。尽管 5′加帽很少产生变异,剪接是真核生物中转录本多样性的主要来源。RNA 测序研究表明,92% 的人类多外显子基因经历可变剪接,这相当于估计 86% 的人类基因产生两个或更多不同的 mRNA 异构体。一些基因可以生成超过十种不同剪接的转录本。这些剪接异构体可能包含不同数量的外显子、互斥外显子、保留的内含子或替代的 5′或 3′剪接位点。
除了少数自剪接内含子外,前 mRNA 剪接通常由一个称为剪接体的大型核糖核蛋白复合物催化。选择替代 5′和 3′剪接位点的选择可以通过与非剪接体 RNA 结合蛋白(如异质核核糖核蛋白(hnRNPs)和富含丝氨酸/精氨酸的(SR)蛋白)的相互作用进行调控。可变剪接在发育过程中是必需的,并且 mRNA 异构体可以特定于某些细胞类型、组织和物种。因此,剪接错误与疾病表型(包括癌症、神经退行性疾病和肌肉萎缩症)相关。丙酮酸激酶基因 PKM 是具有两个可变剪接异构体(PKM1 和 PKM2)的基因的一个例子。这两个异构体包含互斥外显子,这些外显子调节酶的活性。多项研究将 PKM 可变剪接模式的变化以及随后的 PKM2 异构体表达升高与多种不同类型的癌症联系起来。总体而言,可变异构体之间的功能差异通常涉及结合基序或特定翻译后修饰(PTM)位点的包含或排除。
尽管可变剪接被认为是转录本多样性的主要贡献者,但通过转录本 3′端的替代聚腺苷酸化也引入了一些额外的变异。替代聚腺苷酸化可以影响转录本编码区的长度或转录本 3′非翻译区的长度,这反过来可以影响转录本的位置、稳定性和翻译效率。在哺乳动物基因组中,至少 70% 的基因具有多个聚腺苷酸化位点,这些位点在发育和细胞分化过程中表现出组织特异性和严格调控,这表明其对蛋白质组和功能多样性的调控影响。
除了这三个主要加工步骤外,一些前 mRNA 通过选择性脱氨进一步编辑腺苷和胞嘧啶,这通常发生在剪接之前。由于人类前 mRNA 中大多数 RNA 编辑位点位于内含子区域,且突变常常是同义突变,RNA 编辑预计对人类细胞的总体编码潜力影响有限。尽管如此,已报道 RNA 编辑与剪接机制之间的交叉对话以及若干影响蛋白质功能和相关疾病表型的非同义 RNA 编辑事件。例如,编码谷氨酸受体 2 的 RNA 中腺苷转为肌苷的编辑会引起谷氨酸转为精氨酸的替换,从而改变受体的钙离子通透性和恢复能力。然而,RNA 编辑事件的功能影响的整体情况仍有待阐明。
总体而言,从约 20,000 个人类基因注释开始,目前估计人类转录组包含在 GENCODE 数据库中注释的超过 83,000 种蛋白质编码 mRNA 异构体,突显了上述机制在生成转录组多样性方面的相关性。关于 mRNA 异构体的丰富性在多大程度上是随机噪音的产物以及哪些替代转录异构体实际上翻译成功能性蛋白质异构体的问题,仍有很大争议。这些问题不能仅通过研究转录组来解决,而需要进一步了解蛋白质组水平。
蛋白质组的多样性¶
生物系统的蛋白质组由其所有表达的蛋白质分子组成。与转录组一样,同一生物体中不同细胞的蛋白质组可能有很大差异,并且能够动态适应内部和外部刺激。
调节蛋白质表达水平¶
尽管蛋白质合成直接依赖于相应的 mRNA 转录本的表达,转录本浓度之外的因素也影响蛋白质表达水平,从而影响细胞的功能景观。
首先,翻译速率受到 mRNA 序列本身的影响,通过密码子偏好、表转录组修饰、转录本与调控元件(如导致转录后基因沉默的 microRNA)的相互作用以及 tRNA 和未充电核糖体的可用性(缺乏这些可降低翻译效率)等机制。其次,蛋白质组受蛋白质降解率的调节,这些降解率受蛋白质定位、稳定性、三维构象及其整合到稳定的蛋白质复合物中的影响。蛋白质还可以直接成为泛素介导的降解靶标,或成为自噬靶标,进而通过溶酶体介导降解。蛋白质的氨基末端和羧基末端组成可以通过蛋白水解系统识别的降解序列来决定蛋白质的半衰期,从而通过 N- 降解途径或 C- 降解途径导致降解。在真核生物中,这些途径包括蛋白酶体和自噬降解。
在健康细胞和稳态下,蛋白质合成和降解是平衡的。此外,mRNA 丰度的变化常常在蛋白质水平上得到缓冲,这意味着 mRNA 丰度的显著变化不会转化为相应的蛋白质丰度变化。尽管癌症中的基因组拷贝数变化通常影响 mRNA 表达水平,但许多这些变化并不影响相应蛋白质的表达,表明这些异常并不显著影响癌症表型。有趣的是,蛋白质水平的缓冲对于形成稳定蛋白质复合物的蛋白质尤为显著,这表明复合物亚基的化学计量比率可以指导蛋白质水平丰度变化。蛋白质复合物介导的缓冲效应是细胞如何减少基因组和转录组水平变异的不良功能影响的一个主要例子,例如由随机事件或潜在的疾病促进基因型引起的变异。
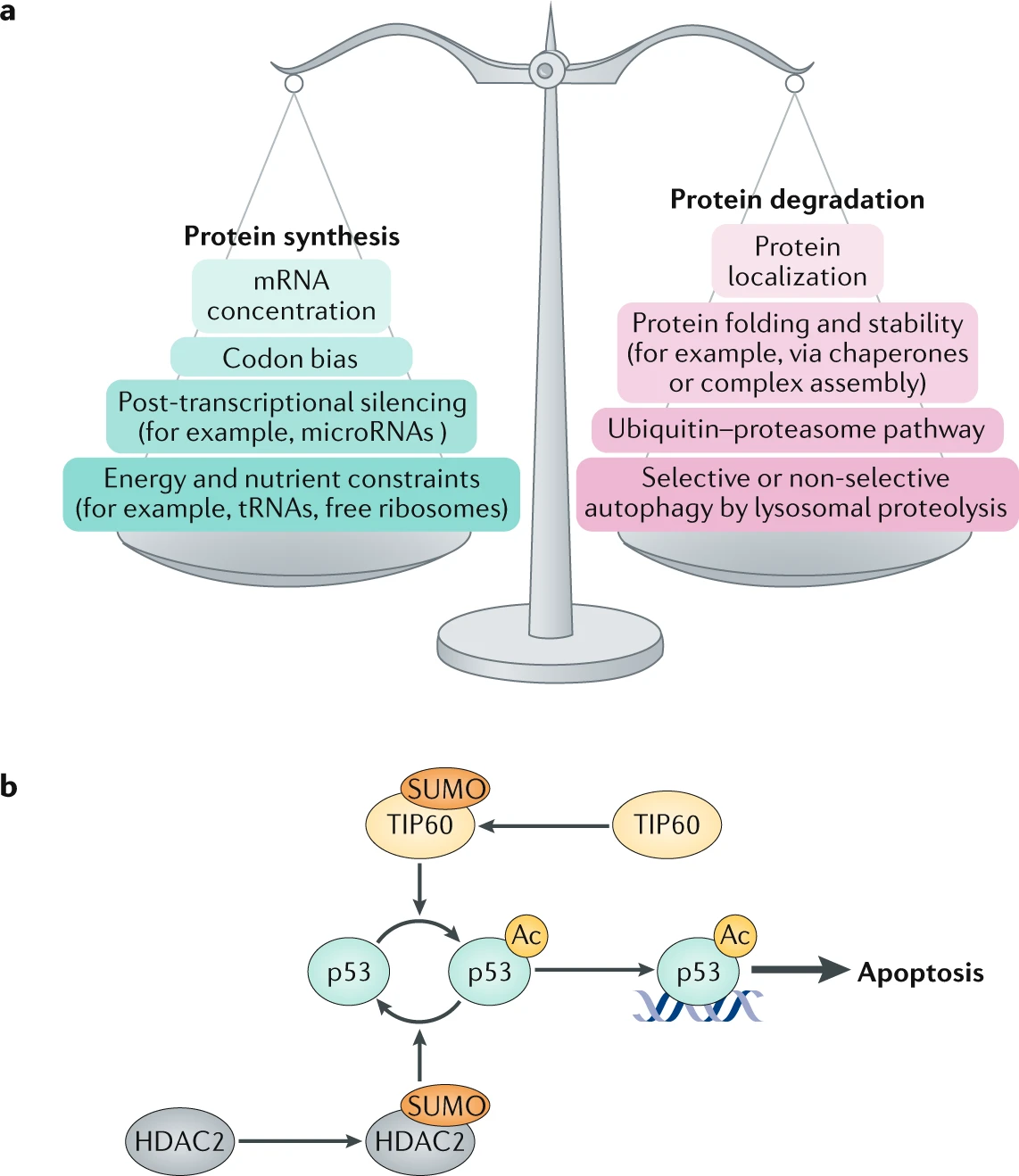
细胞通过多种相互依赖的机制严格调节蛋白质的丰度及其功能活性。
a | 通过蛋白质合成和蛋白质降解之间的精细平衡维持稳态。mRNA 转录本的翻译效率受密码子偏好、转录后调控(如 microRNA 介导的基因沉默)或能量和营养限制(包括 tRNA 和游离核糖体的可用性)的影响。蛋白质降解受蛋白质定位、蛋白质折叠效率(这一过程通常需要伴侣蛋白或整合到蛋白质复合物中)、泛素介导的降解以及选择性和非选择性自噬后溶酶体蛋白水解的影响。蛋白质组降解率预计会缓冲 mRNA 转录本丰度的意外变化,例如由癌症中的基因扩增引起的变化。
b | 翻译后修饰对于调节蛋白质活性非常重要,不同蛋白质上的翻译后修饰通常不是相互独立的。例如,抑癌蛋白 p53 在启动凋亡过程中的活性由其乙酰化(Ac)状态调节。p53 的乙酰化状态和活性由乙酰转移酶 TIP60 增加,而 TIP60 本身通过 SUMO 化激活。相反,组蛋白去乙酰化酶 HDAC2 的 SUMO 化促进 p53 的去乙酰化和失活。
尽管这些影响存在,但 mRNA 丰度通常能够很好地估算稳态下的蛋白质水平(详见上文“转录组指导蛋白质组”)。然而,在细胞过渡阶段(例如细胞周期或分化过程)或快速适应刺激的过程中,mRNA 和蛋白质水平之间的一致性可能会暂时降低。这种下降可能是由于信号从基因到转录本再到蛋白质水平的传递存在延迟,或是由于一些机制允许在不影响 mRNA 水平的情况下快速适应蛋白质组的定量变化;其中一种机制是现有 mRNA 转录本的高翻译率,即所谓的“按需翻译”。另一种机制是快速降解已表达的蛋白质,例如通过泛素 - 蛋白酶体途径,从而无需抑制相应基因的转录。有趣的是,研究表明,在不同条件下,细胞的总体蛋白质浓度保持相对恒定,这意味着如果少数转录本大量上调,相应的蛋白质浓度增加会通过其他蛋白质浓度的下降来补偿。
调节蛋白质水平上的多样性 除了可以直接影响细胞功能景观的定量蛋白质水平外,其他过程还进一步增加了蛋白质组的多样性。蛋白质组多样化的第一级别可以归因于翻译过程中使用替代起始或终止密码子。这些多样化机制较为罕见,因为在约 90-95% 的情况下,5′ AUG 起始密码子的启动发生,而在第一个就位终止密码子的终止效率为约 90-99%。然而,MRPL18 基因中使用替代下游 CUG 起始密码子提供了一个示例,该基因编码线粒体大核糖体亚基蛋白,这种替代起始密码子在这种情况下导致蛋白质的误定位和“杂交”核糖体的形成,促进了耐热性增加。翻译中的错误估计每个氨基酸发生率为约 0.01-0.1%,这些错误也可能导致蛋白质序列变异,从而在应激和衰老中扩展蛋白质组的多样性。然而,大多数随机发生的错误预计会导致非功能性、通常错误折叠的蛋白质,这些蛋白质随后被泛素 - 蛋白酶体系统降解。
蛋白质翻译后修饰(PTMs),可以是共价切割或共价修饰,引入了蛋白质组水平上最多的多样性。蛋白质的主要氨基酸序列的共价切割通常由特定的蛋白酶催化,较少情况下由自催化切割介导。切割可以通过改变蛋白质的定位和活性来改变蛋白质的功能,例如在凋亡过程中胱天蛋白酶介导的前胱天蛋白酶切割就是一个例子。许多蛋白酶以无活性前酶的形式储存,只有在其自身蛋白水解切割后才被激活。截至目前,已知人类细胞表达 460 种不同的具有催化活性的蛋白酶。
共价修饰由酶催化,这些酶将特定的化学基团添加到蛋白质的氨基酸侧链或羧基末端或氨基末端。虽然一些 PTMs 是永久性的,但其他 PTMs 是可逆的,并且可以动态改变。20 种常见蛋白质氨基酸中有 15 种可以被修饰,目前已有约 400 种不同的 PTMs。当前估计三种最普遍的共价 PTMs 是天冬酰胺残基的 N- 连接糖基化、丝氨酸、苏氨酸和酪氨酸残基的磷酸化,以及赖氨酸残基的乙酰化。
已知最大类别的 PTM 酶调节蛋白质磷酸化。尽管>500 种激酶催化蛋白质磷酸化,但只有约 140 种蛋白质磷酸酶催化去磷酸化。粗略估计,至少有 10,000 种不同的磷酸化蛋白质分子形式可以在人类中生成并选择性调控。然而,这个数字可能被低估了,因为磷酸化事件通常在同一蛋白质上组合发生。除了其他功能外,共价修饰是细胞信号通路(如受体酪氨酸激酶信号)的关键组成部分,修饰机制的扰动以及激酶和磷酸酶的特定扰动会导致严重的疾病表型。单个蛋白质或多个蛋白质上的不同 PTM 位点可以通过复杂的分子交叉对话网络连接。例如,关键转录调节因子 p53 的乙酰化状态由乙酰转移酶和去乙酰化酶调节,而这些酶本身受 SUMO 化调节。
总之,转录组和蛋白质组水平上引入变异的机制目前估计基于不同氨基酸序列和 PTMs 的独特组合生成超过 100 万种不同的蛋白形式。因此,蛋白质组学的分析挑战不仅在于检测和量化所有表达的蛋白质编码基因,还在于正确识别和量化每种蛋白形式。尽管最近的技术和方法学发展几乎实现了人类蛋白质组的完整枚举和量化,但蛋白形式的全局识别仍然是一项挑战。然而,最近在顶端蛋白质组学领域的发展使得在人体样本中能够并行识别超过 3000 种独特的蛋白形式。尽管一些蛋白形式的分子功能以及它们相关的表型特征已成功注释,但蛋白形式特定功能的系统评估仍然具有挑战性。
蛋白质组的功能能力通过同一蛋白形式可能存在于多种三维构象中并因此与不同的大分子组件结合而进一步增强。蛋白质的二级和三级结构的组织可以影响其稳定性(例如,通过暴露 N- 降解物或 C- 降解物)、定位和分子功能。目前,大多数系统范围的蛋白质组学研究不考虑这些结构差异,而将蛋白质视为无结构的分子。相比之下,结构重点研究通常从目标前景进行,专注于单个蛋白质或一小部分蛋白质及其结构。探测较大蛋白质组部分中蛋白质构象的方法最近已基于交联质谱、热蛋白质组分析或将有限蛋白水解与目标蛋白质组学相结合进行开发。由于系统范围方法的有限可用性,结构差异在全局尺度上对蛋白质组和功能多样性的影响在很大程度上仍然未知。
动态互作组¶
许多蛋白质作为多分子组件的一部分执行生化功能。这些组件形成了一个庞大的分子相互作用网络,包括相同类型分子之间的相互作用(例如蛋白质 - 蛋白质相互作用,PPI)或不同类型分子之间的相互作用(例如蛋白质 - DNA、蛋白质 -RNA、蛋白质 - 脂质或蛋白质 - 代谢物相互作用)。细胞中的大量分子相互作用被称为其互作组。由于它们在许多生物过程中的核心作用,以及本综述对超越蛋白质编码基因组序列的功能复杂性的关注,我们特别关注 PPI 及其在复杂相互作用网络中的排列。
细胞互作组概述¶
细胞的互作组比转录组和蛋白质组更为多样,并且能够更快地适应环境信号。互作组的多功能性主要原因在于它不需要合成新分子。这在一些 PPIs 的瞬时性质中得以体现,例如激酶与其目标蛋白之间的相互作用。相比之下,其他相互作用更为稳定,参与的蛋白质形成了独特的功能单元,称为大分子蛋白质复合物。显著的这些大分子“机器”的例子是核糖体和蛋白酶体。许多蛋白质在其单体或复合物结合形式中功能不同,例如 14-3-3 蛋白。14-3-3 单体与伴侣蛋白样活性相关,而 14-3-3 二聚体主要作为一个磷酸化依赖的蛋白质支架,重要的组成部分是许多分子信号通路的关键成分。
在过去几十年中,开发了几种实验和生物信息学策略来绘制不同细胞系统的互作组。最全面的单次测定相互作用图谱包含超过 56,000 个相互作用,涉及超过 10,000 个人类蛋白质,这通过系统分析人类开放阅读框并结合串联质谱亲和纯化生成。除了二元 PPI 网络外,还生成了包含有关定义良好的蛋白质复合物及其相关生化功能的信息的数据库;例如,人类蛋白质复合物图谱(hu.MAP)报告了 4,659 个蛋白质复合物。其他蛋白质复合物数据库包括 CORUM 和 Complex Portal。尽管当前数据库提供了人类细胞分子互作组的广泛概述,但随着技术进步,可观察到的相互作用空间预计将显著增加。需要注意的是,PPI 和蛋白质复合物数据库仅提供蛋白质组连接的静态、通用描述,未能解决细胞类型特异性和状态特异性 PPI,这些 PPI 指示细胞的急性生化状态。
影响互作组水平多样性的因素¶
PPI 或蛋白质复合物组装的主要前提是所涉及的蛋白质在所需的化学计量下共表达,并在同一细胞室内共定位。每种蛋白质可能需要处于允许形成并在能量上有利的特定三维构象。相互作用可能依赖于组装伴侣,这些伴侣可以是蛋白质或其他协助相互作用形成的分子,例如作为支架或通过导致相互作用亚基中的任何一个的构象变化。PPI 和复合物组装通常在响应刺激时动态变化,例如暴露于生长因子或在分子过程的整个过程中。一个示例性过程是剪接反应不同步骤中剪接体组成的动态变化。考虑到其许多重排和不同的催化活性,剪接体机械本身提供了一个由互作组水平协调的功能多样性的令人印象深刻的例子。
在互作组水平上研究功能多样化时的一个中心考虑因素是相互作用可能是蛋白形式依赖的,反之亦然。只有当蛋白质亚基具有特定氨基酸序列或 PTMs 时,相互作用才可能形成。此外,某些 PTMs 可能仅在复合物形成时发生。蛋白形式依赖的互作组重排是细胞在响应变化的环境条件下快速适应其功能景观的常见机制。通过磷酸化依赖的 14-3-3 蛋白二聚体与其目标蛋白的结合可以说明这一点,这些目标蛋白通常需要含有一个或两个磷酸化的丝氨酸或苏氨酸残基才能与 14-3-3 蛋白支架相互作用。14-3-3 互作组广泛重排的一个示例原因是缺氧诱导的 AMP 活化蛋白激酶(AMPK)激活,导致若干 AMPK 靶标的磷酸化,这些靶标随后可以与 14-3-3 相互作用,从而引发从合成代谢向分解代谢的适应性转变并促进自噬。
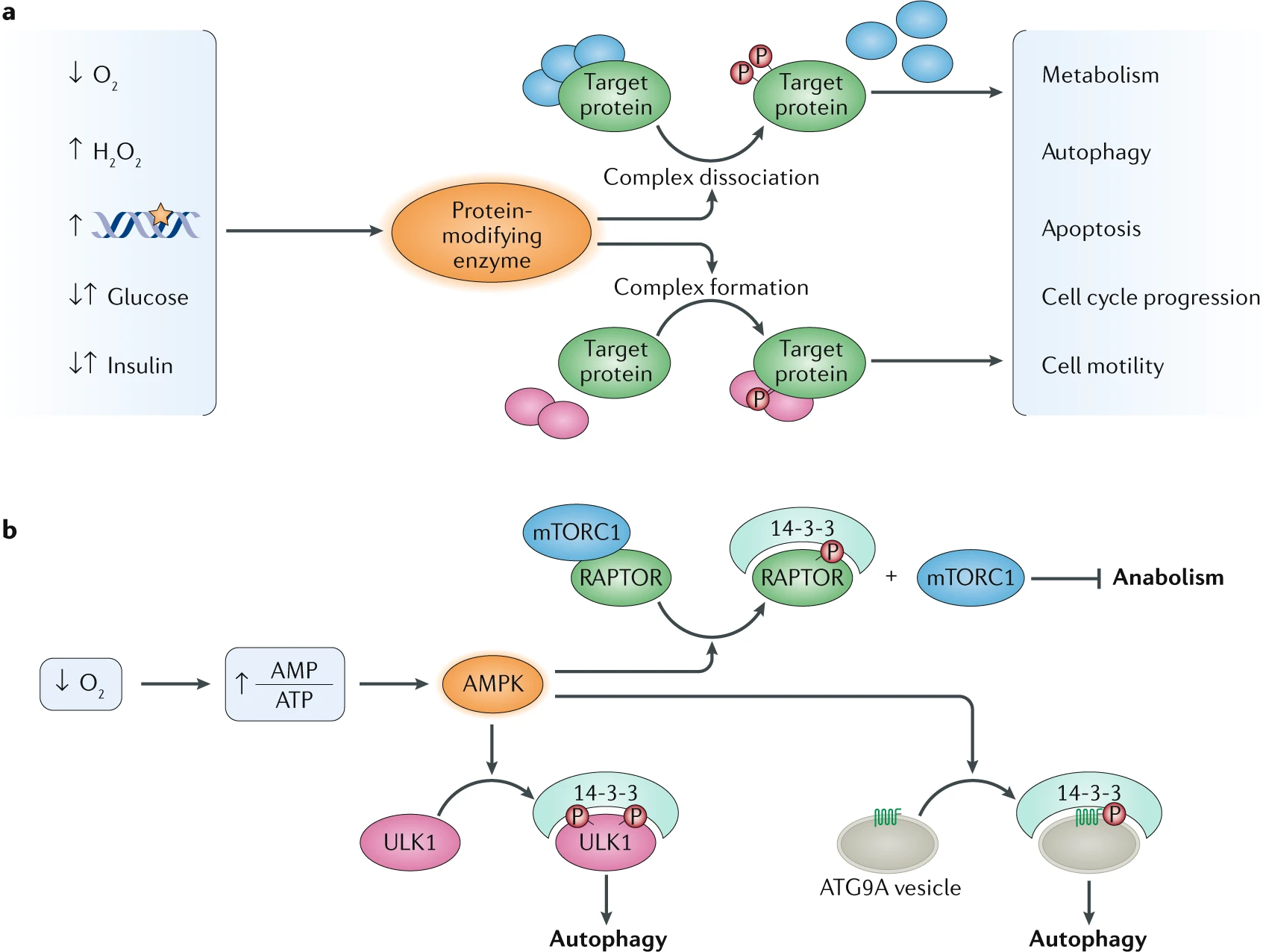
蛋白质复合物的动态组装和解体,通常特定于某些蛋白形式,是调节功能多样性的关键机制。
a | 当细胞暴露于条件变化时,例如缺氧、氧化应激、DNA 损伤、营养供应变化(例如低葡萄糖)或其他信号分子的刺激(例如胰岛素),这通常会导致特定蛋白质修饰酶的激活。随后,酶靶蛋白的翻译后修饰通常与条件特异性蛋白质复合物的形成或解体相关,从而通过调节在合成代谢和分解代谢、自噬、凋亡、细胞周期进展和细胞运动之间的适应性转变来影响细胞的功能景观。
b | 以磷酸化(P)依赖的 14-3-3 与其靶蛋白的结合为例,说明蛋白形式特异性的互作组重排及其功能影响。在缺氧条件下,细胞内 AMP 与 ATP 比率的增加以及随后的 AMP 活化蛋白激酶(AMPK)激活,导致 14-3-3 互作组的磷酸化依赖重排。AMPK 磷酸化 RAPTOR,后者是 mTOR 复合物 1(mTORC1)的关键结合伙伴,随后与 14-3-3 结合。这种结合导致 RAPTOR 释放 mTORC1,促使 mTORC1 抑制,并促进从合成代谢到分解代谢的适应性转变。14-3-3 蛋白还与 mTORC1 下游的其他 AMPK 靶标相互作用,包括 Unc51 样自噬激活激酶(ULK1)和自噬相关蛋白 9A(ATG9A)。14-3-3 与 ULK1 和 ATG9A 的相互作用在饥饿条件下促进自噬,以维持细胞代谢。有关在不同细胞条件下 14-3-3 互作组磷酸化依赖重塑的综合综述可在其他地方找到。
迄今为止,尚未有关于蛋白质复合物形成与特定蛋白质变体之间相互依赖关系的系统性信息。从功能角度来看,研究替代蛋白质功能在多大程度上归因于相互作用组、蛋白质组和转录组或其组合的实际变化是很有趣的。从基因中心角度来看,同样有趣的是研究源自同一基因位点的不同蛋白质变体在多大程度上执行不同的功能,以及哪些剪接异构体或蛋白质翻译后修饰是没有功能影响的随机噪音的产物。近年来,系统地解决这些问题的实验策略已经开始出现,我们将在后文中讨论这些方法。
分子层间的串扰¶
在过去的几十年中,已经发现了许多串扰和反馈回路的例子,其中传统上被认为是“下游”的事件会影响“上游”的目标。这些例子包括正负自我调节,其中基因产物直接或间接调节其自身的生产。一个包含多个转录反馈回路的基因调控网络的典型例子是哺乳动物中的昼夜节律基因网络。核心调控基因 PER1、PER2、CRY1 和 CRY2 的蛋白质产物相互作用,形成一个抑制它们自身转录的蛋白质复合物。另一种负自我调节机制是基于选择性剪接偶联无义介导的 mRNA 降解,这种现象预计会影响约 10-30% 的哺乳动物基因。SR 蛋白和 hnRNPs 在 RNA 处理和剪接中具有各种作用,例如,它们可以结合到自身的转录本上,导致产生的剪接变体被 NMD 降解,从而下调蛋白质水平并维持蛋白质表达的稳态(顺式调控反馈;图 5)。比顺式调控反馈回路更显著的是反式调控机制,由 SR 蛋白和 hnRNPs 激活或抑制其他基因的前 mRNA 剪接所示例。能够调控不同目标基因表达的转录因子是反式调控反馈的最显著形式。迄今为止,细胞内只有一小部分调控网络在详细的机制水平上进行了研究,不同分子层之间的关系才刚刚开始被探索。最近研究不同分子水平上的数量性状位点揭示了多层次分子网络如何调解基因组变异效应的有趣见解。在未来,类似于数量性状位点分析的策略可能会用于研究细胞或有机体内分子和功能多样性的扩展。为了能够全面映射这样的调控网络,并更好地理解不同分子层之间的相互依赖和串扰以及这些如何贡献于表型多样性,高质量的所有“组学”层面的测量是必要的。

剪接由剪接体 RNA 结合蛋白(如富含丝氨酸/精氨酸(SR)蛋白和异质核核糖核蛋白(hnRNPs))调节。SR 蛋白通常作为剪接激活因子,而 hnRNPs 则作为剪接抑制因子。这两类蛋白可以通过反式调控反馈调节不同目标基因的剪接。此外,它们还可以通过负顺式调控反馈调节自身前体 mRNA 的剪接。SR 蛋白可以激活自身前体 mRNA 中携带提前终止密码子的外显子的剪接,从而导致无义介导的 mRNA 降解(NMD)。hnRNPs 则抑制自身前体 mRNA 内部外显子的剪接,导致框移突变并引发 NMD。
生成“组学”数据的方法¶
基于上述机制,真核细胞可以生成多种 mRNA 异构体。通过称为“下一代测序”的技术进步,使得在单碱基分辨率上全面定量分析这些转录本成为可能。最近在基于下一代测序的转录组分析技术方面的发展已经在其他地方得到广泛审查。本文的重点放在蛋白质水平上,比较既有的和最新的基于蛋白质组学和互作组学的技术。
基于质谱的蛋白质组学¶
大多数蛋白质组学研究基于质谱,有两种主要方法:顶端(top-down)和底端(bottom-up)蛋白质组学。在顶端蛋白质组学中,完整蛋白质经过色谱分离、离子化并注入质谱仪。随后分析完整蛋白质及其碎片的质谱,能够直接推导出具有独特初级蛋白质序列和翻译后修饰(PTMs)的单个蛋白形式。然而,复杂蛋白混合物的顶端蛋白质组学分析在分析和专业知识要求方面都具有挑战性,因为蛋白形式分离技术的限制、高分子质量离子的质谱分析问题、复杂的碎片离子模式以及使用现有分析软件进行光谱解释的难度。因此,对于全蛋白质组分析,底端蛋白质组学工作流程更为常用。
在底端蛋白质组学中,首先将蛋白质通过酶解成较小的肽序列,随后通过液相色谱分离、离子化并通过串联质谱进行分析,同时记录肽和碎片离子光谱。由于在酶解过程中肽和蛋白质之间的原始连接丢失,底端蛋白质组学的主要限制是需要进行计算机推理步骤,将测量的肽信号映射回单个蛋白质。蛋白质推理的要求进一步限制了底端蛋白质组学工作流程区分具有相同初级蛋白质结构但不同 PTMs 组合的蛋白形式的能力。尽管可以获得有关单个 PTMs 或序列变体的存在和定位的信息,以及“平均”蛋白形式(即映射到同一标准蛋白质序列的一组肽,包括替代序列变体或 PTMs,其独特组合未解析),但据我们所知,尚未有针对底端蛋白质组学方法的能够自信地分配和区分独特蛋白形式的稳健策略。此外,不同的蛋白质推理方法在其假设上可能有所不同,因此只能在所选模型内提供精确答案,并不一定反映现实。然而,底端蛋白质组学方法的高灵敏度、可重复性和可扩展性仍然使其成为全蛋白质组研究的首选方法。底端蛋白质组学的不同策略在先前已有综述,并在附加框中提供了有关底端蛋白质组学和衍生蛋白形式解析信息的相关机会的进一步详细信息。
互补实验和基于质谱的互作组学¶
在过去几十年中,开发了几种不同的方法来分析瞬时和稳定的蛋白质 - 蛋白质相互作用(PPIs)和蛋白质复合物。尽管许多这些方法(例如,荧光共振能量转移和双分子荧光互补)基于靶向一个相对低通量的小蛋白质组子集,我们特别关注最近开发的用于高通量检测稳定相互作用和蛋白质组装的系统级策略。截至目前,使用了五种主要技术:酵母双杂交筛选(Y2H 筛选)及相关的互补实验、亲和纯化结合质谱(AP-MS)、以 BioID 和 APEX 为代表的邻近标记方法、交联质谱和蛋白质共分级结合质谱(CoFrac-MS)。最近,热蛋白质组分析和有限蛋白水解结合质谱作为一种在一次实验中并行检测多个蛋白质复合物的策略出现。这里,我们重点关注上述更成熟的策略,总结在图 6a 中。
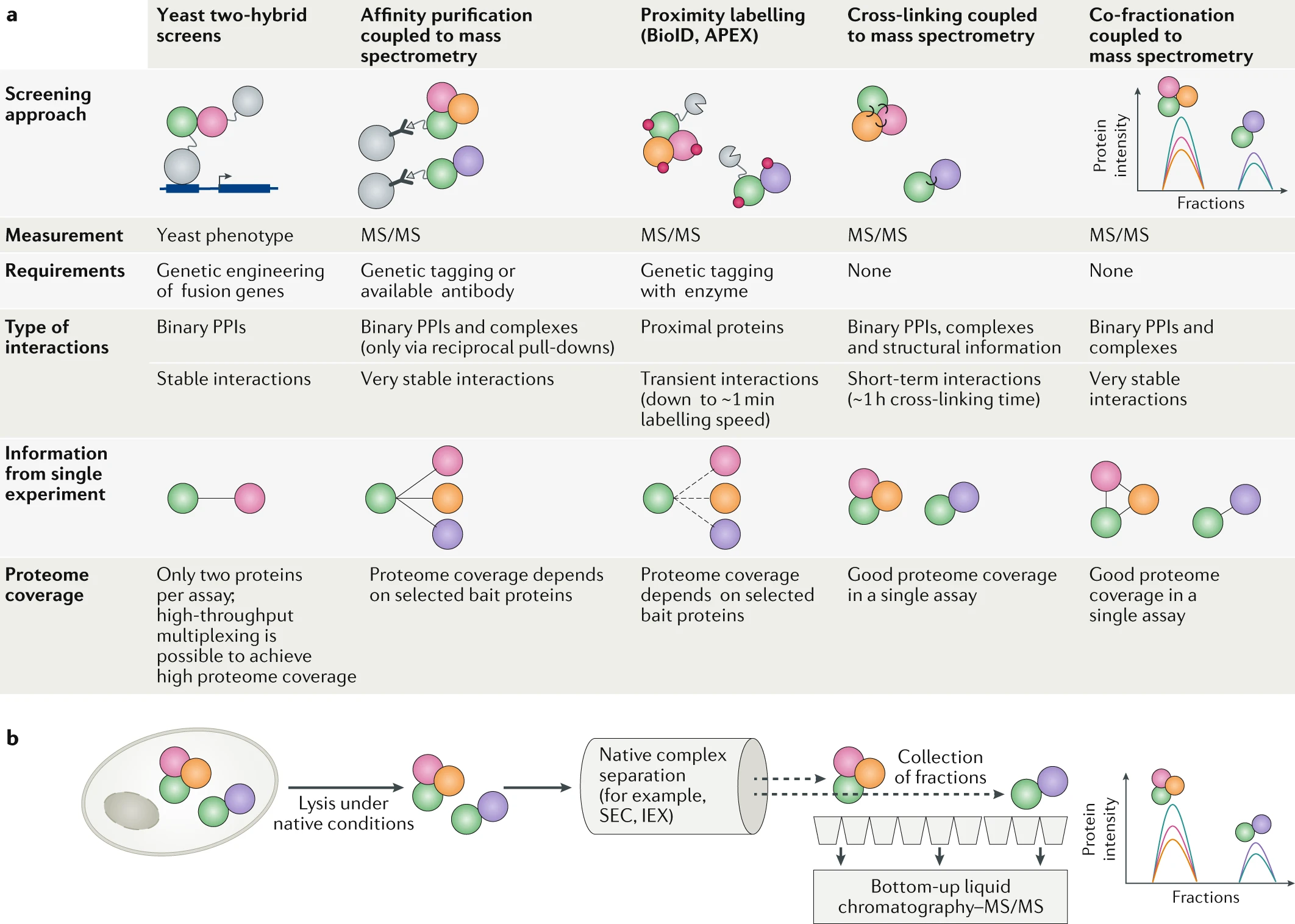
在过去的几十年里,已经开发了几种不同的方法来分析瞬态和稳定的蛋白质 - 蛋白质相互作用(PPIs)及蛋白质复合物。a | 传统上,PPIs(由蛋白质之间的实线表示)通过酵母双杂交策略在中等通量下进行研究。随着基于质谱的蛋白质组学的进步,亲和纯化结合质谱现在是大规模相互作用组图谱绘制的最先进方法。为了捕捉更多的瞬态相互作用和空间接近性,可以采用如 BioID 或 APEX 这样的近距离标记策略,以识别在时空上接近的蛋白质(由蛋白质之间的虚线表示)。交联质谱提供了检测蛋白质复合物及其结构排列的机会(由彼此接触的蛋白质表示)。最近,蛋白质共分级结合质谱已被开发为一种有前景的新策略,可通过一次分级实验高度并行地检测蛋白质复合物。b | 对于蛋白质共分级结合质谱实验,细胞首先在天然条件下裂解以保持蛋白质复合物完整。随后根据其物理化学性质,如体积排阻色谱(SEC)或离子交换色谱(IEX),将复合物分离和分级。所有取样的分级部分分别通过自下而上的质谱(液相色谱与串联质谱(MS/MS))进行分析。可以从蛋白质沿色谱维度的局部共洗脱和高度相关性推断出 PPIs 和蛋白质复合物。实线表示处理步骤的顺序,虚线表示分离过程。
直到最近,Y2H 筛选一直是系统性绘制 PPI 网络的主要技术。尽管经典的 Y2H 筛选实验在许多成功应用和方法扩展方面取得了成果,但该系统仍存在一些局限性。首先,它要求蛋白质在酵母细胞中表达。酵母细胞内环境中的表达可能会影响蛋白质结构、定位和 PTM 模式,从而影响 PPIs。其次,在 Y2H 筛选中,假阳性和假阴性相互作用的比例难以确定,筛选需要验证实验,其中使用正参考集和随机选择的蛋白质对作为对照。第三,要获得全面的 PPI 网络需要大量实验,因为每个可能的蛋白质对都需要进行独立的实验。最后,Y2H 筛选只能生成一般的蛋白质相互作用图,而不能用于确定特定细胞状态下的 PPIs。要实现特定条件下的蛋白质相互作用图,需要其他技术。
随着基于质谱的蛋白质组学的进步,AP-MS 已成为条件解析的大规模 PPI 研究的主要技术。在 AP-MS 中,感兴趣的蛋白质(诱饵)通过特异性针对诱饵的抗体(允许使用非工程化细胞)或特异性针对与诱饵融合的亲和标签的抗体从细胞裂解物中纯化。下拉实验在温和的非变性条件下进行,旨在保持诱饵蛋白及其相互作用伙伴(猎物蛋白)之间的稳定 PPI。随后,通过自下而上的质谱分析诱饵及其共同纯化的猎物蛋白,以识别和量化诱饵的稳定相互作用伙伴。AP-MS 方法通常利用在 AP-MS 数据集中常见的背景蛋白过滤假阳性结果,从而在确定可靠的相互作用伙伴方面实现高选择性和特异性。一个 AP-MS 实验可以并行映射多个相互作用,这是 Y2H 筛选所不具备的优势。通过结合在控制的假发现率下检测 PPIs 的高灵敏度和合理的通量,AP-MS 是研究全球相互作用网络的首选方法。然而,AP-MS 研究受限于常常需要遗传工程,因此可能会改变蛋白质结构和相互作用位点,或者依赖于特定抗体的可用性。此外,AP-MS 实验只能捕捉在整个实验过程中保持稳定的相互作用。尽管存在局限性,AP-MS 技术被应用于创建迄今为止最全面的单次测定相互作用图,包含>10,000 个人类蛋白质之间的>56,000 个关联。
为了捕捉细胞内的瞬态相互作用或一般空间接近性,可以使用以 BioID 和 APEX 策略为代表的近距离标记方法。在此,诱饵蛋白与酶融合,例如 BioID 中的生物素连接酶或 APEX 中的过氧化物酶,该酶以高空间和高时间分辨率标记所有近距离蛋白质。与其他方法相比,报告的蛋白质不必与诱饵物理相互作用,而只需在其附近。与 AP-MS 类似,近距离标记技术可以在稳态下生成全局相互作用网络;然而,如果关注整个细胞系统的相互作用组,研究蛋白质组范围内的 PPI 网络的动态变化仍受限于个别实验的蛋白质组覆盖范围和技术通量。尽管如此,BioID(结合补充的 AP-MS 分析)最近被用于研究在人类中心体–纤毛界面在纤毛发生过程中的动态相互作用图景,绘制了>1,700 个独特成分和>7,000 个相互作用。最近的方法开发进一步提高了结合 AP-MS 和 BioID 的便利性,使用称为 MAC-tag 的单一构建体来同时研究相互作用、细胞定位和蛋白质复合物内的空间距离。
交联质谱主要用于通过生成距离约束来从纯化蛋白质或蛋白质复合物中获取低分辨率结构信息,这提供了分子中氨基酸之间距离的上限。最近,该技术扩展到更复杂的应用,包括交联大肠杆菌、人的 HeLa 细胞系和果蝇胚胎的全蛋白质组,以识别 PPIs 并在系统层面获取结构信息。
获得蛋白质组范围连接信息的另一种方法基于 CoFrac-MS。在典型的 CoFrac-MS 工作流程中,细胞在接近天然条件下裂解以保持大多数 PPI 和蛋白质复合物的完整。生成的细胞裂解物随后根据蛋白质组装体的物理化学性质进行分离和分级,同时尽量在分级过程中保持蛋白质复合物的完整。分离技术的例子包括离子交换色谱(按电荷分离)和体积排阻色谱(按流体动力半径分离)。对于自下而上的蛋白质组分析,每个取样的分级部分被酶切成肽段,随后通过液相色谱与串联质谱进行分析。CoFrac-MS 的基本假设是,相互作用的蛋白质在色谱分离过程中至少部分共洗脱,并且基于液相色谱与串联质谱检测和定量,它们将具有相似的定量蛋白质谱。相互作用的蛋白质因此可以基于它们的高度相关的洗脱信号被识别。与 AP-MS 研究相比,CoFrac-MS 工作流程的主要优点是,原则上,它们允许在不需要遗传工程或特定抗体的情况下分析数千种蛋白质及其相互作用。通过准确的相对定量,该方法允许样品之间的差异分析,并识别不同细胞状态下检测到的 PPI 的变化。CoFrac-MS 数据集通常用于从蛋白质洗脱谱的成对相关性推断新的 PPI 和蛋白质复合物,并使用图分区算法预测蛋白质复合物组装。然而,这种基于相关性的新的蛋白质复合物分配的灵敏度和选择性受到所选色谱分离技术的峰容量的限制,因为如果检测到的蛋白质数量显著超过色谱柱的峰容量,则非相互作用的蛋白质也会色谱共洗脱。为了克服选择性的限制,并在 CoFrac-MS 数据集中实现蛋白质复合物组装状态的更定量评估,我们最近提出了一种新的、针对蛋白质复合物的分析概念。这里,CoFrac-MS 数据直接查询预先定义的蛋白质复合物的证据,使用公共蛋白质复合物数据库的数据,从而显著提高选择性。这种分析策略为探测和量化不同条件下蛋白质组装的重构提供了一种有前途的新方法。
蛋白变体特异性蛋白质相互作用和组装分析¶
蛋白质生物学中的一个主要问题是特定蛋白变体与蛋白质相互作用组的连接性或相互依赖性。在大多数大规模相互作用组研究中,重点放在自下而上的蛋白质组策略上,完全表征的蛋白变体的推断(即精确氨基酸序列和所有 PTM 的位置的表征)及其个体相互作用图景仍然难以实现。然而,个别修饰,如特定肽段甚至特定氨基酸位点的 PTM,已经在大规模研究中与蛋白质相互作用网络相关联。例如,一项人类激酶的 AP-MS 研究整合了 57 个人类激酶的相互作用图景与这些激酶及其相互作用伙伴的磷酸化状态的信息。此外,使用 CoFrac-MS 方法分析了半胱天冬酶介导的蛋白酶切割对 Jurkat 细胞相互作用组的影响。CoFrac-MS 策略是系统评估蛋白变体与蛋白质复合物组装状态之间关系的有前途工具。通过对沿蛋白质复合物分离维度的共洗脱模式进行肽水平分析,可以区分可能源自不同蛋白变体的行为相似的肽段。然而,还没有进行系统研究以了解表达的蛋白变体及其对功能多样化的全球影响,如可以通过组装成不同蛋白质复合物来指示的那样。
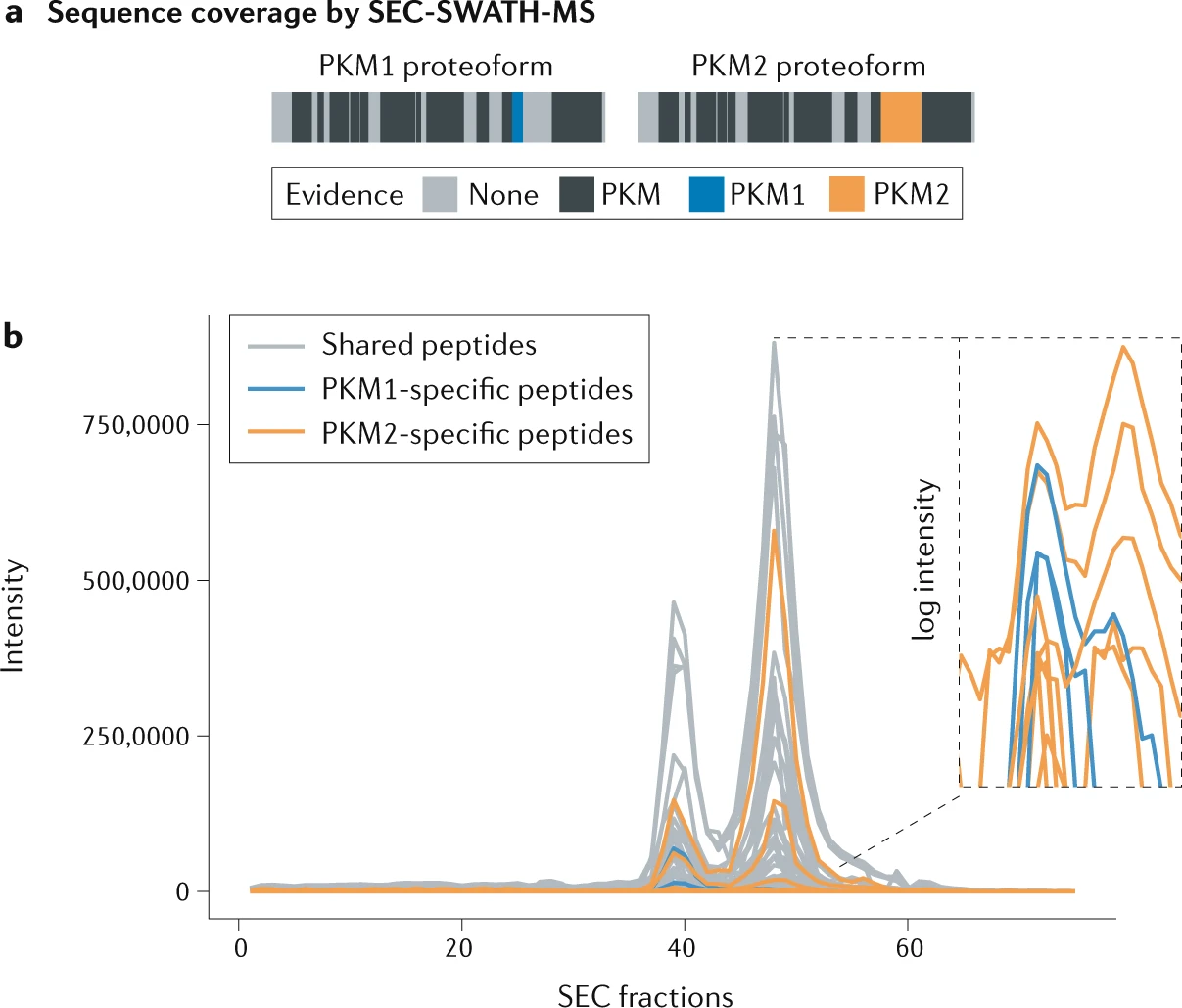
通过肽水平分析从天然复合物共分级结合质谱的数据,确认了 PKM 蛋白形式特异性的组装特性。所示数据通过对人类细胞系裂解物进行尺寸排阻色谱分级,随后进行数据独立采集质谱(SEC-SWATH-MS)生成。
a | 在蛋白质水平评估两种丙酮酸激酶 PKM 异构体显示了 SEC-SWATH-MS 数据中的高序列覆盖率。可以检测到 PKM1(蓝色)和 PKM2(橙色)的特异性肽段。未在所获取数据中检测到的序列区域为浅灰色,而 PKM1 和 PKM2 共享肽段证据的区域为深灰色。
b | 沿色谱维度检测到的 PKM 肽段的洗脱谱显示了两个不同的洗脱峰。这两个峰可能分别对应于 PKM 的二聚体(右)和四聚体(左)。PKM2(橙色)的特异性肽段对两种组装状态都有贡献,而 PKM1(蓝色)的特异性肽段仅在四聚体构象中检测到。此图基于以前未发表的内部数据生成,该数据使用类似于 refs 中描述的 SEC-SWATH-MS 数据采集和分析方法生成。
结论与展望¶
过去几十年的研究揭示,生命系统调整其功能景观的能力来源于多个分子层次,而不仅仅是编码蛋白质的基因组。除了调节转录组和蛋白质组的定量组成,细胞还可以调整转录本和蛋白变体的多样性,以及将蛋白变体整合到不同的蛋白质复合物中。技术进步,尤其是在质谱领域,最近为系统分析细胞系统在所有不同分析水平上的分子多样性铺平了道路,无论是以通用图的形式,还是以差异分析的形式来检测特定状态的差异。
尽管每个基因平均估计有约 100 种蛋白变体,但蛋白质组水平的多样性与细胞功能多样性之间的联系尚未被系统评估。评估不同蛋白变体影响的一个有前途的方向是研究它们与其他特性(如蛋白质定位或蛋白质复合物形成)的相互依赖性。例如,可以通过 CoFrac-MS 数据集的肽水平分析来实现这一方法,从而获得包含关于蛋白质组装状态和特定蛋白变体之间相互作用的有价值信息。
虽然蛋白质复合物水平的信息预计可以为功能多样性的原因提供最佳估计,但仍缺乏系统探测功能多样性的方法。随着“组学”技术的进一步改进,以及方法的融合和整合(如顶 - 下和底 - 上蛋白质组学方法),我们可以推测在未来十年内大多数蛋白变体和蛋白质复合物将被绘制出来。这种绘图将能够更系统地评估不同分子层次之间的相互依赖性和相互作用,并重建复杂的非线性调控网络。主要目标将是解决分子多样性如何转化为功能多样性的问题,以更好地理解在不同实验和临床条件下基因型和表型之间的关系。