Repression of latent NF κB enhancers by PDX1 regulates β cell functional heterogeneity
摘要¶
谱系决定和活性依赖的转录因子之间的相互作用通过尚不完全了解的机制决定了多细胞组织内单个细胞的身份和功能。通过构建人胰岛中染色质状态的单细胞图谱,我们鉴定出了受胰腺十二指肠同源框蛋白 -1 (PDX1) 高或低活性调控的 β 细胞亚型。PDX1 活性降低的 β 细胞在潜在的核因子 κB (NF-κB) 增强子处显示出染色质可及性增加。Pdx1 弱表现型小鼠在夜间表现出 NF-κB 的去抑制和葡萄糖耐受性减弱。三维分析结合染色质免疫沉淀 (ChIP) 测序显示,PDX1 通过涉及 SIN3A 的远程染色质接触在昼夜节律和炎症增强子处沉默 NF-κB。相反,β 细胞中 Bmal1 的缺失会破坏全基因组范围内 PDX1 和 NF-κB 的 DNA 结合。最后,对抗 NF-κB 靶标白细胞介素 (IL)-1β 受体改善了 Pdx1 弱表现型胰岛的胰岛素分泌。我们的研究揭示了由 PDX1 活性梯度定义的功能性单 β 细胞亚型,并确定了 NF-κB 作为胰岛素促分泌治疗的靶点。
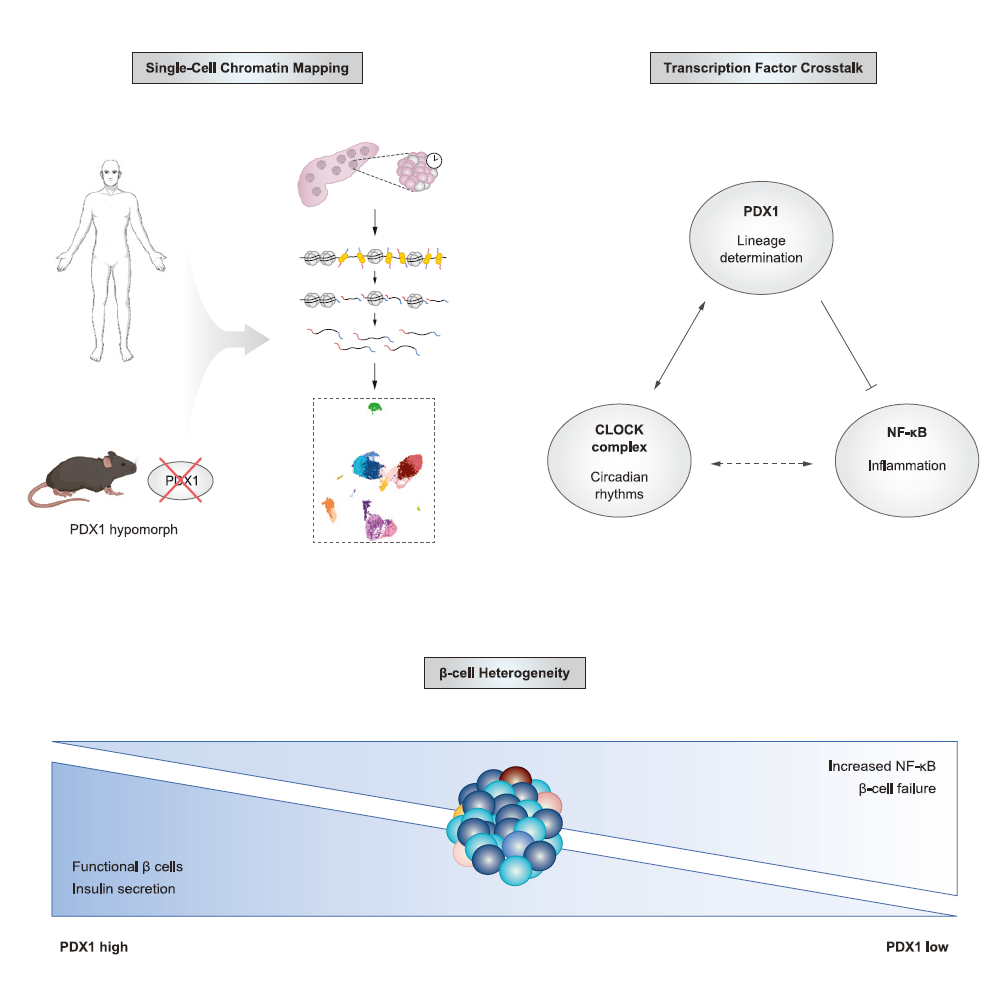
引言¶
谱系决定和信号依赖转录因子(LDTFs 和 SDTFs)之间的协作在应对与禁食 - 进食周期相关的营养环境变化时,维持能量恒定。昼夜节律也在几乎所有代谢组织中协调前瞻性的转录循环,但昼夜节律、营养和谱系依赖的途径在多细胞组织中的单个细胞内如何相互作用仍然未知。
内分泌胰腺是一个异质性器官,其功能在昼夜周期中表现出显著的波动,并由不同的葡萄糖调节细胞类型组成。胰岛素产生的 β 细胞的身份在胰腺内胚层芽的主要转变过程中通过胰腺十二指肠同源框因子 1(PDX1)与 NK6 同源域因子 NKX6.1、基础亮氨酸拉链 MAFA 和肝细胞核因子(HNFs)之间的组合相互作用而启动。PDX1 还通过抑制 α 细胞基因网络(包括由 aristaless 相关同源框(ARX)控制的基因)和抑制如羧酸盐转运蛋白等损害有氧代谢的因子,促进 β 细胞的个体发生。在断奶后,PDX1 诱导由核心时钟转录因子 CLOCK 和 BMAL1 共调控的基因的表达,这些基因在葡萄糖刺激的胰岛素分泌中起重要作用。BMAL1 也被证明通过防止 α 细胞基因表达在白喉毒素诱导的细胞损伤响应再生期间稳定 β 细胞身份。一个尚未解决的问题是 LDTFs 功能受损是否可能导致成年的 β 细胞功能衰退。
在本研究中,我们使用表观基因组分析来识别和解析人类和小鼠胰岛中谱系和信号依赖的转录网络之间的串扰。我们鉴定了由谱系因子 PDX1 活性差异以及炎症和昼夜节律基因网络活动差异所表征的不同 β 细胞亚型。我们的研究揭示了高 PDX1 活性的 β 细胞在涉及胰岛素分泌的基因网络中表现出可及性,这些基因网络也受分子时钟控制。相反,低 PDX1 活性的 β 细胞在核因子 κB (NF-κB) 诱导基因的增强子内表现出更大的染色质可及性。我们还显示,用白细胞介素(IL)-1β 受体阻断肽处理 Pdx1 弱表现型动物的胰岛会增强胰岛素分泌能力。我们的研究揭示了 PDX1 介导的潜在 NF-κB 增强子的抑制丧失导致 β 细胞衰退,并为靶向 NF-κB 以治疗低胰岛素血症提供了依据。
结果¶
单细胞染色质可及性测序在不同的 β 细胞群体中识别了 PDX1 和潜在 NF-κB 增强子的特征¶
胰岛由产生生理上对立激素的多种细胞群组成,这些激素在禁食和餐后葡萄糖代谢中起重要作用。最近的单细胞测序分析表明,在胰岛内,胰岛素产生细胞在顺式调控元件(CREs)内的可及性模式上表现出异质性。识别潜在β细胞亚型的 CREs 也包括与 2 型糖尿病风险相关的非编码变体。为了阐明谱系和信号依赖转录因子之间的组合相互作用如何决定β细胞的异质性和功能,我们使用单核转座酶可及染色质测序(snATAC-seq)对来自综合胰岛分配计划(IIDP)的人的尸体胰岛进行了染色质可及性和转录因子结合基序的分析(图 1A)。我们在 20519 个单胰岛细胞中鉴定了 334116 个可及的 CREs,这些细胞使用统一流形近似和投影(UMAP)降维方法分成 19 个不同的簇(见 STAR 方法)。这 19 个簇包括典型的内分泌细胞类型,具有四个α细胞簇(α1-4)、三个β细胞簇(β1-3)和一个δ细胞簇(图 1B),它们分别由邻近胰高血糖素(GCG)、胰岛素/胰岛素样生长因子 2(INS/IGF2)和生长抑素(SST)编码基因的染色质可及性特征来表征(图 1C)。通过在典型细胞标志物处的可及性识别出的其他胰岛相关细胞亚群包括星形(PDGFRB)、神经嵴(SOX10)、淋巴和髓样免疫(CCL4)、内皮(PECAM1)、导管(KRT19)和腺泡(REG1A)细胞(图 1B 和 1C)。
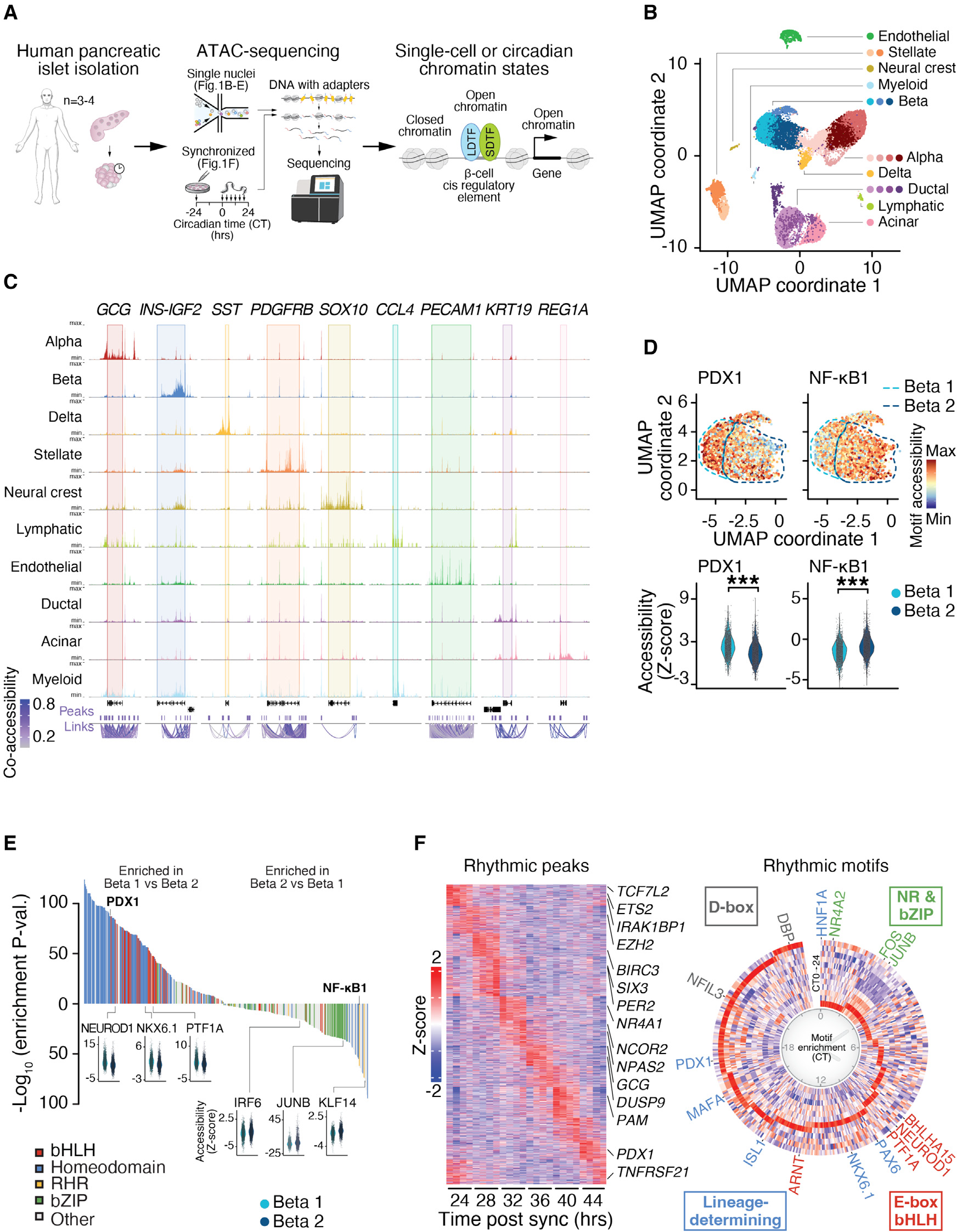
图 1 单细胞染色质可及性测序在不同人胰岛β细胞群体中识别 PDX1 和 NF-κB 增强子特征
(A)通过 ATAC 测序在人的尸体胰岛中分析单核和节律性整体染色质可及性。 (B)通过聚类和降维分析(UMAP)根据可及顺式调控元件的标准化读数识别 19 个不同的细胞亚群(来自 3 个人的 20519 个单胰岛核)。 (C)在谱系特异性标记基因(GCG(α细胞)、INS-IGF2(β细胞)、SST(δ细胞)、PDGFRB(星形细胞)、SOX10(神经嵴)、CCL4(淋巴和髓样免疫细胞)、PECAM1(内皮细胞)、KRT19(导管细胞)和 REG1A(腺泡细胞))处按细胞类型划分的聚合测序片段。共同可及性链接分析连接在共调控区域网络内的单个 snATAC-seq 峰值可及性得分。 (D)在 PDX1(左)和 NF-κB(右)基序处的可及性在β1 和β2 细胞亚群中分别富集。单个细胞 chromVAR Z 得分(上)和跨β1 和β2 亚群的亚群 Z 得分分布(下)展示了在 PDX1 和 NF-κB 基序处染色质可及性的显著变化。 (E)β1 和 β2 亚群之间转录因子基序可及性的差异分析显示,β1 细胞中富含基础螺旋 - 环 - 螺旋(bHLH)和谱系决定的转录因子,而β2 细胞中富含即时早期基础亮氨酸拉链(bZIP)和 NF-κB 转录因子。 (F)在福斯科林冲击后每 4 小时对同步化的整体胰岛进行 ATAC-seq,持续 24 小时(每个时间点每个样本来自 4 个人的 10 个人胰岛等价物 [IEQs])。数据使用 eJTK_Cycle 以 2 小时分辨率分析,揭示了在 ATAC-seq 峰值处染色质开放的 24 小时模式(左)和与细胞身份和节律功能相关的β细胞转录因子基序(右)。详见图 S1 和表 S1 和 S3。
snATAC-seq 相对于单细胞 mRNA 转录组学分析的一个关键优势在于能够检测转录因子(TFs)在基因启动子和远程增强子处的调控活性。为了检查差异基因调控是否在 β 细胞异质性中起作用,我们首先比较了 β1 和 β2 β 细胞亚群的基因活性得分(在基因坐标和上游 2 kb 区域重叠的片段的可及读数总和),它们分别占总 β 细胞群体的 47.2% 和 45.1%。我们鉴定了 600 个基因,这些基因显示出显著的变异(p < 0.05),包括定义 β 细胞的基因如 INS (6.45e−87)、MAFA (2.25e−50) 和 GLP1R (2.37e−36),以及炎症基因如 PTGER3 (1.471551e−05)、IL2 (5.968259e−15) 和 LPAR1 (8.646495e−15)(图 S1A)(表 S1)。使用 snATAC-seq 基因活性得分作为 Seurat 聚类中的传输锚点,分析来自 12 个非糖尿病人类胰岛的单细胞 RNA 测序(scRNA-seq)数据(GEO:GSE114297),揭示了 β1 和 β2 亚群之间 RNA 表达的 log2 倍变化与染色质基因活性得分之间的显著相关性(p = 1.29e−30,Pearson's r = 0.35)(图 S1B)。当比较 α 细胞亚群时(表 S2)以及直接比较整个 α 和 β 细胞群体时(p = 1.0e−16,Pearson's r = 0.75)(图 S1B),也观察到了基因表达变化与染色质可及性之间的类似对应关系。这些数据揭示了单细胞转录本丰度与不同细胞类型间染色质可及性的一致性。
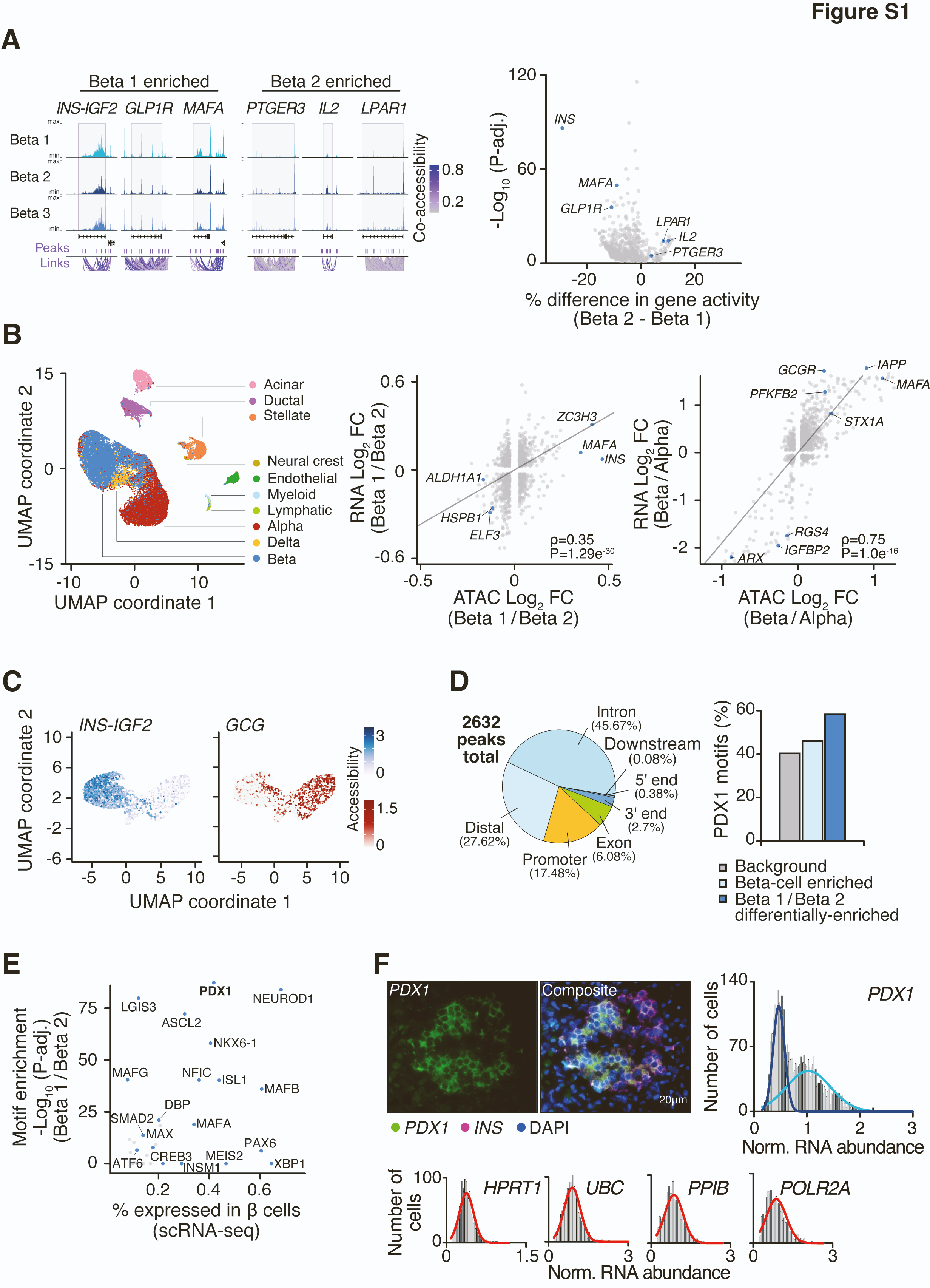
图 S1. 人胰岛中单核 ATAC-seq 揭示 β 细胞亚群中的不同转录特征,相关内容见图 1。
(A)在 β1-3 亚群中 β 细胞和炎症基因处的标准化读数密度(左)和通过评估 β1 与 β2 细胞可及性差异百分比的差异基因活性评分(右)。 (B)单细胞 RNA 测序聚类(GSE114297)和使用 snATAC-seq 染色质可及性进行参考映射注释(左),以及在人 β1 与 β2 细胞(中)和 α 与 β 细胞(右)亚群之间差异调控基因的基因活性评分(ATAC-seq)和 mRNA 表达(RNA-seq)之间的对应关系。 (C)内分泌激素群体在 β 细胞富集的 INS-IGF2 位点和 α 细胞富集的 GCG 位点的不同染色质开放。 (D)在 β1 与 β2 细胞中鉴定出的 2632 个差异可及峰在启动子或增强子区域中的百分比。在 β1 和 β2 的比较中鉴定的差异可及位点中的 PDX1 基序富集(在背景峰集中为 58% 对 40%,超几何检验,P < 0.001,在 β 细胞富集的增强子中为 58% 对 46%,Fisher 精确检验,P < 0.001)。 (E)通过 scRNA-seq 从 snATAC-seq 分析中检测到 TFs 表达的 β 细胞百分比,绘制与通过比较 β1 和 β2 亚群的可及景观计算的基序富集 P 值。 (F)使用 PDX1 和 INS 探针以及 DAPI 染色的人胰岛代表性多重 RNAscope 图像(左上图)。使用 ImageJ 定量单个细胞中每个探针的荧光数据,并将原始值标准化到 DAPI 染色。使用 Kolmogorov-Smirnov 测试对单成分与多成分正态分布进行最佳拟合,揭示了两个不同的 β 细胞群体在 PDX1 丰度方面的差异(右上图)和四个管家基因(HPRT1、UBC、PPIB、POLR2A)的正态分布(下图)。n=3 名来自 IIDP 的人类男性胰腺供体,每个供体分析 13-35 个胰岛(RRIDs: SAMN12227196, SAMN17928660, SAMN25519947)。详见图 1 和表 S1-S3。
为了阐明 β 细胞异质性的表观遗传机制,我们比较了 INS 富集(β1)和 INS 低(β2)β 细胞簇之间的差异 CREs(图 S1A 和 S1C),发现大多数差异可及位点(2,632 个峰中的 73.29%)位于增强子区域(启动子上游超过 3 kb 或下游 1 kb)(padj < 0.05)(图 S1D)。接下来,我们利用每个细胞的强大测序深度(每个细胞 37,400 个高质量片段),使用 chromVAR 测量与 TF 相关的染色质可及性来识别 β1 和 β2 亚群中的差异 TF 活性。比较 β1 和 β2 细胞簇之间的基序 Z 得分,发现多个 β 细胞 LDTF 基序在 INS 旁边具有较高的可及性(图 1D 和 S1A)。在已建立的 LDTFs 中,PDX1 是胰腺发育的主要调节因子之一,是最显著的差异富集基序之一(padj = 3.98e−88),也是成人 β 细胞中表达最丰富的 TFs 之一(图 1D、1E 和 S1E)。我们进一步发现,58% 的 β1 和 β2 细胞之间的差异可及位点含有 PDX1 基序,而背景峰集为 40%(超几何检验,p < 0.001)(图 S1D)。因此,我们分别将这些 β1 和 β2 亚群称为 PDX1 高细胞和 PDX1 低细胞。在 PDX1 高细胞中富集的其他 TFs 是已知的成熟 β 细胞发育所需的因子,包括神经分化因子 1(NEUROD1)、NKX6.1 和基础螺旋 - 环 - 螺旋(bHLH)胰腺转录因子 1A(PTF1A),以及其他 bHLH TFs,它们的 DNA 结合基序包含分子时钟的靶标 E-box 基序(padj < 0.001)(图 1E)(表 S3)。相反,INS 位点低可及性的 β2 细胞簇在 NF-κB1、锌指样因子 KLF14、JUNB 和干扰素调节因子 6(IRF6)TF 基序中高度富集(padj < 0.001)(图 1E)(表 S3),所有这些都在应激、免疫反应和细胞增殖和存活期间的信号诱导炎症基因表达中起作用。这些数据支持最近的 snATAC-seq 研究,这些研究发现了具有独特即时早期 TF 特征的激素高和激素低内分泌胰岛亚型(GEO:GSE160472、GSE160473 和 GSE163610)。我们注意到我们的研究的一个关键优势是使用了 10× Genomics 平台的活组织,这使得每个细胞的测序读覆盖率比之前的研究高约 10 倍,并检测到包括 NF-κB 位点在内的罕见染色质开放事件。总之,我们对染色质活动的单细胞测序在人的胰岛内鉴定出双峰胰岛素产生细胞群体——一个 INS 富集簇具有高 PDX1 活性,另一个 INS 低簇具有低 PDX1 活性但在 NF-κB TF 活性中显著富集。我们还使用 RNAscope 检查了人胰岛中的 PDX1 RNA 丰度,揭示了 PDX1 RNA 水平的双峰分布,与各种管家基因观察到的正态分布相反(图 S1F)。这些观察结果支持两种不同的 β 细胞群体在 PDX1 活性方面的存在。
TFs 之间的协作相互作用定义了成熟 β 细胞的各个亚群,我们的数据表明已知的 β 细胞成熟和功能的调节因子在 PDX1 高 β 细胞中独特地激活。昼夜节律时钟激活因子 CLOCK/BMAL1 被证明有助于胰岛素产生细胞的代谢成熟和身份,尽管其潜在机制仍不清楚。上述发现 PDX1 和时钟调控元件共存于 INS 富集簇中,支持了 PDX1 和节律基因表达可能在 β 细胞功能中发挥作用的可能性。为了检查 β 细胞身份的转录调节因子是否以昼夜节律方式调控,我们通过在先用福斯可林脉冲同步分子时钟的人胰岛中进行 ATAC-seq 来评估 24 小时染色质开放的节律。福斯可林给药后 24 小时开始,每 4 小时进行基因组分析,持续 24 小时,远超过福斯可林对染色质作用的约 1 小时半衰期。我们使用标准化 ATAC-seq 计数在 24 小时时间序列内生成供体内 Z 得分(见 STAR 方法),并使用经验 Jonckheere-Terpstra-Kendall 算法(eJTK_CYCLE)在全基因组范围内鉴定了 3,125 个统计上有节律的 ATAC-seq 峰(Bonferroni p < 0.05)(图 1F)。在胰岛细胞身份(SIX3、PDX1 和 GCG)、昼夜节律表达(PER2 和 NPAS2)和炎症(ETS2 和 TNFRSF21)中起作用的基因增强子附近鉴定了节律位点。TF 基序富集分析在有节律的 ATAC-seq 峰处鉴定出显著富集的 TF(例如 PDX1、NEUROD1 和 PTF1A)(padj < 0.05)(图 1F),这些 TF 也在 PDX1 高细胞中富集(图 1E)。总的来说,这些研究表明 PDX1 和其他 TFs 在一天中的染色质协同激活可能有助于 β 细胞功能。
PDX1 抑制 NF-κB 增强子以控制 β 细胞功能¶
在基因组的非编码调控区域内,谱系和信号依赖 TFs 之间的串扰驱动内分泌胰腺的身份和功能。新兴研究表明,LDTFs 定义了 β 细胞的不同亚群,这些亚群在胰岛素分泌和 2 型糖尿病中很重要。基于我们在不同细胞群体中发现的 PDX1 活性特征,这些细胞群体表现出 NF-κB 和时钟活动的不同模式,我们使用从胰岛中分离的单核的 snATAC-seq 分析,试图解析 PDX1 在炎症和昼夜节律基因网络控制中的功能。由于 Pdx1 的缺失会导致胚胎致死性,我们利用 Pdx1 一个等位基因缺失的杂合小鼠(Pdx1+/-),在有或无第二个等位基因在 Pdx1 的关键调控增强子(区域 IV)内带有突变的情况下(突变的 Pdx1ΔAIV/- 对照 Pdx1+/- 小鼠,分别)(图 2A)。Pdx1ΔAIV/- 小鼠在断奶后会出现低胰岛素血症、葡萄糖耐受不良和 β 细胞增殖减少。使用来自 Pdx1ΔAIV/- 和对照 Pdx1+/- 小鼠断奶后(10-18 周龄)收获的细胞核的合并 snATAC-seq 数据,我们在 9,480 个单胰岛细胞中鉴定了 187,343 个可及 CREs,并将这些细胞分类为 15 个独特的胰岛细胞群体,包括三个 β 细胞群体。这三个小鼠 β 细胞群体根据 PDX1 活性表现出与我们在人的胰岛观察到的类似的变化。比较来自 Pdx1ΔAIV/- 突变体和 Pdx1+/- 基因型的所有三个 β 细胞群体的染色质可及性,发现 1,373 个独特基因的 1,902 个染色质峰发生显著变化(padj < 0.05)。Pdx1 区域 IV 增强子区域是最显著下调的非编码 CRE,这与此增强子的半合子性一致,随后是 Ins1 启动子。来自合并 β 细胞群体的基序分析显示,Pdx1ΔAIV/- 突变小鼠在促进 β 细胞功能的关键 TF 结合位点的染色质可及性降低,包括 NEUROD1、NEUROG2、PTF1A 和 bHLH 因子,提供了遗传证据支持我们的发现,即在人的胰岛中,PDX1 高亚群与与 β 细胞和昼夜节律功能相关的 TFs 相关。相反,在 Pdx1 缺陷突变体中显示出染色质可及性增加的位点包含在促炎信号传导中起作用的 TFs 结合位点,包括 NF-κB1、NF-κB2 和 RELA。在 Pdx1 弱表现型动物的胰岛中发现的具有增加的促炎和减少的胰岛素分泌网络的染色质景观变化,与我们在人的胰岛中观察到的在 PDX1 和 NF-κB 中分别富集或耗竭的独特增强子特征类似。基因本体路径分析进一步表明 PDX1 在诱导 β 细胞中葡萄糖反应胰岛素分泌的基因,同时抑制促炎调控元件方面的作用。在 Pdx1ΔAIV/- 突变 β 细胞中染色质封闭(即在对照组中增加的可及性)附近的峰值与激素外排途径相关,包括 MODY(例如 Iapp、Ins1、Pdx1 和 Mafa)和 2 型糖尿病(例如 Irs1、Mapk10、Mtor 和 Pparg),以及急性炎症反应的负调控。相反,突变 β 细胞中染色质更可及的基因集在形态发生(例如 Egfr、Igf1r 和 Mapt)、突触传递的负调控(例如 Asic1、Mgll、Pla2g6 和 Gabrb3)和在昼夜节律时钟抑制后解除抑制的基因网络(例如 Bhlhe40、Ptger3 和 Cipc)中富集。然而,我们没有检测到 Pdx1 突变和对照内分泌细胞群体之间 α、β 或 δ 细胞的细胞定义基因 Gcg 或 Sst 的可及性显著变化。最后,RNA 测序显示,与对照小鼠相比,从 Pdx1ΔAIV/- 突变小鼠中分离的胰岛中,参与葡萄糖刺激胰岛素分泌的基因(包括 Vamp2、Glp1r 和 Snap25)以及参与昼夜节律的基因(包括 Per 和 Cry 基因)的表达显著减少,这与 Pdx1ΔAIV/- 突变体中的 β 细胞功能受损和昼夜节律中断一致。此外,我们观察到促炎 NF-κB 信号传导和细胞凋亡相关基因的显著富集和上调,支持在没有 PDX1 的情况下 NF-κB 信号传导的去抑制。因此,对 Pdx1 弱表现型小鼠和人胰岛的研究表明,PDX1 在胰腺 β 细胞中在昼夜节律调控程序和 NF-κB 介导的炎症途径的表达之间建立了平衡。
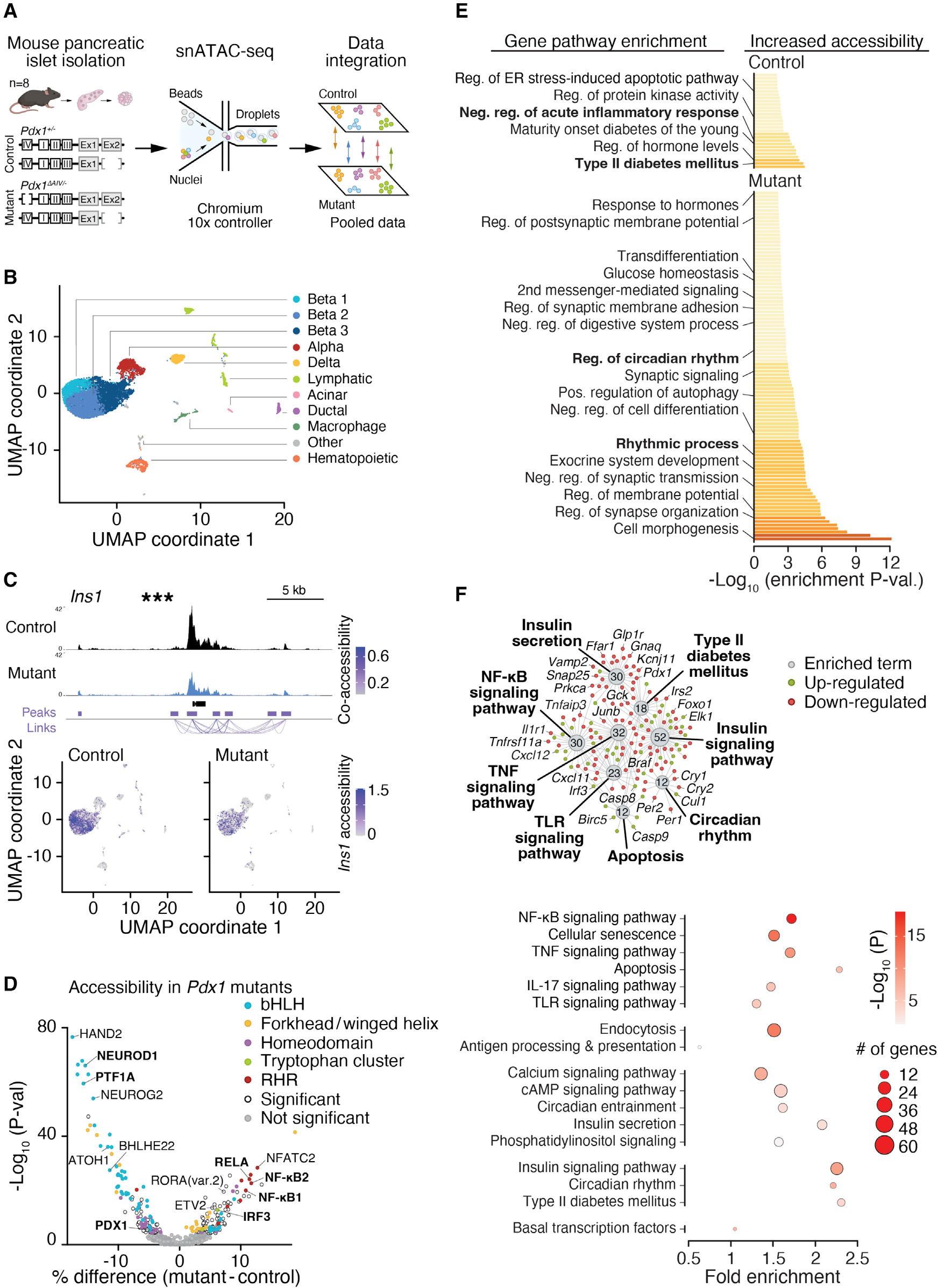
图 2 PDX1 基因缺陷导致 NF-κB 去抑制
(A)在从对照(Pdx1+/−)和 Pdx1 突变(Pdx1ΔAIV/−)小鼠(每个基因型 n = 8)中分离的胰岛中使用单核 ATAC 测序分析单细胞染色质可及性。 (B)对对照和突变胰岛细胞进行聚类和降维分析(UMAP),基于可及顺式调控元件的标准化读数识别了 15 个不同的细胞亚群。 (C)在合并对照与 Pdx1 突变 β 细胞中的 Ins1 基因的累积(上)和单细胞(下)可及读数。可能性比检验 ∗∗∗p < 0.001。 (D)在合并对照与 Pdx1 突变 β 细胞的可及染色质区域中,差异富集的 TF 结合基序。负值表示在对照中更富集,而正值表示在 Pdx1 突变体中更富集。 (E)基因本体分析显示在对照或 Pdx1 突变 β 细胞中上调的注释差异可及峰。 (F)RNA 测序揭示了 Pdx1ΔAIV/− 突变胰岛中昼夜节律、胰岛素分泌和 NF-κB 信号的失调。RNA 测序和路径分析使用 pathfindR 显示昼夜节律、胰岛素分泌和 NF-κB 信号 Kyoto 基因和基因组百科全书(KEGG)路径的富集,显示为在差异表达的转录本(调整后的 p 值 < 0.05)中共享基因富集(上)和带有富集倍数和多重比较 p 值的层次聚类(下),在从 Pdx1ΔAIV/−(n = 5)与 Pdx1+/−(n = 4)小鼠中分离的胰岛中。详见图 S2。
昼夜节律分析揭示 PDX1 对 NF-κB 增强子活性的节律调控¶
昼夜节律首先在出生后的 β 细胞中建立,并在断奶后变得更加明显,此时混合的大量营养素饮食和昼夜节律进食周期的诱导导致 β 细胞的功能成熟。在成人 β 细胞中,强大的昼夜节律时钟活动通过激活与 PDX1 结合位点共定位的预先建立的增强子来促进节律性胰岛素释放。观察到来自 Pdx1 缺陷动物的胰岛在由昼夜节律转录因子结合的增强子内显示出紧凑的染色质(图 2E),促使我们检查 Pdx1 缺陷对 β 细胞增强子的时间调控的影响。为了在内在昼夜节律周期的两个不同阶段检查 Pdx1ΔAIV/− 突变体和 Pdx1+/− 对照胰岛中的染色质可及性,我们将胰岛(每个基因型 3 个生物学独立的胰岛池)暴露于 1 小时福斯可林脉冲,并在节律性胰岛素分泌的低谷(同步化后 36 小时,TPS)和顶峰(同步化后 48 小时,TPS)采集胰岛(图 3A)。我们按照 Omni-ATAC-seq 协议进行适用于低输入样品的 ATAC-seq,并在所有合并样本中鉴定了 132,195 个独特的可及位点(图 3A)。使用 DESeq2 中的标准化 ATAC-seq 计数数据,可能性比检验(LRTs)鉴定了 1,207 个在昼夜节律时间或 Pdx1 基因型方面显著改变的 CREs(padj < 0.05)(图 3B 和 3C)。使用 ATAC-seq 标准化计数的无偏主成分分析揭示了关于基因型和昼夜节律时间的染色质开放的不同模式(图 S3A)。
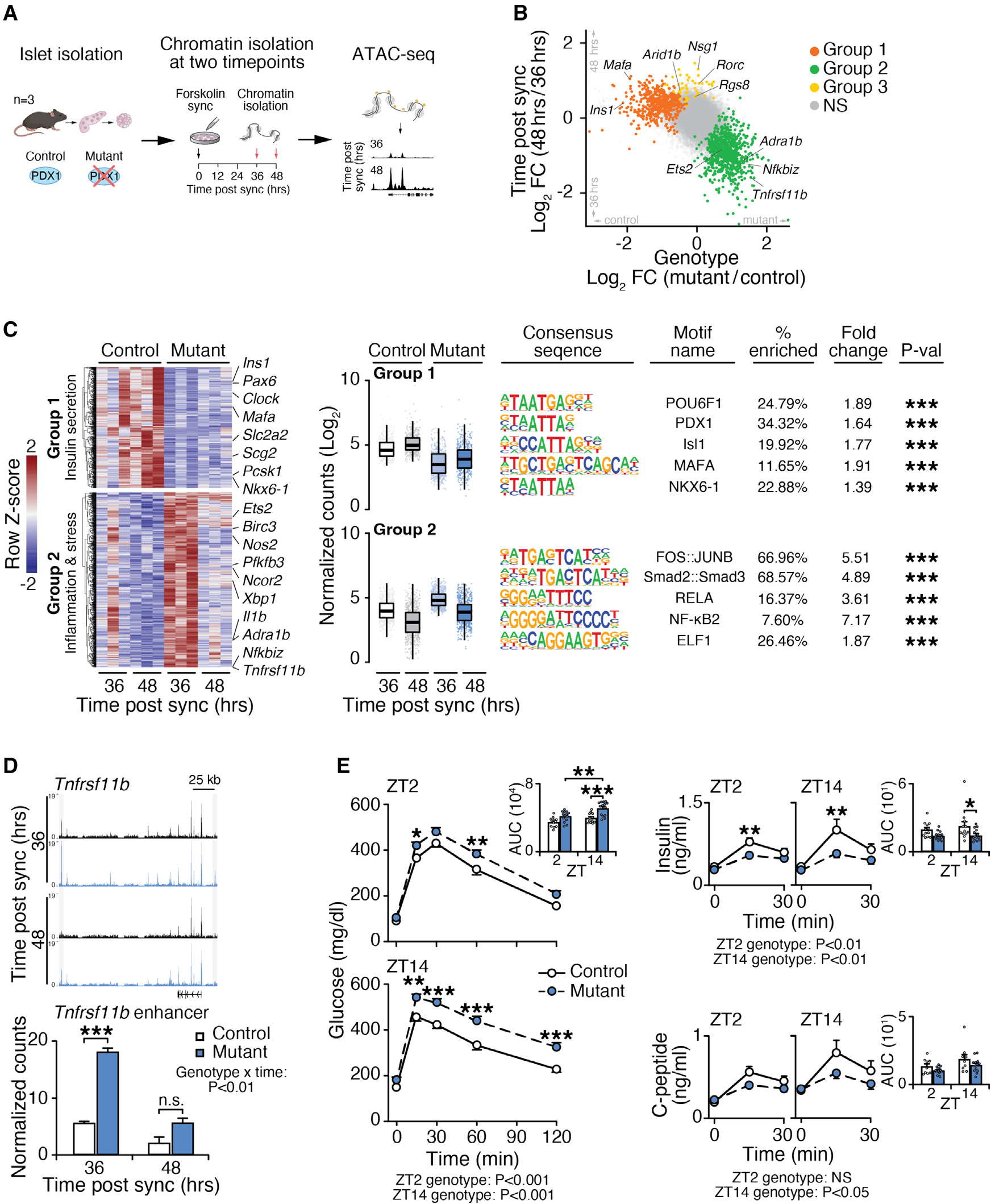
图 3 昼夜节律分析揭示 PDX1 对 NF-κB 增强子活性的节律调控
(A)来自对照(Pdx1+/−)和 Pdx1 突变(Pdx1ΔAIV/−)小鼠的胰岛制备物暴露于昼夜节律同步化脉冲(福斯可林),然后在同步化后 36 小时和 48 小时(分别为胰岛素分泌的低谷和顶峰)进行 ATAC-seq(每个时间点每个基因型 n = 3)。 (B)在使用可能性比检验识别动态峰后,关于昼夜节律时间或基因型的标准化可及读数的 Log2 倍变化。显著峰使用 k-means 聚类分成不同的可及模式组:第 1 组(橙色)—在 48 小时时点(胰岛素分泌顶峰)在对照胰岛中可及性增加但在突变体中可及性降低的位点。第 2 组(绿色)—在 36 小时时点(胰岛素分泌低谷)可及性增加的位点,这些位点在突变体中也增加。第 3 组位点(黄色)—仅依赖于昼夜节律时间而非基因型的位点。 (C)所有样本中标准化可及读数 Z 得分(左,中)揭示了在对照胰岛中邻近胰岛素分泌基因的可及性增加(第 1 组)和在突变胰岛中邻近应激和炎症基因的可及性增加(第 2 组)。差异可及峰内的基序分析(右)显示了第 1 组和第 2 组染色质峰中 β 细胞 TFs 和炎症及即时早期 TFs 的富集。 (D)在同步化后 36 小时,Pdx1ΔAIV/− 突变胰岛中邻近 TNF 相关基因 Tnfrsf11b 的标准化染色质读数增加。 (E)在对照(每个时间点 n = 14 小鼠用于葡萄糖,n = 13 小鼠用于胰岛素和 C 肽)和 Pdx1 突变(每个时间点 n = 12 小鼠用于葡萄糖,n = 9 小鼠用于胰岛素和 C 肽)小鼠中,早晨(ZT2)或晚上(ZT14)口服葡萄糖给药(2 g/kg)后的葡萄糖清除、胰岛素分泌、C 肽水平和曲线下面积,使用混合效应模型分析。主要和交互项显著性由混合效应模型确定。Wald(ATAC-seq)或 Holm-Šídák 检验 ∗p < 0.05,∗∗p < 0.01,∗∗∗p < 0.001,n.s. 不显著。数据表示为平均值 + SEM。详见图 S3。
为了识别这些数据中的染色质可及性调控模式,我们进行了 k-means 聚类,并鉴定了三个依赖于昼夜节律时间和/或基因型的基因簇。第 1 组位点在 Pdx1 突变胰岛的两个时间点的可及性均降低,但在对照胰岛的 CT48 时点表现出更强的开放性,而第 2 组位点在 CT36 时点特异性地获得可及性,并在 Pdx1 突变胰岛中更可及(图 3B 和 3C)。第 3 组位点仅受昼夜节律时间调控,而不受基因型调控(图 S3C 和 S3D)。与时钟和 PDX1 共同促进胰岛素分泌的协同作用一致,第 1 组位点富含 PDX1、MAFA 和 NKX6.1 结合基序,包括促进 Ins1 和 Mafa 表达的典型 PDX1 CREs(padj < 0.001)(图 3B 和 3C)。相反,在 Pdx1 缺陷细胞中富集的第 2 组峰集内的不同 ATAC-seq 峰出现在邻近炎症介质 Tnfrsf11b、Nfkbiz、Il-1b 和 E26 转化特异性(ETS)因子 Etv1 和 Ets2 的位点(图 3B-3D),并在 NF-κB、RELA 和即时早期复合物 FOS::JUNB 的基序中富集(图 3C)。这些发现表明,Pdx1 的丧失导致炎症和应激反应基因的去抑制和节律性重编程。在 Pdx1 突变胰岛中编码 α 细胞富集肾上腺素能受体 ADRB1 和 ADRA1b 的基因也表现出可及性的昼夜节律变化增加,表明 PDX1 通常通过可能涉及昼夜节律因子的机制抑制胰岛 α 细胞的转录(图 3B-3F 和 S3B)。值得注意的是,Pdx1ΔAIV/− 突变体中的淋巴、巨噬和外分泌细胞未显示出差异的 PDX1 活性,这表明第 2 组基因的染色质可及性增加不能归因于非 β 细胞类型的信号(图 S2C)。最后,与第 1 组和第 2 组不同,第 3 组的染色质可及性仅依赖于昼夜节律时间(图 3B、S3C 和 S3D)。这些位点在 D-box 结合蛋白(DBP)和核因子、白介素 3 调节(NFIL3/E4BP4)TF 基序中富集(图 S3D),与先前研究表明 DBP 调节胰岛染色质动态一致。总之,PDX1 水平决定了 β 细胞亚群内的染色质景观,这些亚群的特征是控制昼夜节律和炎症基因程序的增强子区域的独特可及性模式。
为了确定我们同步 ATAC-seq 数据集中识别的不同染色质特征是否与整动物的昼夜胰岛素分泌动力学相关,我们在白天(zeitgeber 时间 2 [ZT2])和晚上(ZT14)对 Pdx1ΔAIV/− 突变体和 Pdx1+/− 杂合对照小鼠进行了口服葡萄糖耐量试验(oGTTs)。与对照小鼠相比,Pdx1ΔAIV/− 突变小鼠表现出显著的葡萄糖耐受不良(p < 0.0001),这种情况在夜间更为明显(图 3E)。使用混合效应模型分析整个葡萄糖漂移曲线还揭示了 Pdx1 基因型、昼夜时间(即 zeitgeber 时间)和葡萄糖水平之间的显著相互作用(p = 0.015)。与对照组相比,这些小鼠在两个时间点的胰岛素和 C 肽水平均降低(双向 ANOVA,p < 0.05)(图 3E),并且在晚上突变小鼠的胰岛素和 C 肽水平相对于对照动物减少的程度更大。这些数据表明,PDX1 在通过改变分泌和促炎基因网络的控制,调节染色质紧凑性、β 细胞成熟和全天葡萄糖稳态方面的必要性。
为了研究分子时钟是否通过控制 PDX1 染色质结合来促进节律性胰岛素分泌,我们在 Bmal1 敲除(KO)小鼠 β 细胞系(Beta-TC-6)中进行了 PDX1 ChIP-seq。我们发现,BMAL1 的丧失导致 β 细胞中 PDX1 DNA 结合全基因组显著减少(p < 0.001)(图 S3F),包括在控制与胰岛素分泌调节相关基因表达的区域,如 Ins1、Neurod1 和 Six2(图 S3F)。因此,分子时钟的丧失导致 PDX1 介导的 β 细胞功能相关基因调控的互补损伤。
PDX1 形成长程染色质环以调节 NF-κB 和昼夜节律增强子活性¶
鉴于我们观察到 PDX1 和 NF-κB 调控元件区分了成体 β 细胞的不同亚群(图 1 和 2),我们试图确定 PDX1 是否在全基因组范围内直接抑制 NF-κB。我们首先使用 p65 ChIP-seq 检查 PDX1 缺失如何影响 p65(一个经典的 NF-κB 亚基)的活性,在 Beta-TC-6 细胞中用针对 PDX1 的小干扰 RNA(siRNA)处理 48 小时,与对照 siRNA 进行比较(图 4A)。我们观察到,尽管 p65 蛋白丰度相似,但 PDX1 的丧失在 5,268 个独特位点上增加了 p65 的全基因组占据。这种结合的增加包括 NF-κB/炎症靶标,如 Tnfrs11a 和编码昼夜节律抑制因子 PER2 的基因,与先前的研究一致。发光分析揭示 Pdx1 弱表现型胰岛显示缩短的周期长度。因此,β 细胞中 PDX1 的丧失增加了 NF-κB 在全基因组的结合,并改变了核心时钟抑制因子的表达,表明 PDX1 对 NF-κB 的抑制反过来改变了时钟功能。
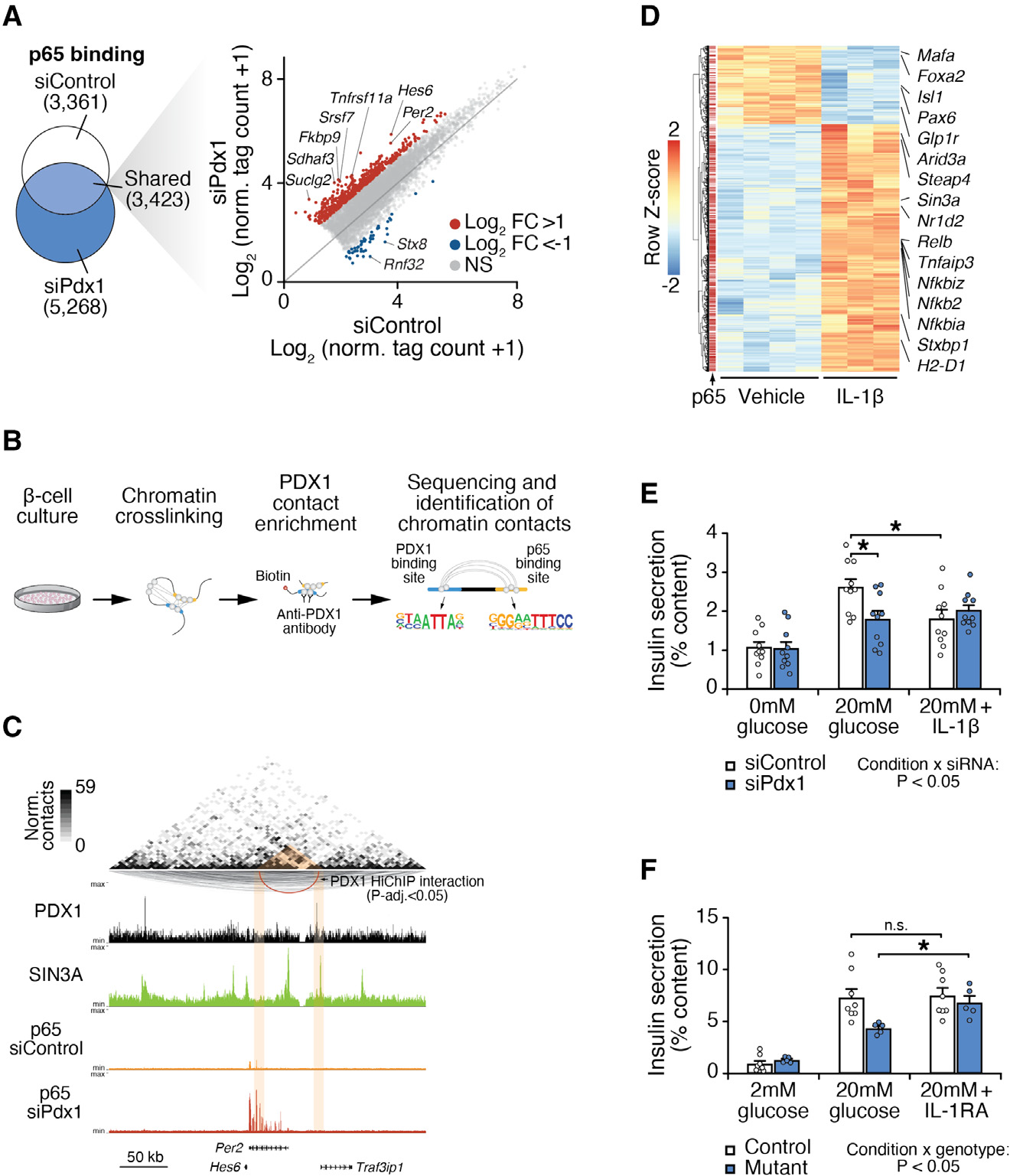
图 4 PDX1 通过与 IL-1β 响应元件共定位的长程染色质接触抑制 NF-κB,以控制胰岛素分泌
(A)在 Beta-TC-6 细胞中使用靶向 Pdx1 的 siRNA(siPdx1)进行 p65 ChIP-seq,揭示了在炎症和昼夜节律(Per2/Hes6)基因区域的全基因组占据增加。 (B)靶向 PDX1 的染色质构象测序结合 ChIP-seq(HiChIP-seq)识别出涉及 PDX1 结合位点的三维染色质接触。 (C)在 PDX1-SIN3A 结合位点之间鉴定出的显著染色质相互作用,这些位点与 p65 结合位点接触。siPdx1 处理的 β 细胞在失去 PDX1 蛋白后表现出增加的 p65 结合,表明 PDX1-SIN3A 染色质环通常在 Per2 附近抑制 p65 结合。 (D)IL-1β 抑制 β 细胞基因表达并在 Beta-TC-6 细胞中激活 p65 介导的炎症基因表达。 (E)急性(2 小时)给予 IL-1β 后,对照 siRNA 处理的细胞中,IL-1β 特异性抑制葡萄糖反应的胰岛素释放,而 siPdx1 处理的细胞对 IL-1β 不响应(每个 siRNA 处理 n = 10 个样本)。数据表示为平均值 + SEM。 (F)使用 IL-1β 受体拮抗剂(IL-1RA)拮抗胰岛 IL-1β 信号恢复了 Pdx1ΔAIV/− 胰岛中葡萄糖刺激的胰岛素分泌(n = 8 Pdx1+/− 小鼠,n = 5 Pdx1ΔAIV/− 小鼠)。双向 ANOVA,Holm-Šídák 检验,∗p < 0.05。数据表示为平均值 + SEM。详见图 S4 和表 S4、S5 和 S6。
由于我们发现 Bmal1 的缺失会削弱 PDX1 的 DNA 结合能力(图 S3F),我们接下来探讨了时钟功能丧失是否反过来改变 p65 在全基因组范围内的结合情况。为此,我们在 Bmal1 敲除(KO)的 Beta-TC-6 细胞中进行了 p65 ChIP-seq。与对照相比(2430 个峰),我们在 Bmal1 KO β细胞中观察到显著较少的 p65 结合事件(177 个峰)(图 S3G),这表明 BMAL1 对于 p65 结合染色质的能力也是必需的。在 Bmal1 KO 细胞中缺失 p65 结合的位点包括细胞存活和抗凋亡基因,如凋亡拮抗转录因子 Aatf、Janus 激酶 1(Jak1)、肌醇多磷酸 -4- 磷酸酶(Inpp4b)和磷酸二酯酶(Pde10a)(图 S3G)。这些数据表明,PDX1 和 p65 的活性都依赖于功能性昼夜节律时钟的三方相互作用。
三维染色质分析已经揭示了 LDTFs 在广泛分离的基因组区域之间形成接触以控制β细胞功能,但 PDX1 是否通过长程接触控制 SDTFs 尚未确定(图 4B)。在 Pdx1 敲低的β细胞中,p65 结合位点在远离(>500 bp)对照细胞类型中观察到的 PDX1 DNA 结合位点的位置上富集(图 S4B)。因此,我们利用三维染色体构象捕获结合 PDX1 ChIP-seq(HiChIP-seq)绘制 PDX1 与远程调控元件之间的长程相互作用图谱(图 4B)。我们鉴定出 4155 个长程(>5 kb)染色质相互作用,涉及 PDX1 与远程顺式调控区域之间的相互作用(见 STAR 方法,错误发现率 [FDR] < 0.05)(图 4C、S4B 和 S4C;表 S4)。其中包括 497 个细胞因子诱导的 NF-κB 结合位点,这些位点在 IL-1β处理的β细胞中通过 p65 ChIP-seq 被识别为 NF-κB 靶点(表 S5)。值得注意的是,IL-1β诱导的 p65 峰在环内的比例比在我们小鼠胰岛 ATAC-seq 峰集所有可及 CREs 中发现的 NF-κB 基序的背景出现率高出 3 倍(12% 对 4%)。在肿瘤坏死因子(TNF)受体相关因子 3 相互作用蛋白 1(Traf3ip1)上游 2.1 kb 处的 PDX1 结合位点和 Per2 基因的 5' UTR 内的 NF-κB 结合位点之间观察到显著的 PDX1 染色质环(图 4C)。我们还观察到 PDX1 和 NF-κB 位点在涉及 Ca2+ 信号传导(Inpp4b)和炎症(Irf2)的基因上共定位(图 S4C),以及在其他 IL-1β诱导的 NF-κB 位点上,如 IL-1β受体(Il1r1)、TNF 受体相关因子相互作用蛋白(Traf3ip2)和 2 型糖尿病相关等位基因 Slc30a8(表 S5)。为了测试 PDX1 是否通过招募共抑制复合物在这些远端增强子处抑制 NF-κB,我们使用转录共抑制因子 SIN3A 进行染色质免疫沉淀,SIN3A 与 PDX1 一起建立了β细胞身份。在 4925 个 SIN3A 结合位点中,有 41% 与 PDX1 调控基因共定位,包括 Traf3ip1 和 TNF 相关基因如 Tnfsf4 和 Traf5(表 S6;图 4C、S4C 和 S4D)。此外,我们发现 PDX1 的丧失会破坏 SIN3A 在全基因组范围内的染色质结合(图 S4E),包括在 Nfkbie 基因附近的 IL-1β诱导的 NF-κB 结合位点的 SIN3A 结合减少(图 S4F)。值得注意的是,该区域还与 PDX1 结合位点进行长程染色质接触(图 S4F),这与 PDX1 通过三维相互作用和共招募 SIN3A 在远距离上抑制 NF-κB 一致。
使用 IL-1β受体拮抗剂抑制 NF-κB 信号增强 PDX1 缺陷β细胞的功能¶
人 PDX1 低和小鼠 Pdx1ΔAIV/− β细胞均显示促炎网络和 NF-κB 靶点(如 IL-1β)的染色质开放性增加特征。IL-1 受体(IL-1R)是胰岛β细胞中表达最高的细胞因子受体之一。暴露于 IL-1β会重塑β细胞染色质景观,并且拮抗 IL-1β代表了一种在人体中已确立的抗炎策略。炎症似乎也通过尚不明确的机制在 2 型糖尿病的β细胞功能障碍中起作用。我们发现 IL-1β刺激野生型小鼠的β细胞会诱导经典 NF-κB 靶点(包括 Nfkbiz 和 Nfkbia)的表达,并抑制 PDX1 靶点(包括 Mafa、Glp1r 和 Pdx1 本身)的表达。IL-1β还抑制了野生型 Beta-TC-6 细胞中葡萄糖刺激的胰岛素释放。相反,用 IL-1β受体拮抗剂(IL-1RA)处理从 Pdx1ΔAIV/−突变小鼠中分离的胰岛会显著增强胰岛素分泌,这与通过阻断 IL-1β诱导的 NF-κB 信号改善 PDX1 缺陷一致。我们使用高血糖钳夹直接研究阻断 IL-1β是否可能在体内克服 Pdx1 低表现型动物的低胰岛素血症。我们评估了在稳定 GIR 后(输注开始后>50 分钟)胰岛素介导的葡萄糖输注率(GIR)的差异,并评估了基因型 - 药物相互作用。我们的分析揭示了 IL-1RA 处理的动物与车辆(PBS)对照之间的基因型依赖性差异,在 IL-1RA 处理的突变体与 PBS 处理的突变体中 GIR 有增加的趋势。因此,高血糖钳夹结果表明,拮抗 IL-1β对 Pdx1 低表现型动物的葡萄糖清除具有基因型特异性影响。最后,使用 IL-1RA 的抗炎治疗增强了 Bmal1 KO 细胞中的胰岛素分泌。总的来说,我们的表观基因组和遗传研究表明,阻断 NF-κB 上游的炎症可能在与 PDX1 和/或时钟功能障碍相关的β细胞衰竭状态中提供一种新的促胰岛素治疗方法。
讨论¶
我们在成人胰岛细胞中鉴定出一种独特的 β 细胞亚型,这种细胞具有炎症转录活性,其特征是全基因组范围内的 NF-κB 网络富集,而主谱系调节因子 PDX1 的活性降低。单细胞分辨率下这种新胰岛亚型的识别得益于染色质可及性的高映射密度和对人胰岛亚群中特定调控元件的解析。通过三维染色体构象捕获,我们进一步表明 PDX1 通过长程抑制 NF-κB 和招募经典共抑制因子 SIN3A 来抑制炎症信号。相反,我们展示了在 Pdx1 缺陷的遗传模型中,单个胰岛细胞中的 NF-κB 去抑制导致β细胞衰竭和糖尿病。这些结果表明 PDX1 保护β细胞免受压力和炎症的侵害,并提供了 PDX1 与促炎/NF-κB 信号交叉调控作为β细胞功能关键要素的证据。
我们的研究还揭示了 PDX1 在成熟β细胞中诱导昼夜节律基因网络,这些网络在代谢稳态中起重要作用。我们观察到 PDX1 通过一种在小鼠和人类中都随着昼夜节律时间尺度变化的机制定位于 CLOCK/BMAL1 调控元件。PDX1 结合位点在 CLOCK/BMAL1 增强子中的富集与先前的研究一致,即时钟因子参与胰岛成熟和对损伤的响应,以及潜在的 NF-κB 介导的炎症网络的抑制。我们的分析进一步表明,PDX1 在促进胰岛素外排基因网络的时间依赖性活动的同时抑制炎症特征。各种条件,如过度营养或衰老本身,已被证明以组织特异性方式改变昼夜节律编程。同样,我们在 Bmal1 KO β细胞中的基因组研究揭示了时钟因子 BMAL1 对 NF-κB 介导基因表达的混合效应。具体来说,Bmal1 的缺失改变了 p65 在细胞存活和抗凋亡基因上的 DNA 结合,而 Bmal1 的缺失并未改变 p65 对细胞因子诱导的炎症基因的结合。先前的研究表明,p65/NF-κΒ对β细胞功能是必需的,因此在 BMAL1 缺失的β细胞中 p65 结合的丧失可能导致这些动物的低胰岛素血症,可能是由于 NF-κB 途径的抗凋亡作用。未来的研究将需要了解在昼夜节律周期中β细胞对炎症刺激的特异性响应以及核心昼夜节律激活因子或下游时钟控制基因如何与 PDX1 相互作用。
人类和动物研究表明,由于肥胖饮食和/或进食时间错误导致的时钟功能紊乱会导致代谢疾病。本文提出的数据表明,IL-1β诱导(已被证明在饮食诱导的肥胖中增加)可能会反过来触发β细胞衰竭。遗传性 PDX1 缺陷导致潜在 NF-κB 增强子的去抑制,并为抑制 IL-1β作为促胰岛素治疗提供了理由。尽管先前的研究表明 IL-1β对葡萄糖稳态的控制涉及中枢神经系统,我们的胰岛培养结果表明,阻断 IL-1β直接在胰岛内增强了胰岛素分泌。由于我们研究中使用的人胰岛来自葡萄糖反应性供体,未来的研究将需要确定在 1 型或 2 型糖尿病中 PDX1 与 NF-κB 之间的拮抗关系是否被破坏。
研究局限性¶
我们的实验在人类胰岛中确定了 PDX1、NF-κΒ和时钟之间的联系,但需要进一步的 snATAC-seq 和 snRNA-seq 研究来确定整个胰岛中染色质可及性的每日节律是否由于单个β细胞与非β细胞的变化以及这些染色质变化是否与单细胞水平的基因表达变化相对应。此外,虽然我们在人的胰岛研究中发现了男女受试者中β细胞异质性的证据,但进一步研究将需要专门检查在 Pdx1 突变动物中性别和年龄的影响。同样,我们的葡萄糖耐量实验提供了昼夜时间、Pdx1 基因型和葡萄糖漂移之间显著相互作用的证据,胰岛素释放在黑暗时期达到最大差异。鉴于这些实验中使用的对照是 Pdx1 杂合子,进一步的研究将需要比较 Pdx1 杂合子和区域 IV 突变体与野生型对照的差异。最后,在临床前背景下的未来研究应探讨时钟或谱系决定因子的丧失是否通过潜在的免疫内分泌机制使动物易于发生不适当的炎症反应。