Single cell analysis unveils B cell dominated immune subtypes in HNSCC for enhanced prognostic and therapeutic stratification
头颈部鳞状细胞癌(HNSCC)的特点是复发率高或远处转移率高,预后具有挑战性。越来越多的证据表明,肿瘤浸润性 B 细胞(TIL-Bs)在肿瘤控制中具有至关重要的、协同的作用。然而,关于 TIL-Bs 在免疫微环境中的作用以及 TIL-Bs 如何影响免疫检查点阻断治疗结果的了解还很少。通过使用来自基因表达综合数据库(GEO)的单细胞 RNA 测序(scRNA-seq)数据,研究发现了 TIL-Bs 中的不同基因表达模式。通过无监督聚类,将 HNSCC 样本分类为 TIL-Bs 抑制组和 TIL-Bs 激活组。这一分类随后通过 TCGA HNSCC 数据得到验证,与患者预后、免疫细胞浸润以及对免疫治疗的反应相关。我们发现,B 细胞激活组展示了更好的预后、更高的免疫细胞浸润和不同的免疫检查点水平,包括提高的 PD-L1。此外,还开发并验证了一个预后模型,突出显示四个基因作为预测 HNSCC 患者生存结果的潜在生物标志物。总体而言,这项研究为基于 B 细胞的 HNSCC 肿瘤分类提供了一种基础方法,为针对性治疗和免疫治疗策略提供了洞察。
引言¶
头颈癌(HNC)由上呼吸道的多种肿瘤组成,是全球第七大常见癌症。在所有 HNC 中,头颈鳞状细胞癌(HNSCC)是最常见的组织学亚型,约占 90% 以上。在过去几十年中,基于手术、放疗和分子靶向治疗的多模式方法为 HNC 患者提供了显著的临床益处。不幸的是,高比例的患者最终仍会出现复发或远处转移,其预后依然不佳。对于进展复发或转移的患者,免疫检查点阻断治疗已成为一线治疗方式,革新了治疗策略。然而,关于如何优化这些药物的治疗,例如提高患者的总体响应率,仍需深入研究。因此,HNSCC 仍是一组极其复杂的疾病,目前需要更好地理解其分子和细胞机制,这可能有助于发现新的治疗策略。
肿瘤微环境(TME)由多种免疫细胞、内皮细胞和成纤维细胞组成,与肿瘤细胞在肿瘤发展过程中持续相互作用。越来越多的证据表明,肿瘤细胞与肿瘤微环境之间的持续互动是肿瘤发生、进展、转移和治疗反应的重要决定因素。例如,在黑色素瘤中,肿瘤核心和边缘的 CD8+ T 细胞密度较高与增强的 PD-1 和 PD-L1 阻断反应相关,而某些 CD4+ Th1 细胞亚群在对 CTLA-4 反应良好的肿瘤中也更为丰富。最近的研究表明,肿瘤内或肿瘤周围 B 细胞与积极的预后和对免疫治疗的响应相关。这些 B 细胞通常形成三级淋巴结构(TLS),建立局部持续的免疫反应,通过分泌识别肿瘤相关抗原的抗体并增强 T 细胞和自然杀伤细胞的杀伤作用来发挥特异性抗肿瘤免疫效应。在 HNSCC 中,肿瘤浸润 B 淋巴细胞(TIBs)是 TME 的重要组成部分,表明它们参与了 HNSCC 的发展。此外,研究表明 TIBs 与 HNSCC 患者更好的生存率相关,B 细胞激活与 PD-1 阻断相关。这些研究表明,B 细胞群体在 HNSCC 中的抗肿瘤免疫中起着关键作用,因此有必要探索 HNSCC 中 B 细胞的基因表达谱及其与患者预后和免疫治疗预测的关系。
最近,单细胞 RNA 测序(scRNA-seq)技术为从肿瘤中获得肿瘤细胞生态系统的高分辨率画像提供了一种有效的方法。更具体地说,对 TME 的细胞类型和细胞状态进行 scRNA-seq 分析可以提供有关疾病分子特征的进一步信息,为个性化免疫治疗铺平道路。基于特定免疫细胞的分子特征建立肿瘤亚型分型可能是预测免疫治疗效果以及患者预后的可靠方法。在这里,我们描述了关于 HNSCC 中 B 细胞的 scRNA-seq 数据集,并基于单细胞差异基因表达建立了两种 B 细胞亚型。我们在单细胞层面和基因层面对这两种亚型进行了表征,并在一个额外的独立样本队列中验证了分型的一致性。此外,我们分析了这两种 B 细胞亚型的差异基因,并构建了一个 B 细胞激活基因签名。然后,我们基于 TCGA 数据库和 GEO 数据库验证了 B 细胞激活基因签名的预后价值,并深入分析了其与免疫浸润和 ICB 反应的关系。此外,我们还在 HNSCC 中首次应用 Cox 比例风险模型建立了一个预后风险预测模型。这项研究为 B 细胞在 HNSCC 中的抗肿瘤免疫作用提供了支持,并为基于 B 细胞的肿瘤分类提供了新的见解。
结果¶
独立 scRNA-seq 数据集中 B 细胞免疫分类的识别与表征¶
经过质量控制和数据过滤后,我们获得了来自 26 个 HNSCC 样本(GSE139324)的 105943 个免疫细胞的单细胞转录组。为了识别肿瘤浸润免疫细胞的主要种群和亚种群组成,我们使用 Seurat 进行了聚类,以识别主要的免疫细胞类型,包括 NK 细胞、CD4+ T 细胞、CD8+ T 细胞、周期性 T 细胞、髓样细胞和 B 细胞(图 1a)。每种细胞类型都基于经典标记基因和文献证据进行识别(图 1b,c)。每个患者样本中的免疫细胞簇组成也显示出来,以观察所有样本中肿瘤免疫环境的状态(图 1d)。
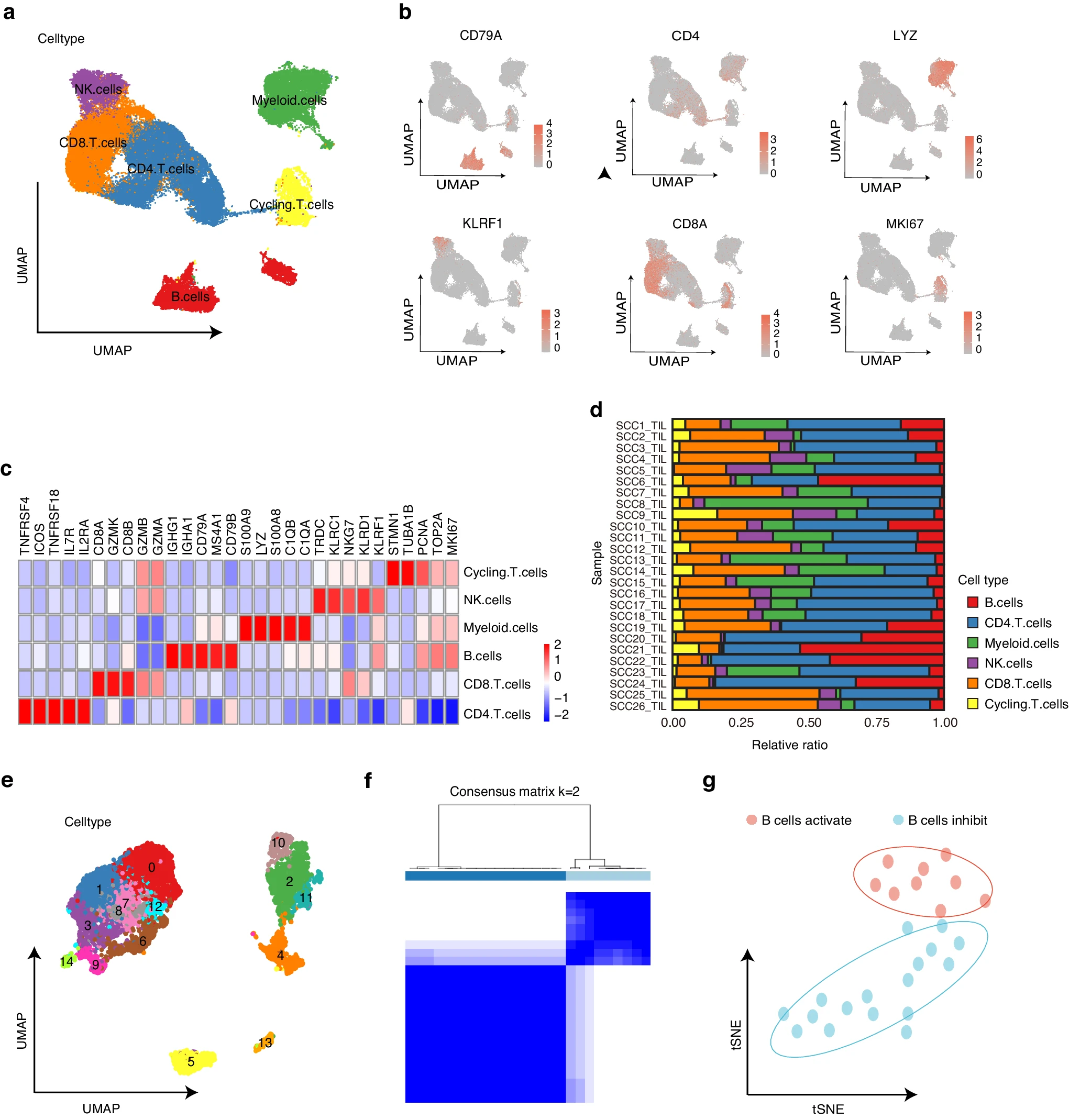
基于单细胞分析确定 TIL-Bs 的免疫亚型。a UMAP 展示了 HNSCC scRNA-seq 中 26 个样本的 105,943 个肿瘤浸润免疫细胞(按细胞簇着色)。b UMAP 图显示了来自免疫细胞簇的经典标记基因的表达。c 为免疫细胞簇显示的特征基因热图。每个细胞簇由五个特异表达的基因代表。d 条形图显示每个 HNSCC 样本中的免疫细胞类型比例。e UMAP 图显示了 15 个 B 细胞簇。f 使用基于标记基因的 ConsensusClusterPlus R 包识别了两个亚组。g t-SNE 图显示了 HNSCC scRNA-seq 队列(26 个样本),按样本组着色。
为了探讨 HNSCC 中 TIL-Bs 的功能亚群和潜在作用,选择在第一级聚类中定义为 B 细胞的细胞,并重新聚类以识别 15 种不同的 B 细胞亚群(图 1e)。然后我们使用 Findallmarkers 功能在这 15 个 B 细胞簇上获得与 B 细胞相关的差异表达基因集(方法)。结果,找到了 440 个标记基因,使用这些基因进行了 HNSCC 样本的新一轮无监督聚类,使用 ConsensusClusterPlus 进行特征选择并重新聚类这 25 个 HNSCC 样本(图 1f 和补充图 1a)。主成分分析还根据基因组将样本分类为两组,称为 B 细胞激活组和 B 细胞抑制组(图 1g)。这两组样本在 CD8+ T 细胞的比例和数量上有最大的差异(补充图 1b–d),表明这两组在抗肿瘤免疫方面可能存在区别。
先前的数据基于 B 细胞特征基因的单细胞样本分组揭示了肿瘤免疫的差异,因此我们在遗传层面分析了两组之间的差异。如火山图所示,使用 log2FC > 1 和 P < 0.05 作为截止阈值,我们在 B 细胞激活组和 B 细胞抑制组之间鉴定了 43 个差异表达基因(21 个上调基因和 22 个下调基因)(图 2a)。我们将 21 个上调基因定义为 B 细胞激活基因签名(BCAGS),并使用此签名进行下一步的验证分析。大多数上调基因与 B 细胞激活有关,而下调基因与 B 细胞抑制有关(图 2a,b)。根据 GO 功能富集分析,上调基因在 B 细胞激活调控、B 细胞受体信号通路和 B 细胞激活通路中高度富集(图 2c)。我们然后对这两组中其他免疫细胞簇的 DEGs 进行 GO 功能富集,发现 B 细胞激活组中上调基因促进了多功能免疫调控,例如白细胞激活的正向调控、抗原结合、细胞杀伤和免疫细胞招募(补充图 2a–e)。
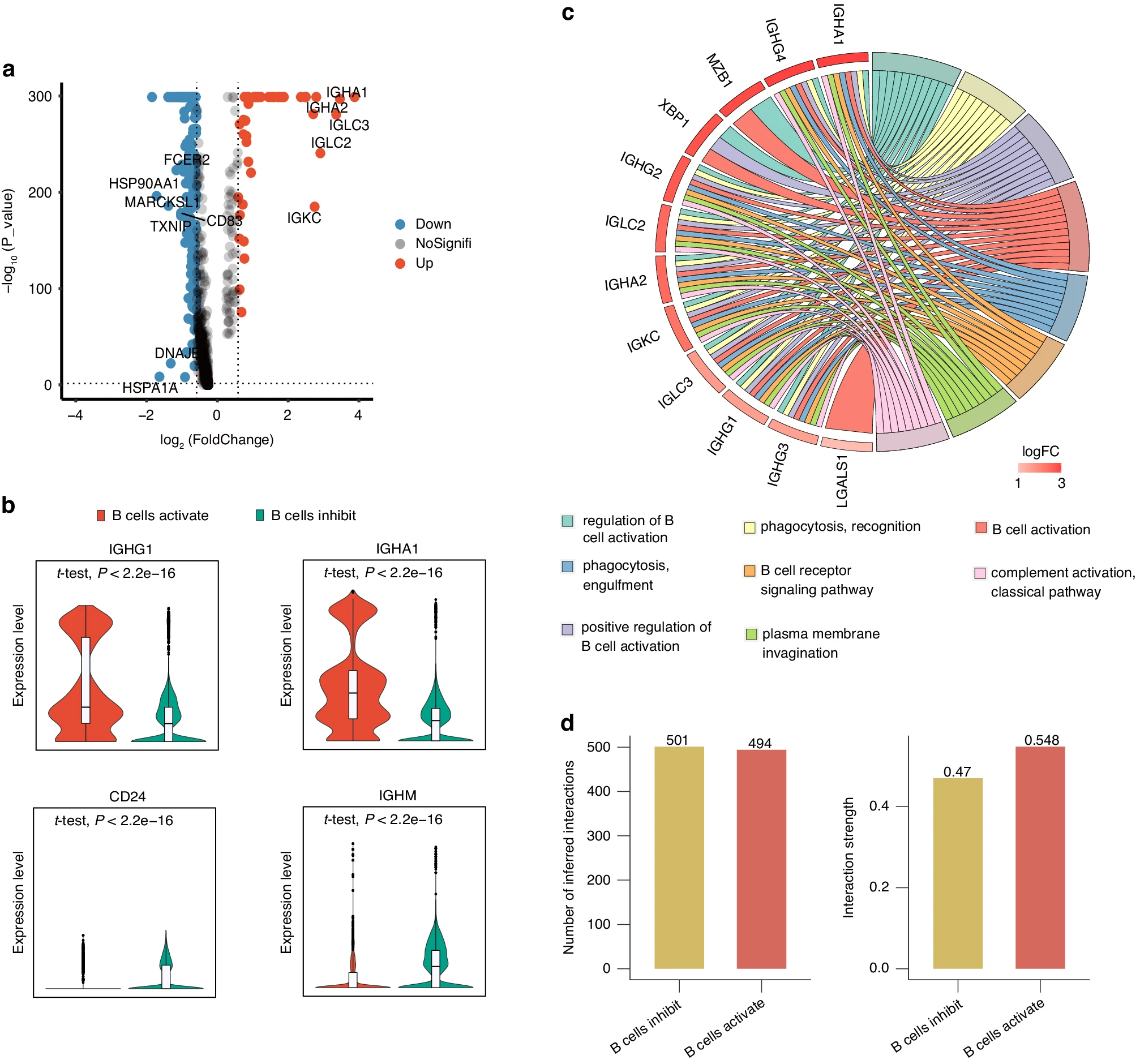
基于 TIL-Bs 的免疫亚型表征: a. 火山图:展示了 B 细胞激活组与 B 细胞抑制组之间差异表达基因。图中标记了下调(Down)、无显著(NoSignifi)、上调(Up)的差异表达基因(DEGs)。 b. 小提琴图:显示了在 B 细胞激活组和 B 细胞抑制组中 IGHG1、IGHA1、CD24 和 IGHM 的表达情况。 c. GO 簇图:展示了基于 B 细胞特征基因分类的 B 细胞中显著上调基因表达谱的聚类和弦树形图。 d. 圆形图:展示了两组之间的细胞 - 细胞通信网络中交互作用数量(左)和交互作用强度(右)的差异。
新兴证据表明恶性肿瘤中的免疫细胞功能或交流偏差。然而,基于这种分组方法在 HNSCC 中的免疫细胞交流的全球轮廓尚未明确定义。为了系统地调查 B 细胞激活组和 B 细胞抑制组的细胞间交流,我们使用 CellChat 进行了无偏见分析,包括整体和差异性交流数量和强度以及“配体 - 受体”交互作用。有趣的是,两组之间的细胞间交流激活强度不同。如图所示,B 细胞激活组的免疫细胞互动总体强度增加(图 2d),T 细胞直接互动更为活跃(补充图 2f)。在配体 - 受体互动中,B 细胞激活组表现出更多的 CSF、CXCL 和 MHC-I 信号通路,而 CD22 和 CD23 被抑制,表明 B 细胞激活组通过细胞间交流改善了抗肿瘤免疫(补充图 2g)。这些细胞间交流数据为基于 B 细胞特征基因的 HNSCC 分类提供了免疫细胞间通信分层的证据。
Validation for B-cell classification suggests the consistency in different scRNA-seq datasets¶
为了进一步展示获得的 B 细胞特征基因在分类 HNSCC 患者中的优势,我们收集了另一份 HNSCC 单细胞数据(GSE164690)作为验证队列。我们选择了肿瘤浸润免疫细胞群体,并根据经典标记或文献证据定义它们,包括 NK 细胞、CD4+ T 细胞、CD8+ T 细胞、髓样细胞和 B 细胞(补充图 3a–c)。同时也列出了所有 18 名患者的免疫细胞群体百分比(补充图 3d)。我们还使用之前获得的 BCAGS(B 细胞激活基因签名),利用 ConsensusClusterPlus 进行特征选择和重新聚类,对这 18 个样本进行了无监督聚类(图 3a)。结果显示,在这个队列中,患者被聚类为两个明显不同的组(图 3b)。此外,在基于 B 细胞特征基因集的分组中,我们也观察到两组之间 CD4+ T 细胞、CD8+ T 细胞和 B 细胞的数量和比例存在差异(补充图 3e, f),这支持了使用这种分类方法可能存在抗肿瘤免疫差距的可能性。
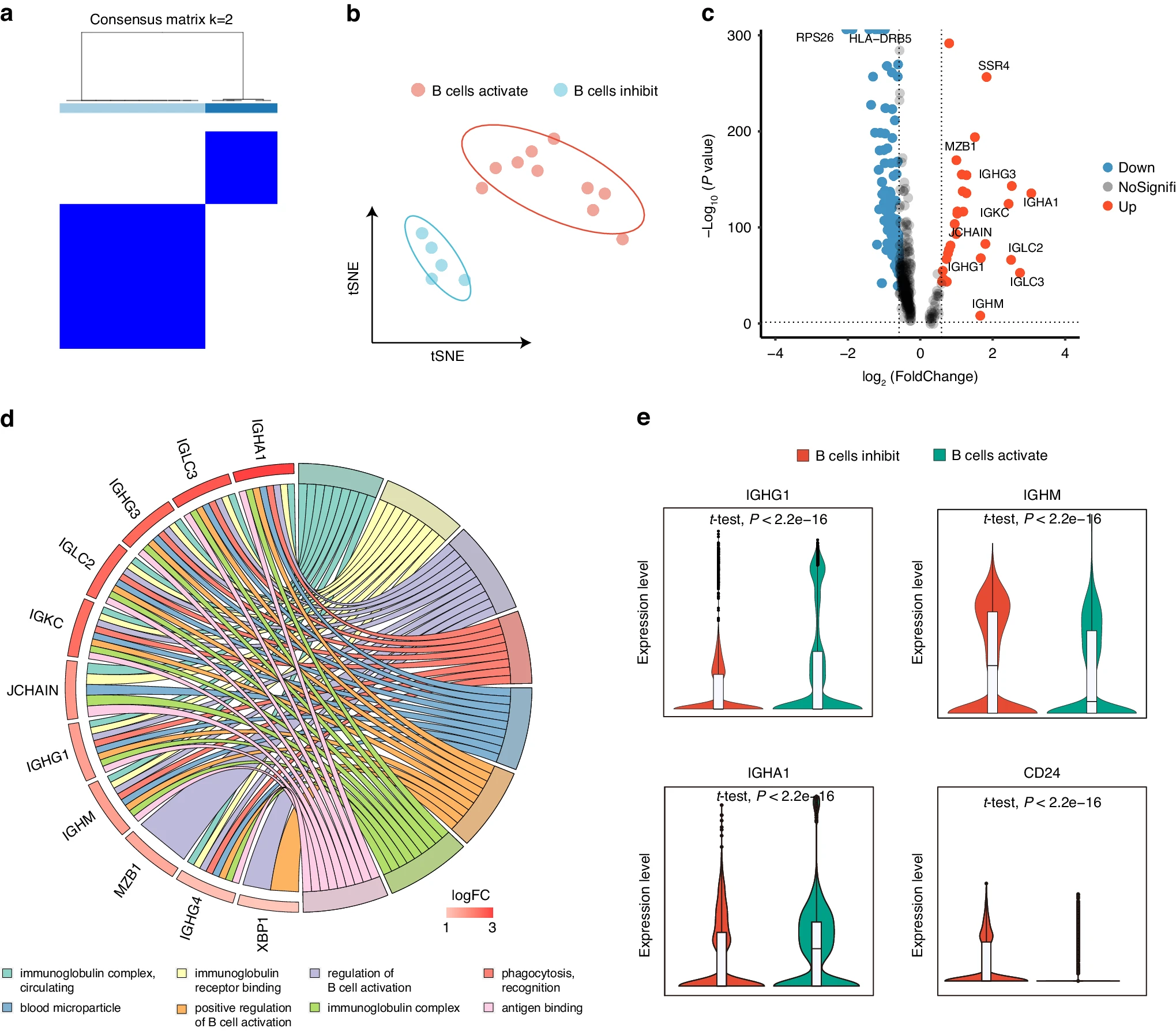
验证基于 TIL-Bs 的分类方法在另一个数据集中的应用。 a. 使用基于 B 细胞特征基因的 ConsensusClusterPlus R 包,识别了两个亚组。 b. t-SNE 图显示了 HNSCC scRNA-seq 队列(15 个样本),按样本组着色。 c. 火山图显示了 B 细胞激活组与 B 细胞抑制组之间的差异表达基因。其中“Down”表示下调的差异表达基因,"NoSignifi" 表示无显著差异,"Up" 表示上调的差异表达基因。 d. GO 簇图显示了基于 B 细胞特征基因分类的 B 细胞中显著上调基因表达谱的聚类和弦树形图。 e. 小提琴图显示了 B 细胞激活组和 B 细胞抑制组中 IGHG1、IGHA1、CD24 和 IGHM 的表达情况。
我们还在基因水平上验证了这种分类方法在单细胞队列中的一致性。火山图显示了 B 细胞激活组与 B 细胞抑制组之间转录组水平的差异表达基因(DEGs),观察到 29 个上调和 101 个下调基因(图 3c)。GO 功能富集显示,上调的 DEGs 在免疫球蛋白复合体、循环、免疫球蛋白受体结合和 B 细胞激活通路的调控中显著富集(图 3d)。与前一队列一致,上调基因主要与 B 细胞激活相关(图 3e)。我们还对这两组中其他免疫细胞群体的 DEGs 进行了 GO 功能富集,显示 B 细胞激活组中上调基因介导了多功能免疫调节,如正向 T 细胞选择、白细胞激活的正向调控、淋巴细胞介导的免疫以及 MHC II 类蛋白复合体(补充图 4a–e)。以上结果表明,基于 HNSCC 的 B 细胞特征基因分类方法在不同单细胞队列中具有一致性。
此外,两组中的细胞间通信具有高度一致性,B 细胞激活组中免疫细胞互动的总数和强度增强(补充图 4e),直接 T 细胞群体互动更为活跃(补充图 4f)。在配体 - 受体互动中,B 细胞激活组显示出更活跃的 CSF、CXCL 和 MHC-I 信号通路,同时 CD22 被抑制,表明 B 细胞激活组通过细胞间互动改善了抗肿瘤免疫(补充图 4g)。这些细胞间通信数据为基于 HNSCC 的 B 细胞特征基因分类在不同单细胞队列中的一致性提供了证据。
B 细胞分类在 TCGA HNSCC 中的应用及与总生存期的关联¶
为了验证这种 B 细胞特征基因分类方法是否适用于 TCGA HNSCC,我们对 501 个患者样本的 RAN-seq 表达谱进行了无监督聚类,基于 BCAGS 获得了两个簇(图 4a),tSNE 聚类显示样本分为两组(图 4b)。如火山图所示,从 TCGA HNSCC 患者的两组中鉴定出 388 个上调基因和 42 个下调基因(图 4c)。对 B 细胞激活组中的 DEGs 进行 GO 功能富集分析,发现显著富集在淋巴细胞激活和免疫激活相关路径中(图 4d)。我们将分组与临床信息相关联,以探索这种肿瘤分类的预后价值。先前的研究表明,B 细胞与肿瘤患者的总生存期改善相关。在我们的研究中,Kaplan-Meier 曲线显示,B 细胞激活组的患者具有显著更好的生存预后(图 4e)。
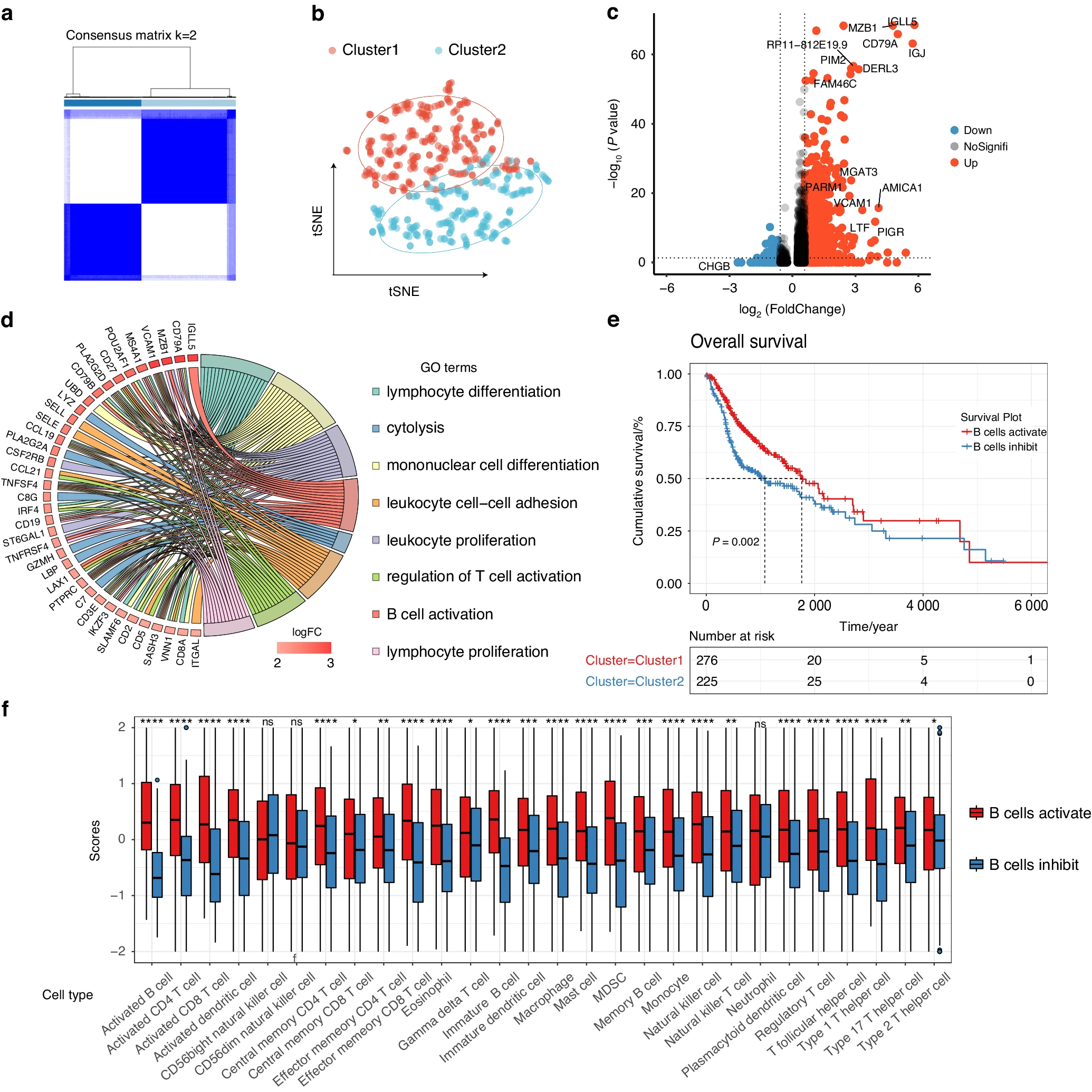
应用基于 TIL-Bs 的分类方法于 TCGA 队列: a. 共识矩阵:TCGA HNSCC 队列(501 个样本)的共识矩阵。 b. t-SNE 图:显示 TCGA HNSCC 样本被划分为两个簇。 c. 火山图:描绘了来自簇 1 和簇 2 的患者之间的差异表达基因。 d. GO 簇图:展示了基于 B 细胞特征基因分类的 B 细胞激活组中显著上调基因表达谱的聚类和弦树形图。 e. Kaplan-Meier 总生存曲线:TCGA HNSCC 患者在 B 细胞激活组与 B 细胞抑制组之间的总生存曲线。TCGA 样本根据 B 细胞特征基因进行分层。 f. 免疫相关细胞的富集水平(通过 ssGSEA 分析)和类型在 B 细胞激活组与 B 细胞抑制组中的差异。
为探索分类方法对免疫细胞浸润分数的影响,我们使用 ssGSEA 来可视化 28 种浸润免疫细胞群体的相对丰度。结果显示,B 细胞激活组区域显示出更高的免疫细胞浸润丰度(图 4f 和补充图 5a)。Pearson 相关性研究揭示了这两组在局部环境中的丰度呈正相关(补充图 5b)。TCGA HNSCC 中 B 细胞激活组的样本,根据 CIBERSORT 估计,具有比 B 细胞抑制组更高比例的肿瘤浸润淋巴细胞(补充图 5c)。综上所述,我们的数据证明了该分类方法在单细胞和 RNA-seq 队列中的一致性。
B 细胞分类准确预测免疫治疗反应¶
我们还使用 ESTIMATE 功能评估了肿瘤中间质和免疫细胞的浸润分数,并推断了肿瘤的纯度。观察到 B 细胞激活组具有更高的免疫分数、间质分数和 ESTIMATE 分数,肿瘤纯度低于 B 细胞抑制组(图 5a-d)。我们之前的数据表明 B 细胞激活组中免疫细胞的丰度更高,这表明可能对免疫治疗有更大的敏感性。同时,我们探索了两组典型免疫抑制分子(PD-1, CTLA4, LAG3, BTLA, CD274, HAVCR2, VSIR, 和 PDCD1LG2)的表达谱。B 细胞抑制组的患者显示出显著的抑制性受体下调(图 5e)。为了进一步探索分类与免疫治疗反应之间的可能关系,我们在两组中计算了肿瘤免疫功能障碍和排除(TIDE)分数,以评估肿瘤免疫逃逸的潜力并预测对免疫反应的响应。结果显示,B 细胞激活组的 TIDE 分数低于 B 细胞抑制组(图 5f)。较低的 TIDE 分数通常与更好的 ICB 治疗效果相关。我们的结果表明,B 细胞激活组的患者可能对 ICB 治疗敏感。
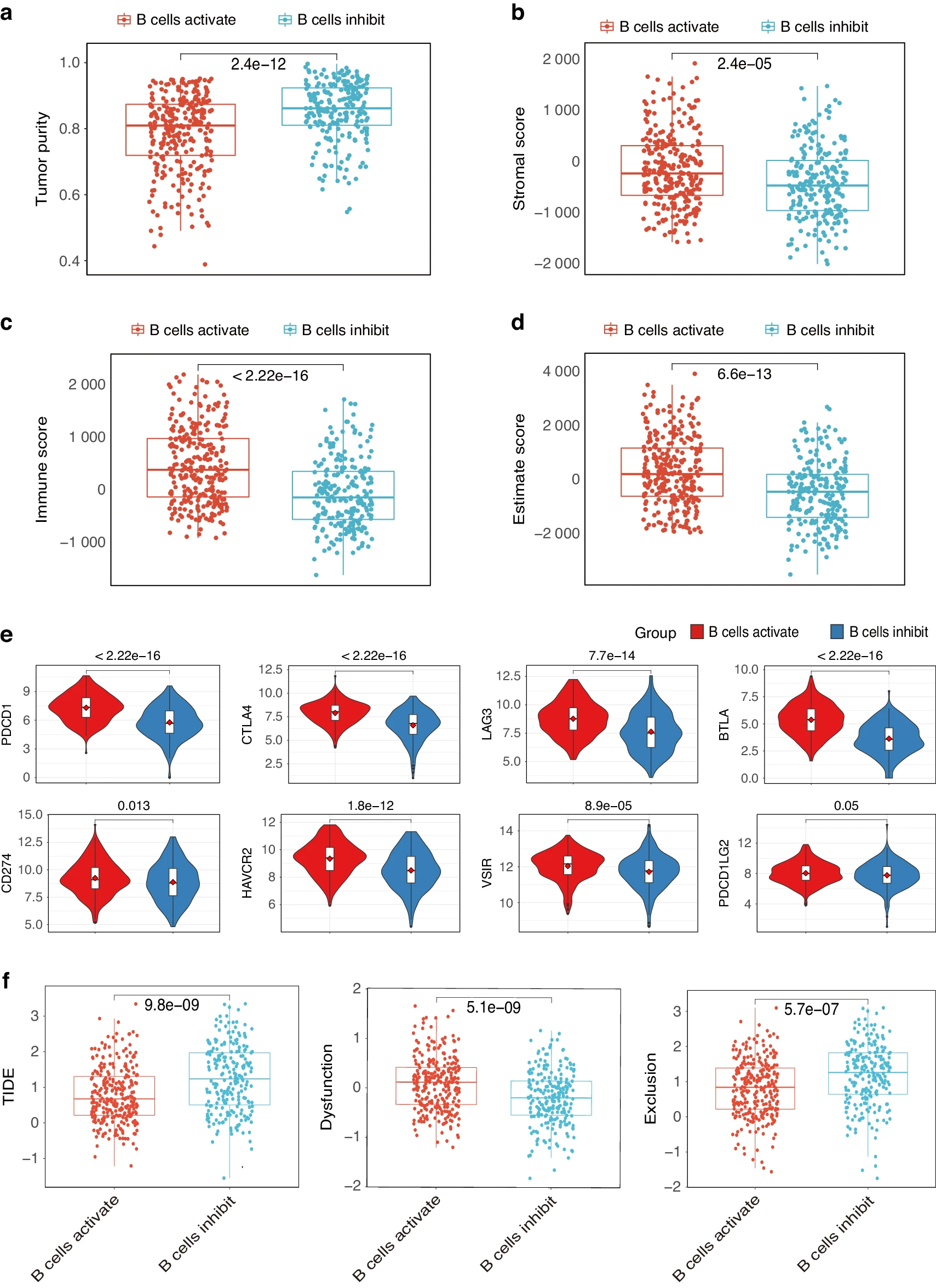
基于 B 细胞的分类准确预测免疫治疗反应。箱线图显示 B 细胞激活组的肿瘤纯度较低(a),间质分数(b)、免疫分数(c)和 ESTIMATE 分数(d)较高。e 箱线图展示了两组间典型免疫抑制受体(PDCD1、CTLA4、LAG3、BTLA、CD274、HAVCR2、VSIR 和 PDCD1LG2)的不同表达水平。f B 细胞激活组的患者显示出较低的 TIDE 分数,较高的 Dysfunction 分和较高的 Exclusion 分。
四基因预后模型的开发与验证于 HNSCC 中¶
我们的研究使用 LASSO 回归分析来优化 BCAGS,发掘出一个由四个基因组成的集合(JCHAIN, GZMB, IGHA1, 和 PRDX4),促进了一个关键的预后风险模型的构建。其中,JCHAIN、GZMB 和 IGHA1 在低风险组中表达显著升高,而 PRDX4 在高风险组中表达明显增加(图 6a)。随后,通过在 TCGA 队列中进行单变量 Cox 回归分析,我们寻求确定风险评分是否可以脱离传统临床因素独立预测患者结果。鼓舞人心的是,我们的发现显示风险评分(HR: 1.9; 95% CI: 1.2–3)作为 OS(总生存期)的独立预兆(图 6b)。值得注意的是,KM(Kaplan-Meier)和对数秩分析揭示了高风险和低风险 HNSCC 队列之间在 OS 方面的明显差异(图 6c)。此外,风险评分在预测 TCGA 队列中的 OS 方面的效果显著(1 年、3 年、5 年和 10 年 OS 的 AUC 分别为 0.622、0.634、0.570 和 0.626),展示了它们在长期预后中的熟练度(图 6d)。为了加强我们模型的预后有效性,我们招募了一个由 20 名异质性 HNSCC 患者组成的临床队列,旨在验证这四个基因的表达模式(JCHAIN, GZMB, IGHA1, 和 PRDX4)。通过 RT-qPCR 对这些基因的 mRNA 进行定量,我们在临床队列中根据基因的表达水平明确区分了低风险和高风险患者。引人注目的是,JCHAIN、GZMB 和 IGHA1 在低风险类别中表达显著增加,而 PRDX4 表达显著下降(图 6e)。随后的 Kaplan-Meier 分析强调了高风险组与 HNSCC 患者预后恶化之间的显著相关性(图 6f)。作为 RT-qPCR 的补充,我们的研究步骤通过免疫组化(IHC)分析更深入地探讨,揭示了这些基因在高风险层中的空间和蛋白水平表达模式(图 6g, h)。这一证实进一步加强了这些基因与不良临床结果之间的联系。值得注意的是,我们的实验结果与我们构建的风险模型完美对齐,确认了这四个基因作为在预测 HNSCC 患者生存结果中的强大预后生物标记物的潜力。
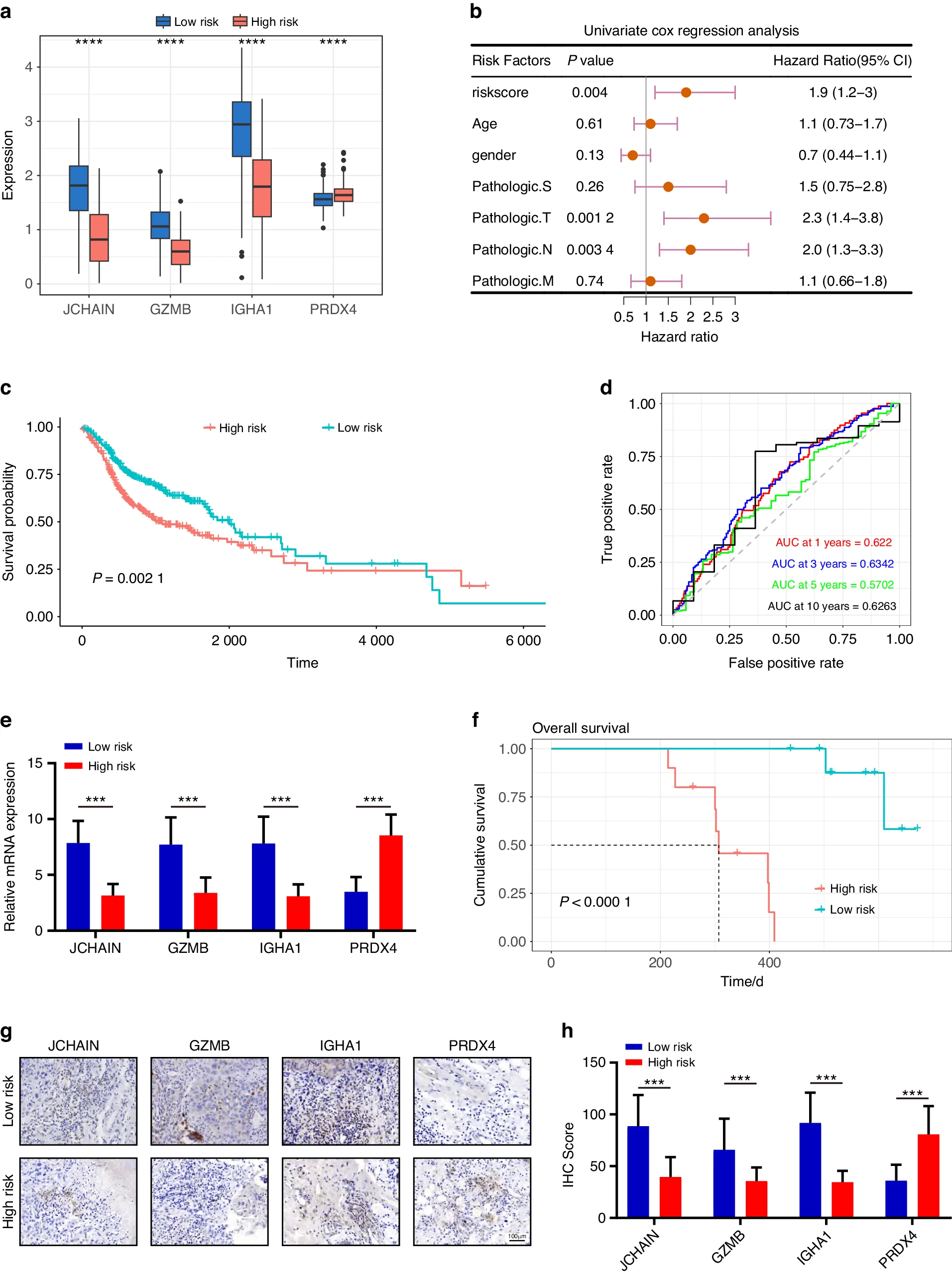
揭示了一个用于口腔鳞状细胞癌(HNSCC)患者预后的四基因风险模型。a 通过比较箱线图描绘了两组中与建模相关的基因表达水平。b 通过单因素 Cox 回归分析绘制出与 OS 相关的临床因素的森林图。c Kaplan-Meier 生存曲线展示了与预后相关的风险评分的影响。d ROC 曲线谱,作为 1、3、5 和 10 年 OS 动态的预测指标。e 比较分析突出了高风险组与低风险组之间的差异基因表达。f 基于四个基因风险评分的 HNSCC 患者 Kaplan-Meier 生存曲线。两组中的代表性 IHC 染色图像(g)和 IHC 评分(h)。比例尺,100μm
材料和方法¶
数据下载
从基因表达 Omnibus (GEO) 数据库(http://www.ncbi.nlm.nih.gov/geo/)下载了来自两个 HNSCC 队列,GSE139324 和 GSE164690 的单细胞转录组数据,并用于建立基于 B 细胞的 HNSCC 分类,构建激活的 B 细胞基因签名以及验证 HNSCC 分类的一致性。我们选择了这些数据集,因为它们广泛覆盖了 B 细胞聚类,这对我们研究的目标至关重要。我们使用多种样本的目的是为了减少可能的偏见,并确认我们的结果适用于不同的患者群体。此外,为了探索激活 B 细胞基因签名预测临床预后的能力,我们从 TCGA 数据库下载了 501 名 HNSCC 患者的转录组和临床数据。我们还从 GEO 数据库下载了有关 HNSCC 患者队列的信息,以进行进一步与生存相关的基因筛选。
HNSCC 患者样本
在中山大学附属第一医院口腔颌面外科部接受手术的 20 名患者提供了 HNSCC 肿瘤组织样本。研究开始前,所有参与者都签署了同意书。中山大学附属第一医院的伦理审查委员会批准了所有与患者相关的研究。
scRNA-seq 数据处理
我们使用 R 中的 Seurat 4.0 软件包进行了 scRNA-seq 数据分析。简要地说,通过使用 Read10X 函数将样本表达矩阵导入 R 来创建 Seurat 对象,并整合相关的临床信息。质量控制过程涉及几个步骤。首先,我们选择保留仅表达基因计数在 200 到 10,000 之间的细胞。此外,我们筛选掉将其读数的 10% 以上映射到线粒体基因组的细胞,因为这可能表明细胞质量较差。此外,我们采取措施从每个样本中去除双倍体(错误地识别为单个细胞的细胞),以确保数据集的纯度和准确性。经过质量控制后,我们继续鉴定高度可变基因(HVGs),因为这些基因通常在区分不同细胞类型方面起关键作用。我们选择了前 2000 个 HVGs 进行进一步分析。使用这些基因,我们进行了主成分分析(PCA)来减少数据集的维度,并识别主要的变异轴。根据 PCA 结果,我们使用前 1-20 个主成分进行聚类分析。我们主要鉴定了以下主要细胞类型:B 细胞、CD4+ T 细胞、CD8+ T 细胞、循环 T 细胞和髓样细胞。
获取一致性聚类的基因集
为了探索 B 细胞在 HNSCC 中的作用,我们获得了所有身份 B 细胞亚群之间的差异表达基因集。简言之,应用 Findallmarkers 函数来识别与 B 细胞相关的亚群,以计算每个细胞亚群的差异表达基因(DEGs),并使用 Log2FC > 1,P < 0.05 的阈值。这个基因集随后用于一致性聚类,包含 B 细胞亚群的所有可能的特征基因。
BCAGS 鉴定
为了获得代表 B 细胞亚群的特征基因的精确集合,使用“limma”方法识别了 B 细胞伪块的 DEGs。从 B 细胞激活组的 DEGs 中选择了 22 个基因(Log2FC > 1,P < 0.05)用于构建 B 细胞激活基因签名(BCAGS)。
GO 富集分析 为了在基因水平探索 BCAGS,我们使用“clusterProfiler” R 包对 B 细胞激活组进行了基因本体(GO)富集分析。我们选择了人类作为研究物种,并考虑了生物过程,设置了 P 值阈值<0.05。B 细胞激活组的 GO 富集分析结果显示,上调基因富集于免疫细胞激活相关通路中。
ssGSEA 引入单样本基因集富集分析(ssGSEA)来量化两个不同亚组肿瘤微环境中 28 种免疫相关细胞类型的相对浸润。每种免疫细胞类型的特征基因面板来自最近的一项出版物。在 ssGSEA 分析中,每种免疫细胞类型的相对丰度由富集分数表示。ssGSEA 分数被归一化为统一分布,其中零是每种免疫细胞类型的最小分数,一是最大分数,并且直方图基于归一化的 ssGSEA 分数。通过多维尺度(MDS)和高斯拟合模型估计了免疫细胞浸润的生物相似性。
CIBERSORT 本研究中使用 CIBERSORT 评估了基于 BCAGS 亚组的 HNSCC 队列中免疫浸润的丰度差异,来自 TCGA 和 GEO 数据库的 HNSCC 的转录组数据基于“CIBERSORT” R 包量化了每种免疫细胞亚群的得分。CIBERSORT 的白细胞特征矩阵(LM22)预测了不同亚组中的所有免疫细胞类型。
细胞间通信 使用“CellChat” R 包进行细胞间通信分析,并基于两组(基于 BCSGS 的组)基因表达矩阵创建 CellChat 对象。使用 mergeCellChat 函数合并两组 CellChat 对象,并分析推断的相互作用数量和相互作用强度的差异。netVisual_diffInteraction 用于可视化推断的相互作用数量和相互作用强度。在不同的免疫细胞群体中比较了两组之间的细胞间通信,涉及“分泌信号”和“受体 - 配体”。
建立 BCAGS 预后风险模型 为了构建预后风险模型,我们对 TCGA HNSCC 病例进行了精细分析。利用 R 中的“glmnet”包,进行了严格的最小绝对收缩和选择算子(LASSO)回归,以识别 BCAGS 中与预后最相关的基因。随后,利用“forest plot”包,进行了单因素 Cox 回归分析,将具有危险比(HR)>1 的基因定义为风险因素,HR<1 的基因定义为保护因素。为患者计算了个性化风险评分,如下所示:风险评分=h0*e^∑i=0nexp(),并通过 Kaplan-Meier 方法绘制分层生存曲线。Uni-cox 分析进一步探索了风险评分、临床病理特征和 HNSCC 总生存期(OS)之间的复杂关系。