Single cell analysis of anti BCMA CAR T cell therapy in patients with central nervous system autoimmunity
嵌合抗原受体(CAR)T 细胞免疫疗法在治疗神经类自身免疫疾病方面前景广阔,但治疗后 CAR T 细胞的动力学和免疫变化仍不清楚。在此,我们对接受抗 B 细胞成熟抗原(BCMA)CAR T 细胞治疗的视神经脊髓炎谱系障碍(NMOSD)患者的成对脑脊液(CSF)和血液样本进行了单细胞多组学测序。结果显示,增殖的细胞毒性样 CD8+ CAR T 细胞克隆被确定为自身免疫的主要效应细胞。具有增强趋化特征的抗 BCMA CAR T 细胞有效穿过血脑脊液屏障,消除了脑脊液中的浆母细胞和浆细胞,并抑制了神经炎症。输注产品中表达 CD44 的早期记忆表型可能与 CAR T 细胞在自身免疫中的持久性相关。此外,与血液系统恶性肿瘤相比,来自 NMOSD 患者的 CAR T 细胞表现出独特的抑制细胞毒性特征。因此,我们提供了有关神经类自身免疫疾病患者 CAR T 细胞功能的机制见解。
简介¶
视神经脊髓炎谱系疾病(NMOSD)是一种中枢神经系统(CNS)的炎性自身免疫性脱髓鞘疾病,其特征是反复发作的视神经炎和脊髓炎,这些复发会导致严重的后遗症【1】。抗水通道蛋白 4(AQP4)免疫球蛋白 G(IgG)通过诱导 CNS 脱髓鞘损伤,引发疾病的发病机制,包括抗体依赖的细胞介导的细胞毒性和补体依赖的细胞毒性【1】。尽管 NMOSD 的治疗取得了重大进展,仍有一部分患者对当前最先进的治疗方法反应不佳,表现为难治性和反复发作的疾病【1, 2】。这可能是因为当前的治疗方法未能有效针对 CNS 驻留的免疫失调【3】。
嵌合抗原受体(CAR)T 细胞疗法在长期控制血液癌症方面显示出新兴的潜力【4-6】。多项研究已证明其在若干难治性自身免疫疾病病例中的治疗效果【7-9】。我们医疗中心目前正在进行一项针对 B 细胞成熟抗原(BCMA)的 CAR T 细胞疗法的 I 期研究,该抗原主要在复发或难治性 NMOSD 的浆母细胞(PBs)和浆细胞(PCs)中表达。中期分析显示其初步临床疗效【10】。与目前批准用于治疗 CNS 自身免疫疾病的疗法相比,CAR T 细胞具有若干优势,包括直接诱导靶细胞死亡、自我扩增特性以及穿越血脑屏障的能力【3, 11, 12】。然而,CAR T 细胞在自身免疫疾病中的分子机制尚不清楚。
在此,我们对五名接受抗 BCMA CAR T 细胞治疗的 NMOSD 试验参与者的纵向血液和脑脊液(CSF)样本进行了单细胞多组学分析,以研究 CAR T 细胞在自身免疫疾病中的体内特性。我们发现,增殖的细胞毒性样 CD8+ CAR T 细胞克隆是自身免疫中的主要效应细胞,而在制造后的 CD8+ 效应 T(TE)细胞效应特征减少以及随后细胞毒性效应表型的抑制可能是 CAR T 细胞在自身免疫中的独特特征。此外,富含趋化基因程序的抗 BCMA CAR T 细胞可能能够穿过血 -CSF 屏障,消除 CSF 中异常扩增的 PCs,并减少 NMOSD 患者 CNS 中的促炎环境。
结果¶
抗 AQP4 IgG 阳性 NMOSD 患者血液和脑脊液中 B 细胞谱系异常扩增¶
为了全面了解 NMOSD 患者的免疫变化,我们从五名难治性/复发性抗 AQP4 IgG 阳性 NMOSD 患者和五名年龄和性别匹配的对照组中生成了外周血单核细胞(PBMC)和脑脊液(CSF)细胞的单细胞结合转录组、T 细胞受体(TCR)和 B 细胞受体(BCR)数据(图 1A 和表 S1)。我们获得了来自 10 个血液样本的 113,766 个免疫细胞的单细胞转录组数据和来自 10 个 CSF 样本的 34,098 个细胞的单细胞转录组数据。在对 T 细胞和 B 细胞进行基本注释后,我们在 80,998 个 T 细胞中的 89,070 个 T 细胞(91%)和 13,303 个 B 细胞中的 14,215 个 B 细胞(94%)中鉴定出了成对的 TCR 和 BCR 序列(图 1A 和图 S1)。难治性 NMOSD 患者的 B 细胞在 CSF 中增加,但在血液中没有增加。相比之下,NMOSD 患者的 PBs 和 PCs 在两种体液中都比对照组高(图 1B)。
由于 AQP4 特异性 IgG 是 NMOSD 病理的直接效应因子,我们分析了特定 B 细胞和 PCs 类别的组成变化(图 1C 和图 S2,A 至 C)【13, 14】。与对照组相比,NMOSD 患者的 B 细胞,特别是 PBs/PCs 在血液和 CSF 中都有更大的克隆扩增(图 1C)。CSF B 细胞库高度克隆,具有大量的 IgG 亚型(图 S2D),表明选择克隆的抗原特异性增殖发生在 CSF 中。此外,NMOSD 患者不同 B 细胞子群中的 IgG1 亚型比例升高(图 S2E)。一致的是,与 Ig 生产、Ig 介导的免疫反应、B 细胞激活和 BCR 信号通路相关的基因高度上调(图 S2F)。因此,NMOSD 患者的外周和 CNS 中都有增强的体液反应。此外,CSF 可能是克隆进化的潜在场所【1】。
为了研究 NMOSD 患者循环和 CSF B 细胞之间的关系和差异,我们比较了每个 B 细胞子类中的单细胞 BCR 数据中的 Ig VH(免疫球蛋白重链可变区)序列库(表 S2)。在 CSF 中检测到 13% 的所有 B 细胞中有重叠的 B 细胞 Ig VH 序列,包括 21% 的未成熟 B 细胞、15% 的初始 B 细胞、10% 的记忆 B 细胞、22% 的双阴性记忆 B 细胞和 7% 的 PBs/PCs(图 1D,图 S2G 和表 S2)。这表明这些血液和 CSF 中的子类可能有不同的起源。例如,CSF B 细胞可能起源于循环前体或 CNS 特异性的 B 细胞驻留位点【15, 16】。单样本基因集富集分析(ssGSEA)揭示了 CSF B 细胞中补体、白介素 -6(IL-6)和趋化因子通路比循环 B 细胞更为丰富(图 1E)。在最近发布的 NMOSD 患者数据集中也发现了类似的 CSF B 细胞转录签名,这支持了我们发现的稳健性和可重复性(图 S3)。
由于可溶性 BCMA(sBCMA)是由膜 BCMA 直接脱落产生的【18】,主要在 PBs/PCs 中表达,我们使用酶联免疫吸附试验(ELISA)在独立队列中测量了抗 AQP4 IgG 阳性 NMOSD 患者(n = 55)和年龄和性别匹配的对照组(n = 55)中的血液和 CSF 中的 sBCMA(图 1F 和表 S3)。尽管这两组之间的血液 sBCMA 没有显著差异,但 NMOSD 组的 CSF sBCMA 显著高于对照组(图 1F),与年龄和性别无关(图 S4,A 和 B)。NMOSD 患者的 sBCMA 商(QsBCMA)和 QsBCMA/白蛋白商(QAlb)比对照组显著更高。受试者工作特征(ROC)分析表明,QsBCMA 和 QsBCMA/QAlb 具有潜在的诊断价值,高表达与 NMOSD 诊断相关(ROC 曲线下面积(AUC)为 0.7441,95% 置信区间(CI)为 0.6531 至 0.8351,P < 0.0001;AUC 为 0.6175,95% CI 为 0.5110 至 0.7241,P = 0.0336)。计算的截断值为 4.6073(对于 QsBCMA)和 0.7406(对于 QsBCMA/QAlb)。此外,还检测到几种已知的神经炎症和神经损伤的生物标志物,包括 QAlb(血 -CSF 屏障标志物)【19】、神经丝轻链蛋白(NFL)(神经损伤标志物)【20】和可溶性髓系细胞 2 型受体(sTREM2)(小胶质细胞激活标志物)【21】。正如预期的那样,NMOSD 患者 CSF 中 NFL、QAlb 和 sTREM2 的表达水平高于对照组,并与 CSF sBCMA 水平正相关(NFL,P < 0.001,R = 0.390;QAlb,P < 0.001,R = 0.472;sTREM2,P < 0.001,R = 0.398)(图 1F 和图 S4C)。因此,CSF 中升高的 sBCMA,代表 CNS 中的 PBs/PCs,是 NMOSD 中神经炎症和连续神经损伤的可靠生物标志物,可能作为潜在的药物靶点。
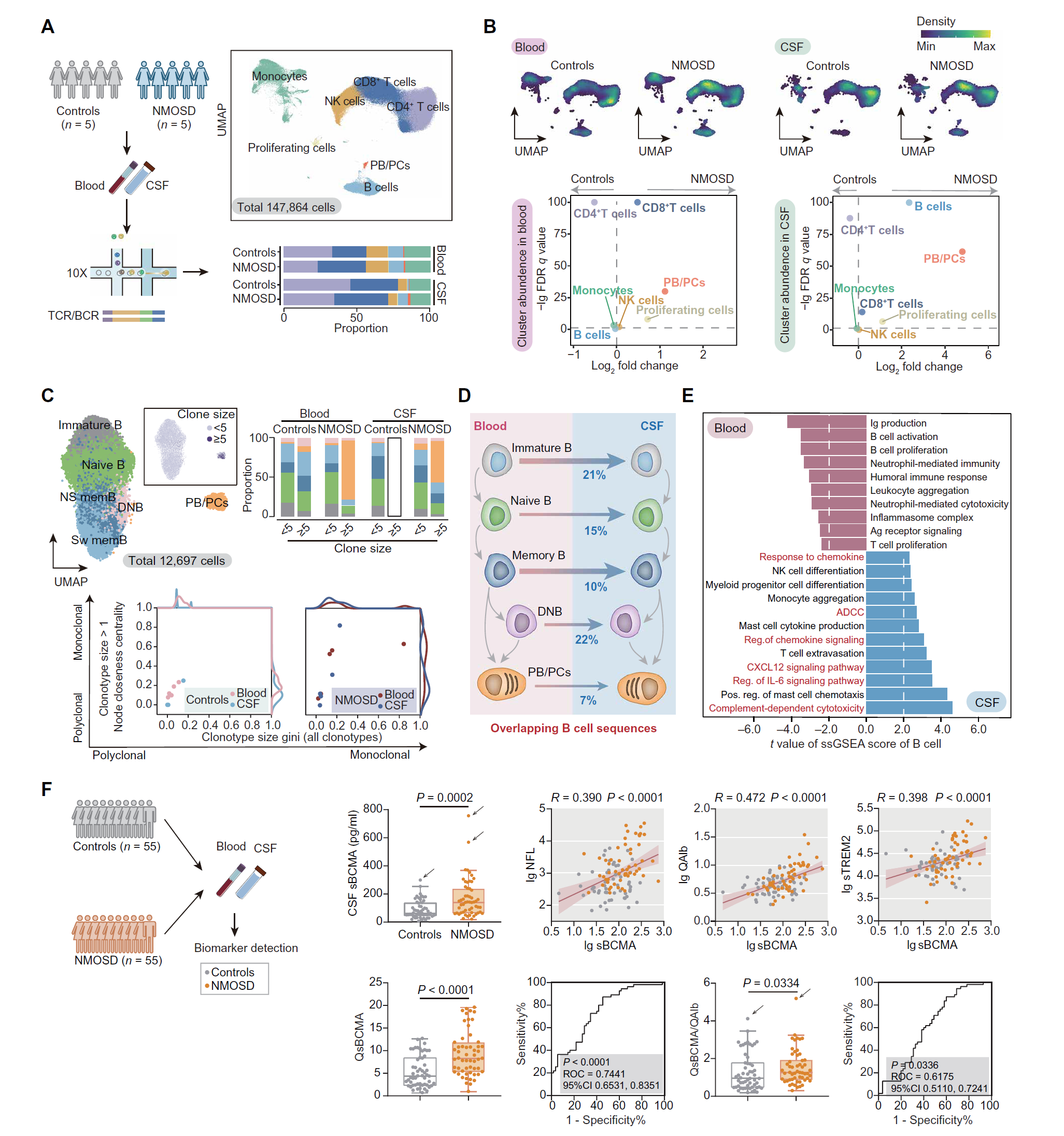
图 1. NMOSD 患者脑脊液中克隆性扩增的 B 细胞和分区 B 细胞库。
(A) 实验设计和生物信息学流程的示意图。UMAP 图显示从 PBMC 和 CSF 细胞中衍生的 147,864 个细胞。条形图显示 NMOSD 患者和对照组的血液和 CSF 中细胞类型的比例。 (B) UMAP 图显示对照组和 NMOSD 患者的细胞密度。火山图显示 NMOSD 患者与对照组之间的簇丰度差异。 (C) B 淋巴细胞的 UMAP 图重新聚类为六个亚群:未成熟、初始、未切换记忆(NS mem)、切换记忆(Sw mem)和双阴性(DN)B 细胞以及 PBs/PCs。UMAP 图按克隆大小着色。条形图显示克隆 B 细胞的频率。通过节点接近中心度基尼指数显示克隆类型大小的散点图,边缘直方图指示分布。每个点代表一个个体(n = 5)。 (D) 血液和 CSF 中 B 细胞亚群的示意图,显示每个亚群内重叠的 Ig VH 序列。 (E) 指示途径的条形图显示由 ssGSEA 得分得出的 t 统计值。 (F) 验证队列的示意图。箱线图描绘了 CSF sBCMA、QsBCMA 和 QsBCMA/QAlb 的中位数和四分位范围水平。使用 Mann-Whitney U 检验进行两组比较。箭头指示离群值。散点图描绘了 CSF sBCMA(lg)与 CSF sTREM2(lg)、QAlb(lg)和 CSF NFL(lg)之间的相关性。使用年龄和性别作为固定效应进行部分相关分析。实线表示回归线和 95% 置信区间(CI)。根据 QsBCMA 和 QsBCMA/QAlb 计算的 ROC 曲线,显示特征数的 AUC 和 95% CI。每个点代表一个个体(n = 55)。
抗 BCMA CAR T 细胞在 NMOSD 患者中的克隆动力学和转录分析¶
目前正在我们医疗中心进行一项针对复发/难治性 NMOSD 的抗 BCMA CAR T 细胞疗法的 I 期临床试验【10】,随访工作仍在进行中。在本研究中,我们研究了五名连续患者的新鲜采集的血液和 CSF 样本,并调查了 CAR T 细胞的进化轨迹及随后的免疫细胞变化。患者 5 在第 45 天经历了右眼视力下降的复发。尽管发生了这一事件,所有五名患者在超过 15 个月的时间内显示出神经功能的逐步改善(图 2A)。
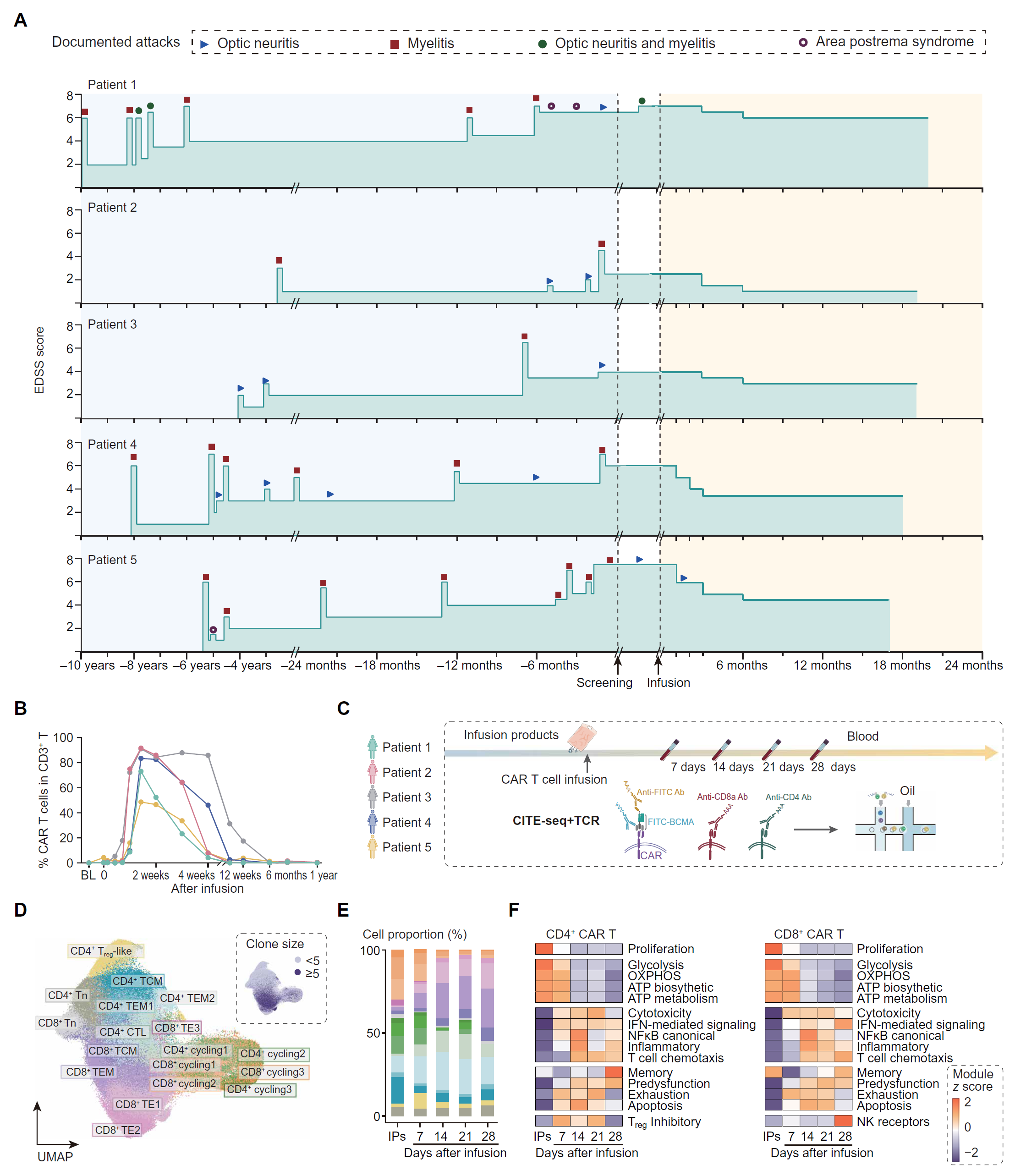
图 2. NMOSD 患者中抗 BCMA CAR T 细胞的时间演变。
(A) 图表展示了 NMOSD 患者个体的临床结果,以攻击和扩展残疾状态量表(EDSS)评分在 CAR T 细胞治疗前后的变化为代表。 (B) 输注后不同时点通过流式细胞术检测到的五名患者血液中 CAR T 细胞在淋巴细胞中的百分比变化。 (C) CITE-seq 策略的示意图,用于检测输注产品(IPs)和五名患者输注后不同时点的血液样本中 CAR T 细胞表面的 CAR、CD8α和 CD4。 (D) 重新聚类成子集的 CAR T 细胞的 UMAP 图(左)和按克隆大小着色的细胞(右)。 (E) 在 IPs 和输注后不同时点的 CAR T 细胞类型比例的条形图。 (F) 热图显示了 CAR T 细胞在 IPs 和输注后不同时间点的增殖、记忆、功能失调前状态、耗竭、凋亡、细胞毒性、IFN/NF-κB 信号、炎症、T 细胞趋化、糖酵解、氧化磷酸化(OXPHOS)、ATP 生物合成和 ATP 代谢特征的表达。
通过流式细胞术测量的抗原驱动的 CAR T 细胞扩增在输注后约 10 天达到峰值,然后在 1 个月内大幅下降(图 2B)。在输注前和输注后不同时点(7、14、21 和 28 天)对输注产品(IPs)和 PBMCs 进行转录组和表位测序(CITE-seq)以及成对的单细胞 TCR 测序(图 2C 和图 S5A)。我们对来自五个 IPs 和 20 个血液样本的 68,603 个 CAR T 细胞进行了聚类分析(图 2D 和图 S5,B 至 E,以及图 S6)。IPs 主要由 MKI67+ 循环 T 细胞组成,具有 CD4+ 和 CD8+ 表型的组合。输注后,循环 T 细胞主要被 TE 细胞(CD8+ TE 细胞和 CD4+ 细胞毒性 T 淋巴细胞(CTLs))以及 CD4+ 和 CD8+ T 效应记忆(TEM)细胞取代(图 2,D 和 E)。
差异表达基因(DEG)分析显示,IPs 中的 CD4+ 和 CD8+ CAR T 细胞表达高水平的与增殖和腺苷三磷酸(ATP)及葡萄糖代谢通路相关的基因,这些基因在输注后逐渐下降(图 2F)。输注后早期时间点(7 至 14 天内),CAR T 细胞表达高水平的与炎症和细胞毒性相关的基因,这些基因在晚期时间点(21 至 28 天)下降。CD4+ T 细胞逐渐表达越来越高的记忆相关基因。CAR T 细胞逐渐显示出越来越多的前功能障碍基因,表明早期功能障碍 T 细胞类似于人类癌症中的功能障碍 T 细胞【22】。
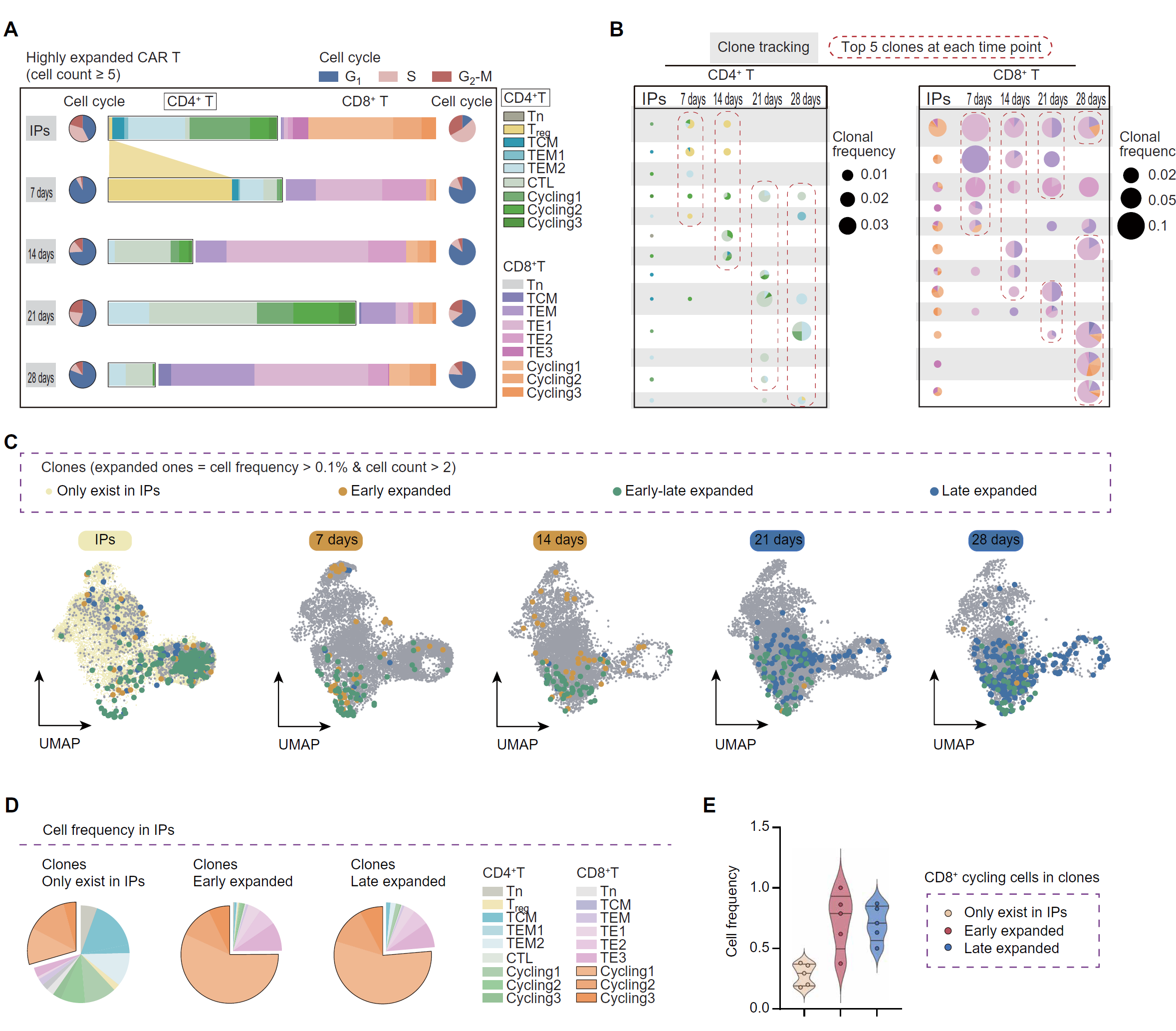
图 3. NMOSD 患者中 CAR T 细胞的克隆动力学。
(A) 条形图显示扩展克隆 CAR T 细胞的频率,扩展克隆表示在≥5 个细胞中观察到的克隆类型。 (B) 显示在输注后和输注产品(IP)中识别出的最普遍的前五大 TCR 克隆,分别针对 CD8+ 和 CD4+ CAR T 细胞子集。每个饼图显示克隆在每个时间点的归属,其大小对应于样本中克隆的频率。 (C) 输注后按扩展 TCR 克隆着色的 CAR T 细胞的 UMAP 图。扩展克隆表示细胞频率>0.1% 且在≥2 个细胞中观察到的克隆。显示在第 7 天或第 14 天扩展的克隆(早期扩展,黄色)、在第 21 天或第 28 天扩展的克隆(晚期扩展,蓝色)以及在两个阶段都扩展的克隆(早晚期扩展,绿色)。 (D) 饼图显示仅存在于 IPs 中的克隆、在早期时间点扩展的克隆以及在输注前晚期时间点扩展的克隆的细胞类型频率。 (E) 小提琴图显示仅存在于 IPs 中的克隆、在早期时间点扩展的克隆以及在输注前晚期时间点扩展的克隆中的 CD8+ 循环细胞频率。每个点代表一个个体(n = 5)。
CD4+ CAR T 细胞在扩增峰值(第 7 天)时显示出克隆性扩增的调节性 T 细胞(Treg)样细胞的增加,并在第 21 天时独特地扩增了 CTLs 和 TEM 细胞(图 3A)。Treg 抑制分子在第 7 至 21 天显示出强烈的激活,并在第 28 天略微下调(图 2F)。相比之下,TE 簇是输注后早期和晚期时间点扩增最显著的 CD8+ CAR T 细胞(图 3A)。此外,CD8+ CAR T 细胞中与自然杀伤(NK)受体相关的标志物表达逐渐增加,在第 7 至 14 天时表现为中等表达,并在第 28 天时达到最高表达,表明向 NK 样 T 细胞的转变增加(图 2F)。在第 7 至 14 天时,CD4+ CAR T 细胞中的前五大 TCR 克隆主要由 Tregs 组成,而在第 21 至 28 天时主要由 CTLs 组成,并且在 IPs 中具有多样的主要起源(图 3B)。然而,在 CD8+ CAR T 细胞中,每个时间点的前五大 TCR 克隆与 CD4+ CAR T 细胞相比显示出更大的细胞频率,主要起源于 IPs 中的循环 T 子群,并在输注后分化为 NKG7+ 细胞毒性 TE 亚群、NKG7+KLRF1+ NK 样 TE 亚群和 TEM 细胞(图 3B 和图 S5B)。此外,早期和晚期阶段的所有扩增克隆都被追踪并在 IPs 中主要表现为 CD8+ 循环表型(图 3,C 至 E)。因此,循环 CD8+ CAR T 细胞簇似乎在 NMOSD 的治疗中起着显著作用,至少在前 4 周内。
表达 CXCR3 的 CAR T 细胞渗透进 CSF¶
成像流式细胞术揭示了抗 BCMA CAR T 细胞与血液和 CSF 中的 CD138+ 浆细胞(PCs)直接接触,这表明 CAR 介导的 PC 杀伤可以在这两种体液中发生(图 4A)。输注后 3 个月内,血液和 CSF 中的 sBCMA 和免疫球蛋白(Igs)水平显著下降(图 4B),这表明 PCs 正在被消耗,体液自身免疫在外周和中枢神经系统(CNS)中均受到抑制。
为了理解这一观察结果的机制,我们试图在单细胞水平上表征 CSF 中 CAR T 细胞的克隆性和转录组。通过将单细胞 RNA 测序(scRNA-seq)读取映射到 5' CAR 序列,我们在 3 个月时鉴定了 287 个 CAR T 细胞(图 4C)。对 scRNA-seq 数据的重新聚类进一步揭示了四个不同的簇,这些簇以趋化性(CCL4、CCL5 和 CXCR6)、细胞毒性(PRF1 和 GZMH)、记忆(IL7R 和 TCF7)、NK 受体(KLRK1 和 KLRC4)以及溶酶体(CSTA、CSTH 和 CST3)相关基因为特征(图 4D 和图 S7)。此外,血液中的 CAR T 细胞显示出与对干扰素 -γ(IFN-γ)反应、氧化应激反应、核因子κB(NF-κB)和肿瘤坏死因子(TNF)信号以及细胞杀伤相关的基因表达增加,而 CSF 中的细胞则表现出增强的细胞趋化性特征和 CXCR3 的高表达(图 4E 和 4F)。
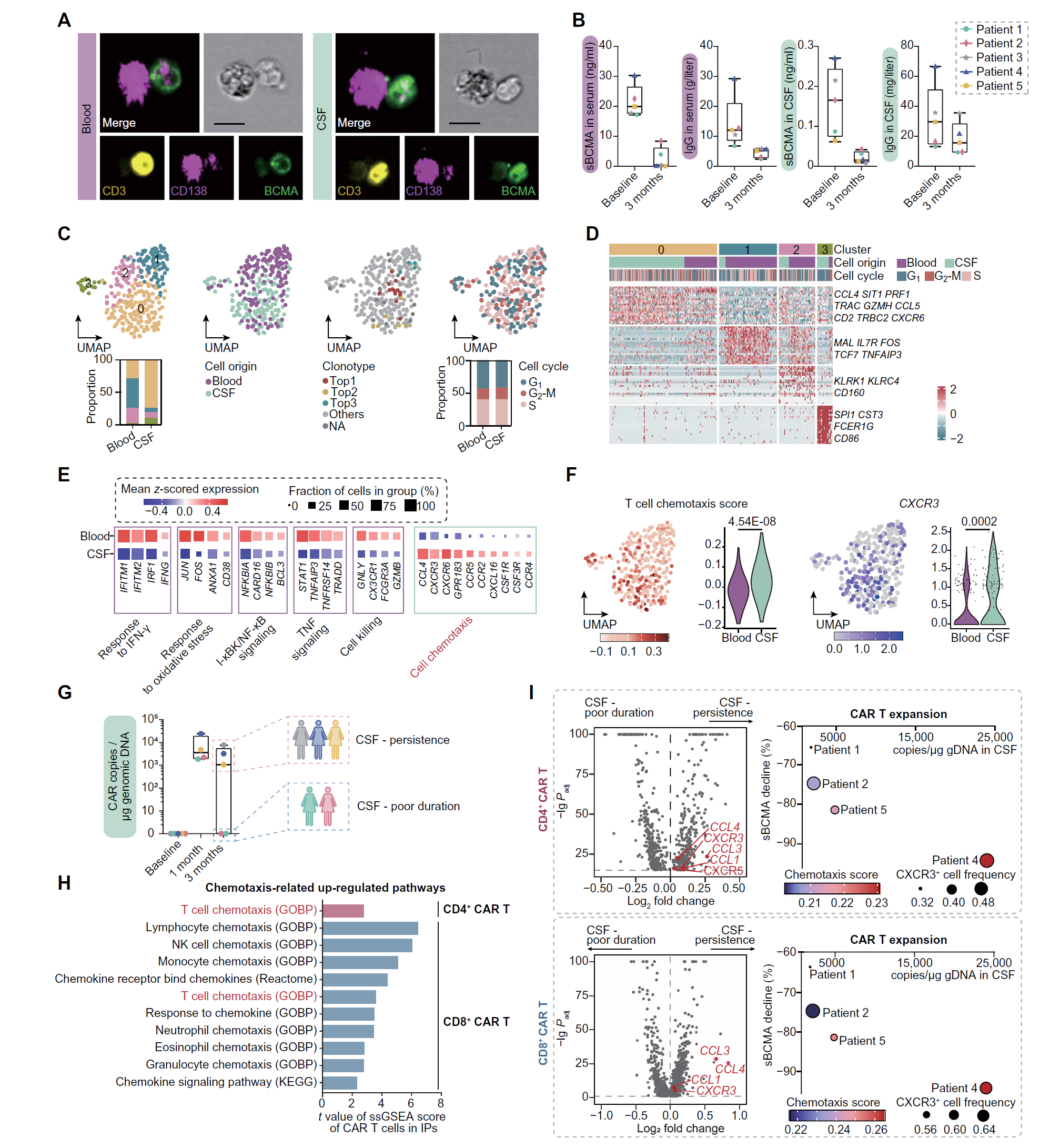
图 4. CAR T 细胞渗透 CSF 的特性。
(A) 成像流式细胞术显示抗 BCMA CAR T 细胞(CD3,黄色;BCMA 蛋白,绿色)和 CD138+ 浆细胞(PCs,CD138,洋红色)在血液和 CSF 中直接接触的代表性图像,在输注后第 10 天在患者 5 中检测到。比例尺,10 微米。 (B) 箱线图显示五名患者在基线和输注后 3 个月时血液和 CSF 中的 sBCMA 水平和 IgG 水平。每个点代表一个个体(n = 5)。 (C) UMAP 图显示输注后 3 个月的 CAR T 细胞,按子簇、样本类型、前三大 TCR 克隆和细胞周期阶段着色。 (D) 热图显示 CAR T 细胞子簇之间的差异表达基因(DEGs)。 (E) 显示输注后 3 个月血液和 CSF 中 CAR T 细胞在指示途径中的 DEGs。 (F) UMAP 和小提琴图显示输注后 3 个月 CAR T 细胞中的 T 细胞趋化性特征和 CXCR3 表达的特征富集。使用 Benjamini-Hochberg 校正的双侧 Mann-Whitney U 检验。 (G) 使用数字 PCR 检测的五名患者在输注后指示时间点 CSF 中 CAR 转基因拷贝的动力学变化。患者 3 在输注后 1 个月拒绝腰椎穿刺。在输注后 3 个月 CSF 中检测到 CAR 拷贝的患者表示为 CSF 持久性,而在 CSF 中未检测到 CAR 拷贝的患者表示为 CSF 短暂性。 (H) 比较 CSF 持久性和 CSF 短暂性患者输注前 CAR T 细胞的趋化性相关途径富集图。 (I) 比较 CSF 持久性和 CSF 短暂性患者输注前 CAR T 细胞的 DEGs 火山图,突出显示与趋化性相关的标志物。气泡图显示 IP 中 CXCR3 表达 T 细胞频率、T 细胞趋化性特征评分、sBCMA 下降和 CAR T 细胞在 CSF 中扩展之间的关联。
我们随后测试了输注前 CAR T 细胞的这些分子特征是否与其在 CNS 中的效果相关。在输注后 3 个月内,在患者 3、4 和 5 的 CSF 中检测到 CAR 转基因拷贝(CSF 持久性),但在患者 1 和 2 中未检测到(CSF 短暂性)(图 4G)。正如预期的那样,来自 CSF 持久性个体的输注前 CAR T 细胞表现出趋化性相关通路的显著富集,以及在 CD4+ 和 CD8+ CAR T 细胞中 CCL1、CCL4 和 CXCR3 基因表达的增加(图 4G 至 4I)。我们然后使用 scRNA-seq 数据对输注前的 CAR T 细胞进行 T 细胞趋化性和 CXCR3 特征评分,并分析特征评分和 CXCR3+ 细胞比例与 CAR T 细胞在 CSF 中的表现的相关性(图 4I)。与之前的结果一致,CAR T 细胞在 CSF 中的扩增与早期 CNS PB/PC 耗竭效应之间存在明显关联,如 CSF 中 sBCMA 水平的降低所确定的那样。患者 4 的输注前 CAR T 细胞具有高度富集的 T 细胞趋化特征和高表达的 CXCR3,显示出在 CSF 中的增强扩增,并与输注后 sBCMA 的显著减少相关,而患者 1 的 CAR T 细胞显示出较低的 CXCR3 频率和较低的 T 细胞趋化评分。因此,提高输注产品中 CXCR3+ 群体的频率可能会增强 CAR T 细胞在 CNS 自身免疫中的疗效。
CAR T 细胞逆转 NMOSD 患者 CSF 中的免疫失调¶
接下来,我们研究了 CAR T 细胞在 CSF 中消除扩增的 PBs/PCs 是否会影响 CSF 中的免疫环境(图 5A)。NMOSD 患者基线时的 CSF 免疫细胞显示出增强的相互作用潜力,这在输注后 3 个月显著改善(图 5B)。在输注后 3 个月,CSF 中有相当比例的 B 谱系细胞(包括 PBs/PCs)被消耗(图 5C)。相应地,CSF 中剩余的免疫细胞,包括 CD4+ T 细胞、CD8+ T 细胞、NK 细胞和髓系细胞,表现出改善的免疫反应、炎症激活和趋化性的转录特征(图 5D)。因此,CAR T 细胞治疗后,CSF 中免疫细胞的炎症特性受到抑制。
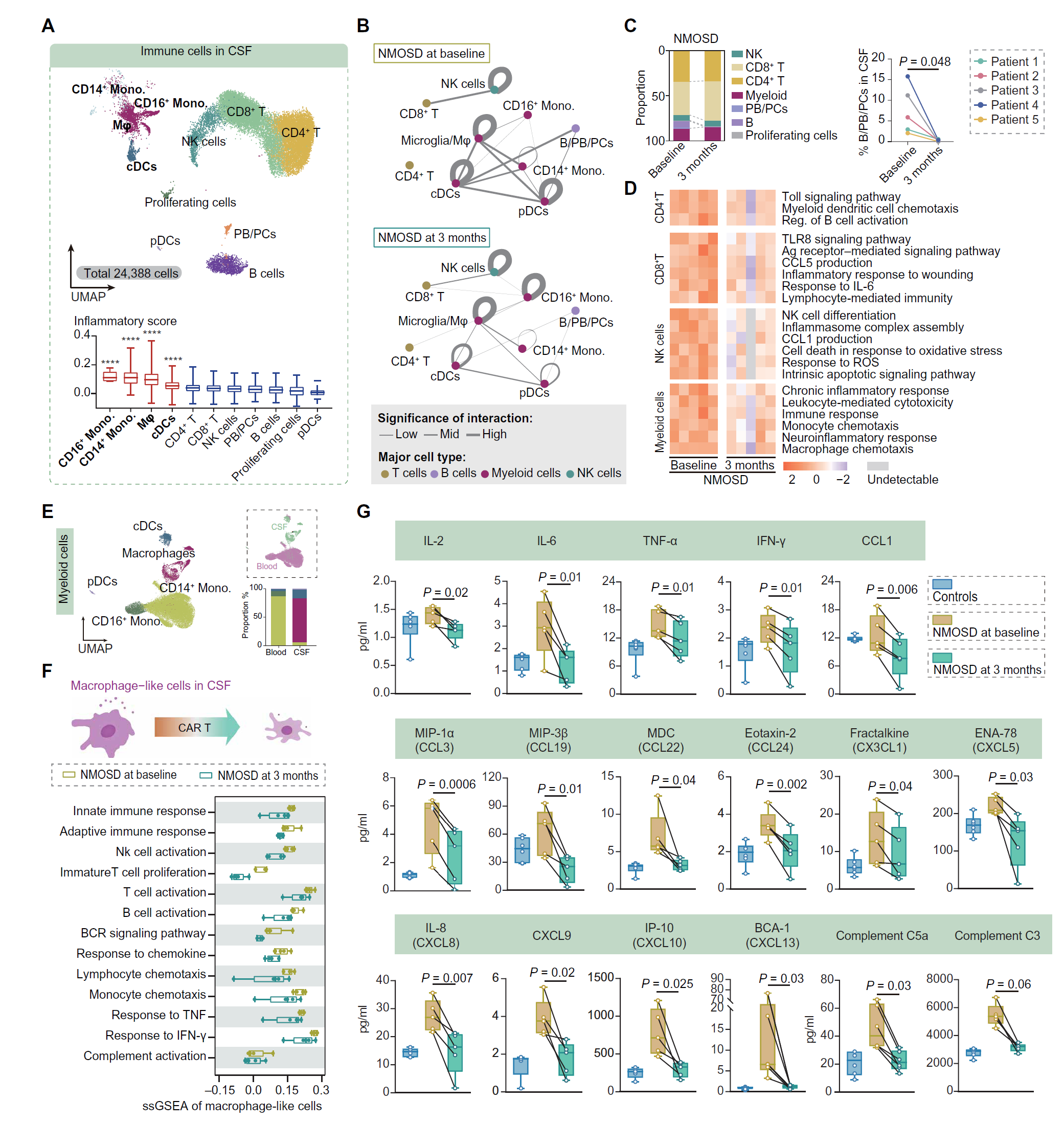
图 5. CAR T 细胞治疗后 NMOSD 患者 CSF 中的免疫异常被逆转。
(A) UMAP 图显示 CSF 中的免疫细胞,按主要细胞亚型着色。细胞亚型的炎症评分箱线图。通过 Wilcoxon 秩和检验将一种类型的细胞与 CSF 中的所有其他细胞分别进行比较来评估显著性。****P < 0.0001。 (B) 基于 CSF 细胞的 scRNA-seq 数据推断的 CSF 免疫细胞在基线(左)和输注后 3 个月(右)的细胞 - 细胞相互作用网络。显示 q 值<0.1 的相互作用。显著性:−log10(q 值)。 (C) 显示 NMOSD 患者在基线和输注后 3 个月 CSF 中细胞类型频率的图表。两组比较使用配对 t 检验。每个点代表一个个体(n = 5)。 (D) 热图显示 NMOSD 患者在基线和输注后 3 个月 CSF 中的 CD4+ T 细胞、CD8+ T 细胞、NK 细胞和髓系细胞的单样本 GSEA 特征。 (E) UMAP 图显示来自血液和 CSF 样本的髓系细胞重新聚类为五个亚群(左),按样本类型着色的细胞(右上)。血液和 CSF 中细胞类型比例的条形图(右下)。 (F) 显示基线和输注后 3 个月 CSF 中类巨噬细胞特征的单样本 GSEA 箱线图。每个点代表一个个体(n = 5)。 (G) 显示在基线和输注后 3 个月 CSF 中对照组和 NMOSD 患者的促炎细胞因子、趋化因子和补体水平的箱线图,通过 Luminex 和 ELISA 检测。配对比较使用 Wilcoxon 配对符号秩检验。每个点代表一个个体(n = 5)。
与其他细胞类型相比,四种髓系细胞亚型中检测到显著更高的炎症评分(图 5A)。NMOSD 和其他 CNS 自身免疫中报告了导致脑组织损伤的髓系细胞过度炎症【23】。髓系细胞的转录组分析识别了五个簇,其中类巨噬细胞在 CSF 中更为丰富(图 5E 和图 S8)。这些类巨噬细胞的炎症、趋化性和补体相关特征在 CAR T 细胞治疗后减少(图 5F)。Luminex 和 ELISA 进一步确认了 CAR T 细胞治疗后几种促炎细胞因子(包括 IL-2、IL-6、IFN-γ和 TNF-α)、多种趋化因子和补体系统成分(C3 和 C5a)的减少,与对照组相当(图 5G)。因此,CAR T 细胞逆转了 NMOSD 患者 CSF 中的免疫功能失调,这可能是 CAR T 细胞治疗后恢复的基础。
输注前 CD4+ CAR T 细胞特性与 NMOSD 中的持久性相关¶
接下来,我们通过对 9 名接受抗 BCMA CAR T 细胞治疗并随访超过 1 年的 NMOSD 患者的可用输注产品(IP)样本进行单细胞转录分析,研究了与 CAR T 细胞持久性相关的潜在因素(图 6A 和表 S4)。在输注后超过 1 年仍可在外周血中检测到 CAR 拷贝的患者被定义为长持久性,而在 2 个月内无法检测到 CAR 拷贝的患者被定义为非常短暂性(图 6A)。长持久性患者在输注前的 IP 中含有较大比例的 CD4+ CAR T 细胞(图 6B 和 6C)。此外,与短暂性组相比,长持久性患者的 IP CD4+ CAR T 细胞在输注前上调了 CD44 和多个细胞毒分子,包括 GZMA 和 GNLY,但仅表达低水平的 L 选择素(SELL)(短命效应细胞的标志物)(图 6D 和 6E)。
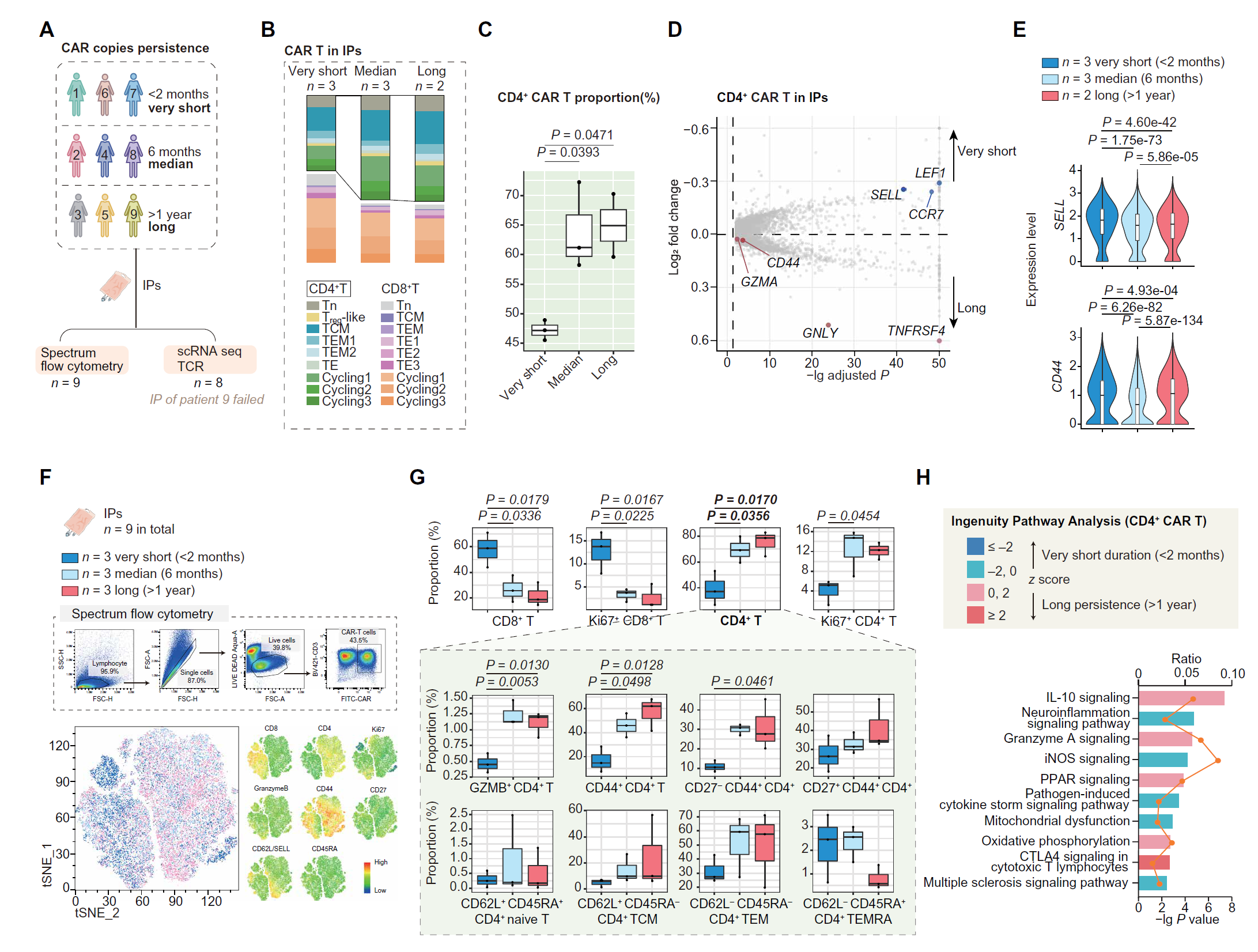
图 6. 与 NMOSD 中持久性相关的 CD4+ CAR T 细胞潜在特性。
(A) 示意图概述了来自 NMOSD 患者的九个 IP 样本的分组。输注后 1 年以上检测到 CAR 拷贝的患者被定义为长持久性,2 到 6 个月之间 CAR 拷贝减少的患者被定义为中持久性,输注后 2 个月内 CAR 拷贝不可检测的患者被定义为非常短暂性。 (B) 不同持久性 IP 中 CAR T 细胞子集的频率。 (C) 根据持久性分层的 CD4+ CAR T 细胞簇的相对频率。使用 Tukey 事后检验的一元 ANOVA 进行两组间的多重比较。 (D) 火山图显示长持久性患者和非常短暂性患者的 IP 中 CD4+ CAR T 细胞的差异基因表达。 (E) 小提琴图显示指示基因的表达。使用 Tukey 事后检验的一元 ANOVA 进行两组间的多重比较。 (F) 示意图和代表性图像显示使用全光谱流式细胞术对 IP 中 CAR T 细胞的淋巴细胞亚群分析的门控策略。t 分布随机邻居嵌入(tSNE)图显示按不同组(深蓝色表示非常短暂性,浅蓝色表示中持久性,红色表示长持久性)或按标志蛋白表达着色的 CAR T 细胞。 (G) 根据 CAR 持久性分层的指示子群的相对频率。使用 Tukey 事后检验的一元 ANOVA 进行两组间的多重比较。 (H) IPA 显示长持久性患者和非常短暂性患者的 IP 中 CD4+ CAR T 细胞比较的 DEGs 调控的相应信号通路。每个 z 得分反映了预测的激活水平。黄色曲线表示 DEGs 数量与每个通路中基因总数之间的比率。
这些转录发现随后通过全光谱流式细胞术得到确认。具有较高持久性的患者在其 IP 中具有更高比例的总 CD4+ T 细胞和 Ki67+ 增殖 CD4+ 细胞,但总 CD8+ T 细胞和 Ki67+ 增殖 CD8+ 细胞的比例显著较低。此外,具有较高持久性的个体在其细胞毒和记忆 CD4+ T 细胞簇中具有更高频率的颗粒酶 B(GZMB)和 CD44 标志物(图 6F 和 6G)。我们接下来应用 Ingenuity 通路分析(IPA)并比较了短暂性组与长持久性组 IP 中的 CD4+ CAR T 细胞,确定长持久性组中的激活功能程序为颗粒酶 A 信号传导和氧化磷酸化(OXPHOS)(图 6H)。因此,CD4+ CAR T 细胞的某些功能和代谢特征可能是其更大持久性的基础。
NMOSD 患者生成的 CAR T 细胞的独特特征¶
不同来源的 CAR T 细胞可能具有不同的表型和功能特征【24】。为了进一步识别 NMOSD 患者生成的 CAR T 细胞的独特特征,我们将 NMOSD 患者的输注前 CAR T 细胞和基础内源性 T 细胞与接受相同方式制造的 CAR T 细胞治疗的多发性骨髓瘤(MM)患者(n = 5)的输注前 CAR T 细胞和匹配的内源性基础 T 细胞进行比较(抗 BCMA CAR T 细胞,CT103A)(图 7A)。
与 NMOSD 患者相比,MM 患者 IP 中的 CD8+ 和 CD4+ 簇具有更高的与细胞周期和细胞毒性相关的特征评分(图 7B 和表 S5)。MM 患者 IP 中富含 MKI67+ 循环 CD8+ CAR T 细胞(图 7A 和 7C)。MM 的 CD8+ 循环簇上调了与增殖(MKI67 和 TOP2A)、IFNs(IFNG 和 IFITM3)和细胞毒分子(GNLY)相关的基因(图 7C)。IPA 结果一致地展示了 MM 的 CD8+ 循环簇中与效应模块(GZMA 信号和 CD27 信号)和细胞周期相关的激活途径(图 7D)。
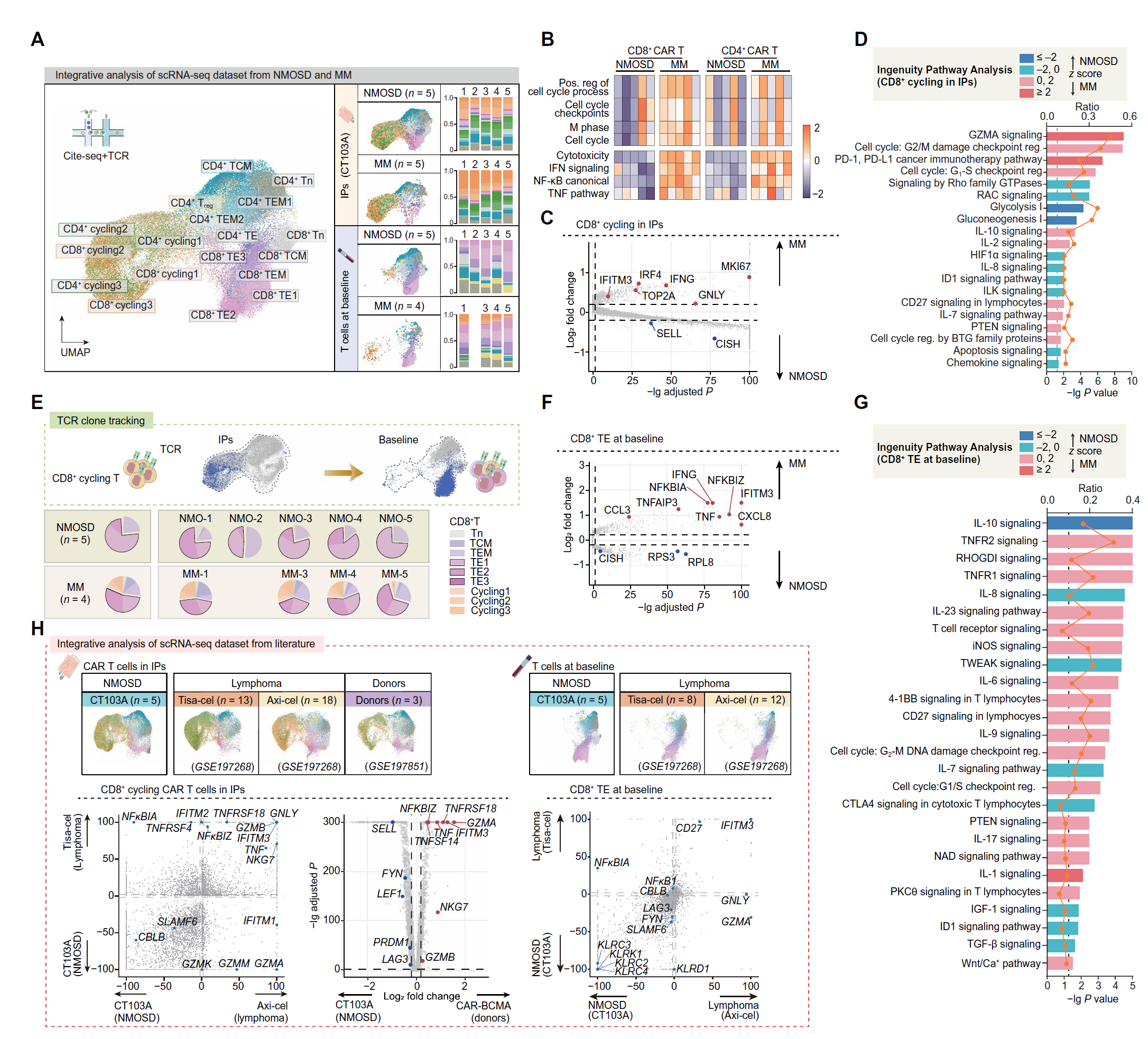
图 7. NMOSD 患者 CAR T 细胞的细胞毒性特征受抑制。
(A) NMOSD 患者和 MM 患者基线的 CAR T 细胞和 T 细胞的 UMAP 图(患者 MM-2 的 PBMC 不可用)。显示各个细胞类型比例的条形图。 (B) 显示细胞周期和细胞毒性特征表达的热图。 (C) 显示 NMOSD 和 MM 之间 CD8+ 循环 CAR T 细胞中 DEGs 的火山图。 (D) IPA 比较 NMOSD 和 MM 之间 CD8+ 循环 CAR T 细胞的相应信号通路。每个 z 得分反映预测的激活水平。黄色曲线表示 DEGs 数量与每个通路中基因总数之间的比率。 (E) 饼图显示通过克隆追踪制造成 IP 中的 CD8+ 循环 T 细胞的基线细胞分布。 (F) 显示 NMOSD 和 MM 之间基线 CD8+ TE 细胞中 DEGs 的火山图。 (G) IPA 比较 NMOSD 和 MM 之间基线 CD8+ TE 细胞的相应信号通路。 (H) 验证设计的示意概述。UMAP 图显示细胞子集。比较 NMOSD 和淋巴瘤基线 CD8+ TE 细胞以及 Tisa-cel 和 CT 103A、Axi-cel 和 CT 103A 的 IP 中 CD8+ 循环 CAR T 细胞的 DEGs。散点图显示 Tisa-cel 与 CT 103A 和 Axi-cel 与 CT 103A 的 IP 中 CD8+ 循环 CAR T 细胞之间的签名 P 值(-lg)(左上)以及 NMOSD 与淋巴瘤之间基线 CD8+ TE 细胞的签名 P 值(底部)。火山图显示 Tisa-cel 与 CT 103A 和 Axi-cel 与 CT 103A 的 IP 中 CD8+ 循环 CAR T 细胞的 DEGs(右上)。
为了确定原始 T 细胞资源是否与 CAR T 细胞的功能特征相关,我们使用 TCR 克隆追踪定义了基线时自体 T 细胞的来源(图 7E)。IP 中的 CD8+ 循环 T 细胞主要起源于 MM 和 NMOSD 中的内源性 GNLY+NKG7+ TE 细胞子簇。与我们在 IP 中的观察一致,NMOSD 患者基线 TE 细胞中的标志物(TNF 和 IFNG)表达减少和途径(TNFR1/2 信号和 CD27 信号)抑制也被观察到(图 7F 和 7G)。因此,NMOSD 中 CAR T 细胞的效应表型减少可能是由 CAR T 细胞制造前 TE 细胞的基础抑制激活状态所导致的。
为了验证自身免疫中 CAR T 细胞的固有特征,我们将 NMOSD 患者的 CAR T 细胞转录谱与之前生成的用于治疗淋巴瘤的 CD19 靶向 CAR T 细胞(Tisa-cel 和 Axi-cel)【25】和健康供体生成的抗 BCMA CAR T 细胞(图 7H)【26】进行比较。与淋巴瘤患者的 CAR T 细胞或健康供体生成的 CAR T 细胞相比,NMOSD 患者的 CAR T 细胞中基因如 GZMB、GNLY 和 NKG7 一致下调(图 7H 和图 S9)。在比较 NMOSD 患者和淋巴瘤患者的基线 TE 细胞时也观察到类似的模式(图 7H)。因此,与血液系统恶性肿瘤患者和健康对照相比,NMOSD 患者的输注前 CAR T 细胞和基线 TE 细胞具有较低的效应特征。
讨论¶
在中枢神经系统(CNS)自身免疫中,已经鉴定出分区 B 细胞【14, 27】,但其功能相关性和神经炎症的预测生物标志物的证据仍然稀缺。CSF 是表征脑部疾病中免疫系统的理想材料【28, 29】。我们观察到,NMOSD 患者的 CSF B 细胞的 Ig VH 序列与血液样本中的 B 细胞有约 13% 的重叠,表明大多数 CSF 中的 B 细胞可能来源于 CNS 驻留,并且是 CNS 自身免疫的潜在靶点。
抗 CD19/CD20 单克隆抗体 B 细胞消耗在与自身抗体相关的自身免疫疾病(包括 NMOSD)中显示出很大的治疗效果。然而,一部分患者对当前的治疗仍然具有耐药性和抵抗性【2, 30】。CAR T 细胞已显示出比单克隆抗体更彻底渗透组织的能力,并在实验性自身免疫性脑脊髓炎小鼠模型的 CNS 中诱导深远且持久的效果【3, 31】,这强化了 CAR T 细胞对 CNS 自身免疫患者的巨大潜力。在这项人类样本研究中,我们展示了 NMOSD 中 CAR T 细胞的动态特征,显示出高频率的扩展 CD4+ CAR T 细胞,在峰值阶段表现出 Treg 样表型,并且在晚期缓解期的 CD8+ CAR T 细胞通过表达 NK 受体获得了先天特征,而不是耗竭的 T 细胞。输注后 1 周在非应答的淋巴瘤患者中观察到 CAR Tregs 的存在,与克隆限制和较低毒性相关,并可能触发 NK 细胞转变程序【32】。然而,具有 CD28 信号域的 CAR T 细胞有利于 Treg 增殖,而 4-1BB(与我们的抗 BCMA CAR T 细胞 CT103A 类似)则阻止 Treg 增殖【25】。CAR Tregs 的扩展是否是 CAR T 细胞在自身免疫中的独特特征,尚需通过使用更多技术方法和在更大人群中的验证,并与血液系统恶性肿瘤患者中的 CAR T 细胞动态变化进行比较。此外,我们发现晚期缓解期的 CD8+ CAR T 细胞通过表达 NK 受体获得了先天样特征,并具有早期功能障碍特征,而不是耗竭的 T 细胞。抑制性 NK 受体的表达最初被认为是一种反馈机制,用于抑制过度刺激信号以避免激活诱导的细胞死亡。然而,这最终可能导致耗竭,并在慢性抗原暴露后限制 CAR T 细胞的寿命,如在胰腺癌中所报道的那样【33】。在我们的队列中,CD8+ CAR T 细胞表现出向 NK 样 T 细胞转变的增加,而没有显著上调耗竭表型。然而,晚期缓解期 CD8+ CAR T 细胞中高表达的前功能障碍基因表明早期阶段的功能障碍细胞,正如在癌症中报道的那样【22】。
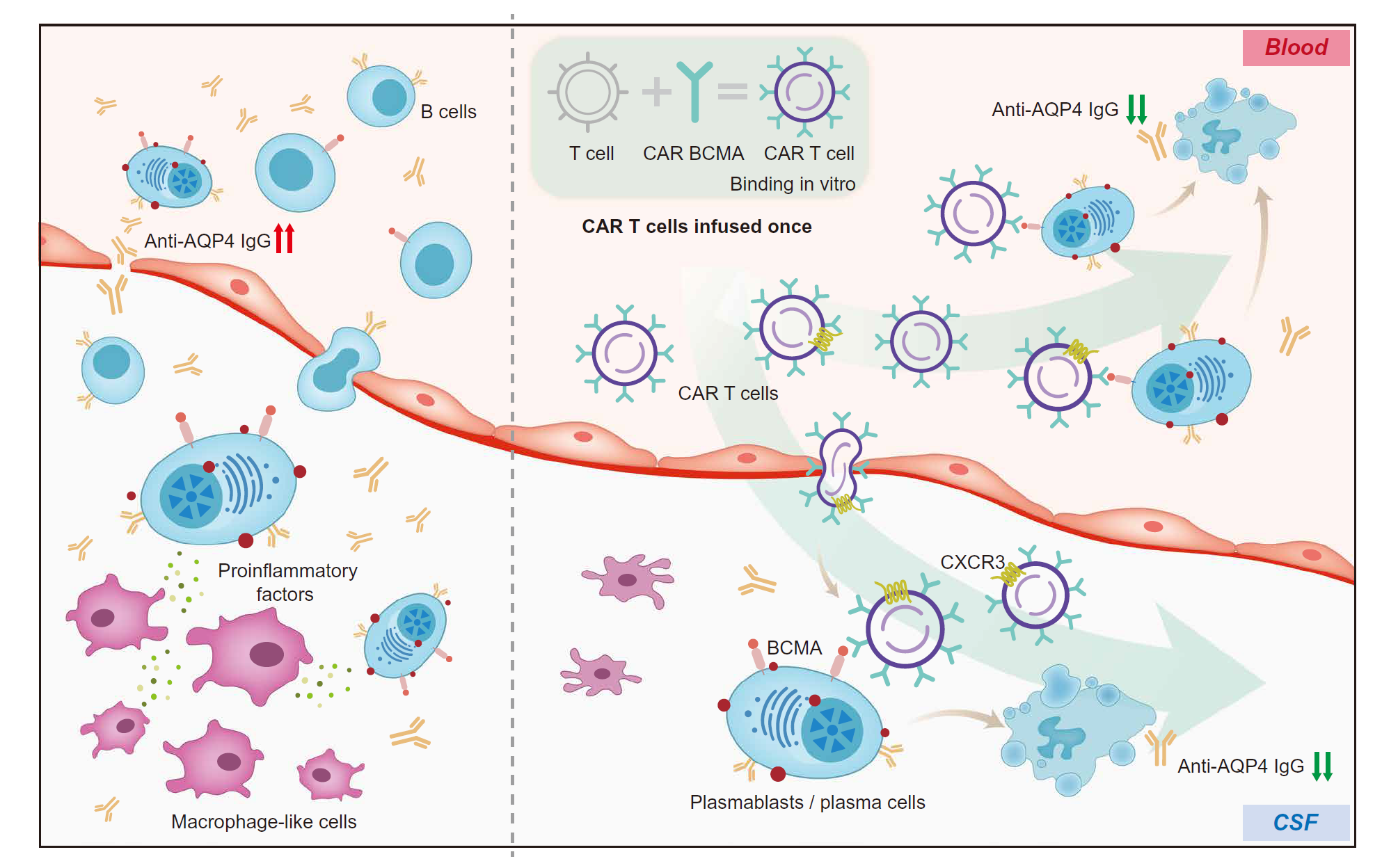
图 8. CAR T 细胞逆转了 NMOSD 患者 CSF 中的免疫功能失调。NMOSD 患者 CSF 中克隆性扩增的浆母细胞(PBs)和浆细胞(PCs)增加,与神经炎症和随后神经损伤呈正相关。具有增强趋化特征和高表达 CXCR3 的抗 BCMA CAR T 细胞高效穿越血脑屏障,消除 CSF 中扩增的 PBs/PCs,并抑制 NMOSD 患者中髓系细胞的激活和神经炎症。
我们还观察到,CAR T 细胞介导的 BCMA+ 细胞杀伤在外周和 CSF 中都直接发生,并且两种体液中的 sBCMA 水平显著降低。表现出富集的 T 细胞趋化特征和高表达 CXCR3 的输注前 CAR T 细胞与更高的 CAR 拷贝扩展和更好的 PB/PC 消耗效率相关。支持这一观点的是,已报道 CXCR3 在黑色素瘤和其他类型癌症背景下成功迁移血液 T 细胞至肿瘤中是必要的【34, 35】。此外,通过定制趋化因子受体来利用 T 细胞趋化性可能会改善小鼠脑肿瘤模型中采用性 T 细胞疗法的疗效【36, 37】。我们提供了开发富含 CXCR3 表达的 CAR T 细胞用于 CNS 自身免疫治疗的证据。由于 CSF 中异常扩展的 PB/PCs 与神经炎症呈正相关,CAR T 细胞输注后 CSF 中 PB/PCs 的消除可能导致免疫细胞活化和相关炎症反应的抑制(图 8)。尽管在长期随访中应注意 CAR T 细胞穿越血脑屏障的潜在安全问题【38】,但迄今为止在我们的队列中尚未观察到神经毒性。
在 NMOSD 患者中抗 BCMA CAR T 细胞的相对较短持久性(中位持续时间小于 6 个月)【10】—类似于系统性红斑狼疮队列中抗 CD19 CAR T 细胞的有限持久性【7】—仍需克服,以便成功使用 CAR T 细胞治疗自身免疫疾病。我们的分析表明,CD4+ CAR T 细胞 IP 中升高的 CD44 和下调的 SELL(CD62L)与长持久性(>1 年)患者共享分子特征。已知 CD44 在癌症转移过程中促进细胞迁移和存活【39】,并可作为识别干细胞样 T 细胞记忆群体的标志物【40】。在一项临床前研究中,CAR T 细胞治疗的狼疮小鼠模型在体内积累了一种 CD44+ CAR T 细胞群体,被怀疑在晚期缓解期持续 B 细胞消耗中起作用【41】。我们还表明,表达 CD44 的早期记忆 CAR T 细胞的输注前可能与 NMOSD 中的 CAR T 细胞持久性相关。这个观察背后的机制值得在未来的研究中进一步调查。
通过克隆追踪,我们证明 CD8+ 循环 CAR T 细胞在自身免疫中起主导作用,主要来源于制造前的基线 CD8+ TE 细胞。NMOSD 患者内源性 TE 簇的效应特征受抑可能解释了 IP 中 CAR T 细胞的细胞毒潜力降低,以及 NMOSD 患者中观察到的较低级别和较少的细胞因子释放综合征事件(相比于使用相同产品治疗的 MM 患者)【5, 42】。然而,需要将以相同方式制造的健康供体的 CAR T 细胞与 NMOSD 患者的 CAR T 细胞进行比较,以更好地描述其内在特征。总之,我们的结果提供了 CAR T 细胞在 CNS 自身免疫患者体内机制作用的独特见解,可能为未来优化 CAR T 细胞免疫疗法的研究提供指导。
材料和方法¶
人体参与者¶
参与本研究的患者包括在 I 期试验(NCT04561557)中接受抗 BCMA CAR T 细胞治疗的难治性或复发性抗 AQP4 IgG 阳性 NMOSD 患者【10】以及在 I/II 期研究(NCT05066646)中接受抗 BCMA CAR T 细胞治疗的 MM 患者【5】。患者签署了使用其 IP、血液和 CSF 样本以及在研究中使用和披露去标识健康信息的知情同意书,这些协议获得了同济医院机构审查委员会(TJ-IRB20220101)的批准。
为了进行单细胞测序分析,提供了来自五名 NMOSD 患者的纵向 IP 和血液样本,这些样本在基线和输注后 1、2、3、4 周以及 3 个月时采集。来自这五名 NMOSD 患者的 CSF 样本在基线和输注后 3 个月时采集。本研究还招募了五名年龄和性别匹配的对照个体(一个原发性头痛,三个无炎症性周围神经病变个体和一个特发性颅内高压),并采集了血液和 CSF 样本。此外,还包括五名接受 CAR T 细胞治疗的年龄和性别匹配的 MM 患者,采集了基线的 IP 样本和血液样本。还包括另外四名 NMOSD 患者的 IP 进行单细胞测序分析。患者和对照的详细信息显示在表 S1、S6 和 S7 中。对患者和对照的单细胞测序分析获得了同济医院机构审查委员会(TJ-IRB20220101 和 TJ-IRB20190502)的批准。
CAR T 细胞制造¶
CT103A 由南京亚索生物技术有限公司制造。使用 CD3 微珠(Miltenyi Biotec)从入选患者的白细胞单采产品中分离 T 细胞,按照制造商的协议进行。这些细胞随后使用 Dynabeads Human T-Activator CD3/CD28(Invitrogen)在 CTS OpTmizer 培养基(Gibco)中激活,补充有 20 mM L-GlutaMAX(Gibco)和重组人 IL-2(200 IU/ml;SL PHARM)。为了生产 CAR T 细胞,T 细胞经过慢病毒载体转导。该载体包含全人抗 BCMA 单链抗体可变片段(scFV)、CD8α铰链和跨膜域、4-1BB 共刺激域和 CD3ζ T 细胞激活域。转导后,细胞在第 5 天洗涤和去珠,然后在 G-Rex(Wilson Wolf)中培养。到第 10 或第 11 天,收集 CAR T 细胞,洗涤并重新悬浮在冷冻保存液中(上海拜特医疗产品有限公司的多种电解质注射液;广东双林生物制药有限公司的人白蛋白;上海拜特医疗产品有限公司的 10% 葡萄糖注射液;费森尤斯卡比华瑞制药有限公司的 18AA-II 复方氨基酸注射液;上海现代哈森商丘制药有限公司的维生素 C 注射液;含有 7.5% 二甲基亚砜的 Origen)。这些准备好的 CAR T 细胞被分装到冷冻袋(Miltenyi Biotec)中,并使用赛默飞世尔科技的 7451TF CRF#4 冷冻至 -90°C。最终产品存储在气相液氮罐(BIOBANK-22K)中,温度低于 -130°C。
样本收集和处理¶
单细胞 RNA 测序(scRNA-seq)由 OE Biotech 有限公司(上海,中国)进行。简而言之,NMOSD 患者在基线和输注后 3 个月以及对照个体的血液和 CSF 样本新鲜收集并立即处理用于 scRNA-seq 和 V(D)J 测序,使用 10x Genomics 平台。使用 Ficoll-Paque PLUS(Cytiva)和密度梯度离心法获得 PBMC,按照制造商的说明进行操作。CSF 样本以 300g 离心 10 分钟。沉淀的细胞重新悬浮在 60μl 的磷酸盐缓冲盐水中。然后将单细胞悬液加载到 10x Chromium 平台上,随后进行 5' 基因表达库和免疫库的准备。所有文库随后在 NovaSeq 6000(Illumina,圣地亚哥,美国)上进行 2×150 配对末端读取测序以获取序列信息。
在输注前收集五名患者的 CAR T 细胞 IP 和输注后 1、2、3 和 4 周的血液样本,并进行 CITE-seq 处理。使用含有人 TruStain FcX(BioLegend)的抗体染色缓冲液对 PBMCs 进行封闭,然后用标记有荧光素异硫氰酸酯(FITC)的人的 BCMA/TNFRSF17 蛋白(Acrobiosystems)染色。洗涤三次后,将细胞与抗 CD4、抗 CD8A 和抗 FITC 抗体及同型对照(BioLegend TotalSeq 目录号 300567、301071、408311 和 400283)在 4°C 下孵育 30 分钟。染色后的细胞洗涤三次并封装进行 CITE-seq。单细胞悬液加载到 10x Chromium 平台上,随后进行 5' 基因表达库、免疫库和细胞表面蛋白库的准备。所有文库按上述方式进行测序。
在输注前收集五名 MM 患者的 IP 和四个可用的基线血液样本。每个样本的细胞悬液分裂、用“哈希抗体”条形码标记并与标签寡核苷酸(HTOs)结合。孵育后,将九个分装样本进行哈希处理并合并为三个对象。然后将哈希样本用 CITE-seq 抗体染色,并加载到 10x Chromium 平台上进行文库构建,如上所述。
数据处理用于 scRNA-seq、CITE-seq 和单细胞 V(D)J 测序¶
原始 RNA 数据和抗体衍生标签序列在 Cell Ranger v5.0.0 中使用 Cell Ranger mkfastq 管道进行去重和比对,其中进行了读取比对、数据过滤、条形码和唯一分子标识符(UMI)计数以及假定细胞的识别。然后在 R 中使用 Seurat v4.2.0 包对通过该管道生成的特征条形码矩阵进行分析,如下所述。对于 HTO 数据,使用 Seurat v4.2.0 中的 HTODemux 方法根据建议的分析管道将细胞去重到其原始样本来源。BCR/TCR 序列通过 Cell Ranger vdj 识别,然后使用 Loupe VDJ 浏览器 v3(10x Genomics)进行分析。
Seurat 工作流程用于 scRNA-seq、CITE-seq 和单细胞 V(D)J 测序¶
Seurat v4.2.0【43】用于下游分析。简而言之,细胞过滤保留至少具有 200 个基因,UMI 在 1000 到 40000 之间,新颖度评分(log10GenesPerUMI)高于 0.7,线粒体 RNA 少于 10% 的细胞。在少于三个细胞中检测到的基因也被过滤掉。然后对每个样本应用 DoubletFinder v2.0.3【44】以去除双重体。质量控制后,将所有样本合并以执行对数归一化、高度可变基因的识别、z 得分缩放和主成分分析。为了消除批次效应,我们使用 Harmony v0.1.0【45】。使用 Harmony 生成的降维(dims = 1:30)计算 UMAP 可视化。使用 Seurat 中的 FindNeighbors 和 FindClusters 函数以 0.8 的分辨率进行聚类。根据已知的标记基因(CD3E、CD4、CD8A、KLRC1、MKI67、MS4A1、MZB1、S1008A 和 LYZ)对簇进行注释。使用 CITE-seq 蛋白标记(CD4、CD8A 和 BCMA-FITC)对 CAR T 细胞进行额外标记,其表达主要与 RNA 表达一致。
在输注后 3 个月收集的血液和 CSF 样本中的 CAR T 细胞通过识别映射到 5' CAR 序列的 scRNA-seq 读取来识别,该序列使用 Cell Ranger mkref 管道集成到人类参考基因组中。此外,将克隆型信息添加到元数据中。含有 BCR 和 TCR 序列的细胞被移除。对于 TCR 克隆型追踪,使用α链或β链克隆型识别基线的内源性 T 细胞、IP 中的 CAR T 细胞以及治疗后不同时点的分选 CAR T 细胞之间的重叠。
基因富集和通路分析¶
为了说明指示细胞组之间的潜在差异功能,使用 GSVA v1.44.0 包【46】基于从 MsigDB v7.5.1 下载的基因集进行基因富集和通路分析。为了评估感兴趣通路的差异活动,分别为每个样本计算 ssGSEA 分数,然后使用 limma v3.52.0【47】识别两个特定簇之间显著差异的通路,阈值为|t 值|> 2。使用条形图可视化显著差异通路的结果。
细胞发育轨迹和拟时间分析¶
使用 Monocle v2.24.1【48】分别推断 B 细胞、PBs/PCs 和单核细胞的细胞谱系轨迹。首先按照 Monocle2 教程创建一个 CellDataSet 对象,并使用参数“min_expr = 0.1”过滤低质量细胞。使用“differentialGeneTest”函数导出每个细胞簇的 DEGs,并使用 q 值<0.01 的基因进行拟时间分析的细胞排序。
CSF 和外周 B 细胞 Ig 库重叠分析¶
基于 scRNA-seq 和单细胞 V(D)J 测序的数据,根据之前报道的定义【15】,将每个 NMOSD 患者的 CSF 和外周 B 细胞 Ig 库视为相关:(i) 相同的 VJ 基因片段,(ii) 相同的 CDR3 长度,以及 (iii) ≥75% 匹配的 CDR3 核苷酸序列。分别计算每种细胞类型的重叠,并显示在表 S2 中。
特征分数计算¶
使用 Seurat 中的 AddModuleScore 函数计算每个细胞与增殖、细胞毒性、炎症、记忆、功能失调、凋亡、共刺激、ATP 生物合成、ATP 代谢、糖酵解、氧化磷酸化(OXPHOS)、能量代谢、Treg 身份、NK 受体表达、耗竭、T 细胞趋化性、IFN 信号、NF-κB 信号、TNF 信号、M 期、细胞周期和细胞运动相关的基因的签名分数,并基于先前报道的相关基因表达进行定义【22, 33, 49-52】。这些分数用作指示器,评估每个细胞对这些特征的潜在贡献。
细胞 - 细胞相互作用分析¶
使用 CellChat v1.4.0【53】推断 PBs/PCs 与其他细胞类型之间的细胞 - 细胞相互作用,遵循官方教程。简而言之,上传表达矩阵的标准化计数以创建 CellChat 对象。下一步,识别过表达的配体 - 受体相互作用。基因表达被投射到相互作用网络上。使用 CellChat 的 computeCommunProb 函数计算通信概率,默认参数控制群体大小。我们过滤了少于 10 个细胞的通信。接下来,使用默认参数计算信号通路水平和聚合网络的通信概率。为了在 CSF 中创建三维伪空间【35】,我们使用了生物信息学工具 CSO_map【35】。我们为每个数据集选择不同细胞类型之间显著的相互作用(q 值<0.1)。
NMOSD 患者先前报道的 scRNA-seq 数据的重新分析¶
我们重新整合并重新分析了最近报告中的四个 CSF 样本和五个 NMOSD 患者血液样本的单细胞转录组数据【17】,以验证我们的结果。根据文献【17】,去除少于 600 个基因和超过 10% 线粒体 RNA 的细胞。后续过程与上述 Seurat 和 Monocle 工作流程一致。
来自文献的 CAR T 细胞相关 scRNA-seq 数据集的综合分析¶
从基因表达综合数据库(GEO 访问号:GSE197851 和 GSE197268)获取先前发布的数据集的基因表达矩阵,包括健康供体生成的 IPs【26】、CD19 靶向 IPs(Axi-cel 和 Tisa-cel)以及基线时采集的相应 PBMCs【25】。去除少于 200 个基因和超过 10% 线粒体 RNA 的细胞。按照上述数据处理流程进行双重体过滤和样本级别的 harmony 批次校正,以识别常见群体。使用 FindAllMarkers 函数识别两个非配对比较组的 DEGs,并使用 ggplot2 进行可视化。
流式细胞术¶
为了分析 CAR T 细胞的频率,使用 FACSCanto II 流式细胞仪(BD Biosciences;NOVOCYTE-D3000 仪器)和以下抗体进行流式细胞术:过氧化物叶绿素蛋白(PerCP)- 标记的人 CD45(BD Biosciences,347464),藻红蛋白(APC)-Cy7- 标记的人 CD3(BioLegend,344818),FITC- 标记的人 BCMA Fc 标签蛋白(Acrobiosystems,BCA-HF254)。为了分析 IPs 中 CAR T 细胞的细胞比例,使用全光谱流式细胞术,使用以下抗体和试剂:BV421 标记的人 CD3(BioLegend,300434),BV605 标记的人 CD62L(BD Biosciences,562719),BV750 标记的人 CD4(BioLegend,344644),PE 标记的人 CD44(BD Biosciences,550989),PE/CF594 标记的人 CCR7(BD Biosciences,562381),PE-Cy5.5 标记的人 GZMB(Invitrogen,GRB18),PE-Cyanine7 标记的人 CD45RA(BioLegend,304126),Alexa Fluor 700 标记的人 Ki67(BD Biosciences,561277),APC-H7 标记的人 CD27(BD Biosciences,560222),APC/Fire 810 标记的人 CD8 抗体(BioLegend,344764),FITC- 标记的人 BCMA Fc 标签蛋白(Acrobiosystems,BCA-HF254),和 Fixable Aqua Dead Cell Stain Kit(Invitrogen,L34966)。所有抗体均按制造商推荐的浓度使用。详细信息见数据 S5。使用 Cytek Northern Lights-CLC 进行数据采集,并使用 SpectroFlo 1.4.1 和 Flowjo 10.8.1 进行分析。
Ingenuity 通路分析¶
为了表征特定簇的差异分子和细胞功能特征,将 DEGs 及其相应的 P 值和倍增变化值导入 IPA 软件(QIAGEN)进行核心和比较分析。使用 Ingenuity 知识库作为参考,并通过 z 得分表示特定经典通路的激活或抑制水平。
液滴数字聚合酶链反应¶
在不同时间点进行 CAR T 细胞输注前后进行载体拷贝数分析。将裂解缓冲液(BD Biosciences)加入收集的血液样本,然后使用 DNA Blood Mini Kit(目录号 51104;QIAGEN,Valencia,CA)提取基因组 DNA。在指示时间点使用探针和引物靶向 scFV 序列进行液滴数字聚合酶链反应(ddPCR)测量每个样本的 CAR 拷贝数,如之前报道的那样【54】。
成像流式细胞术分析¶
成像流式细胞术用于显示 CAR T 细胞与血液和 CSF 中的靶细胞之间的直接接触。按照上述方法收集 PBMCs 和 CSF 样本,并用 100μl 荧光激活细胞分选(FACS)缓冲液(BD Biosciences,目录号 554656)重悬。然后加入 Fc block 并在冰上孵育 10 分钟。加入抗体(PE 标记的人 CD3,FITC 标记的 BCMA 蛋白和 APC 标记的人 CD138)并在黑暗中孵育 30 分钟。详细的抗体信息见数据 S5。洗涤和离心后,将细胞重悬于 50μl FACS 缓冲液中。使用 AMNIS ImageStreamX Mark II 流式细胞仪(AMNIS/Luminex,西雅图,WA,美国)进行可视化。
Luminex 检测¶
按照制造商的说明使用 Luminex 检测(R&D Systems)评估 CSF 样本中的细胞因子和趋化因子水平。检测的人细胞因子和趋化因子包括:CCL1、CCL3、CCL19、CCL22、CCL24、CX3CL1、CXCL5、CXCL8、CXCL9、CXCL10、CXCL13、IL-2、IL-6、IFN-γ和 TNF-α。
酶联免疫吸附测定¶
使用人 BCMA/TNFRSF17 DuoSet ELISA Kit(R&D Systems,DY193)、人 TREM2 ELISA Kit(Abcam,ab224881)、人补体 C3 ELISA Kit(Abcam,ab108823)和人补体 C5a ELISA Kit(Abcam,ab125963)按照制造商的说明测量 CSF 中的这些标志物水平。
CSF NFL 检测¶
使用超敏 Simoa 技术(Quanterix,MA,美国)在自动化 Simoa HD-X 平台上按照制造商的说明量化 CSF 中的 NFL 水平。
统计分析¶
使用 GraphPad Prism 8.4.1 版和 R 4.2.0 版进行统计分析。除非另有说明,否则数据以均值±SD 表示。使用未配对学生 t 检验、Mann-Whitney U 检验或一元方差分析(ANOVA),在各个图例中指示,使用事后 Tukey 检验进行多重比较校正。所有统计检验均为双尾检验,显著性水平为 0.05。