Single cell and spatial transcriptomics analysis of non small cell lung cancer
摘要¶
肺癌是第二大常见的癌症,也是全球癌症相关死亡的主要原因。肿瘤生态系统包含多种类型的免疫细胞,其中髓系细胞尤为常见,并在促进疾病方面发挥了重要作用。在我们的研究中,通过单细胞和空间转录组学,我们对来自 25 名未接受治疗的腺癌和鳞状细胞癌患者的约 90 万个细胞进行了分析。我们注意到抗炎巨噬细胞与 NK 细胞/T 细胞之间存在逆相关关系,且肿瘤内 NK 细胞的细胞毒性降低。虽然在腺癌和鳞状细胞癌中观察到类似的细胞类型组成,但我们发现各种免疫检查点抑制剂的共表达存在显著差异。此外,我们揭示了肿瘤中巨噬细胞的转录“重编程”证据,使它们转向胆固醇输出并采用类似胎儿的转录特征,从而促进铁外排。我们的多组学资源提供了肿瘤相关巨噬细胞的高分辨率分子图谱,增强了我们对其在肿瘤微环境中作用的理解。
引言¶
肺癌是全球第二常见的癌症,也是癌症死亡的首要原因,最晚期患者的 5 年生存率约为 6%。非小细胞肺癌(NSCLC)是最常见的肺癌类型(约占总病例的 85%),其次是小细胞肺癌(15%)。肺癌是一种复杂的疾病,肿瘤微环境在其中起着关键作用,而巨噬细胞(Mɸ)与疾病的进展密切相关。特别是肿瘤相关巨噬细胞(TAMs)可以表现出双重作用,通过抑制免疫反应、促进血管生成和帮助组织重塑来促进肿瘤,但也可以通过促进炎症和参与对癌细胞的细胞毒性活动来抑制肿瘤。肺癌与巨噬细胞之间错综复杂的相互作用突显了了解其动态关系的重要性,以便开发更有效的治疗策略。
在 NSCLC 中,腺癌(LUAD)是最常见的组织学亚型,其次是鳞状细胞癌(LUSC)。肺叶切除术(即解剖性肺叶切除)目前是早期 NSCLC(I 期/II 期)治疗的金标准,而对于不可切除的 III 期或转移性 IV 期 NSCLC 患者,则采用化疗和新辅助靶向血管内皮生长因子(VEGF)或免疫检查点抑制剂(ICIs)如 PD1、PDL1 和 CTLA4 的组合治疗。过去十年中在发现预测性生物标志物方面的进展为基于肿瘤组织学和 PDL1 表达的靶向治疗和免疫治疗领域带来了新的治疗前景。
许多研究利用单细胞技术探索了 NSCLC 中的转录变化。他们深入研究了肺肿瘤微环境,揭示了与患者预后相关的多样化 T 细胞功能、B 细胞多样性在 NSCLC 中对抗肿瘤治疗的重要性、肿瘤浸润髓系细胞的多种状态,并提出这些细胞是免疫治疗的新靶点,以及组织驻留中性粒细胞与抗 PDL1 治疗失败的关联。他们进一步揭示了晚期和转移性肿瘤的肿瘤异质性和细胞变化,以及肿瘤治疗诱导的癌细胞向原始细胞状态的转变。在许多这些研究中,每个患者分析的细胞数量有限,并且通常没有系统地收集患者匹配的非肿瘤组织,从而限制了对肿瘤和邻近非肿瘤组织中生物异质性的剖析。此外,除了少数例外,LUAD 和 LUSC 通常被视为一个整体,从而阻碍了对这两种在分子和病理水平上截然不同的癌症类型的特定特征的研究。尽管单细胞 RNA 测序(scRNA-seq)可以在组织中识别细胞类型及其状态,但它缺乏定位它们的空间分布或捕捉局部细胞 - 细胞相互作用以及介导这些相互作用的配体和受体的能力。因此,限制了我们全面探索肿瘤微环境(TME)及其中复杂的细胞 - 细胞相互作用的能力。
为了克服上述限制,我们结合了来自 25 名未接受治疗的 LUAD 或 LUSC 患者的约 90 万个细胞的 scRNA-seq 数据和来自 8 名患者的空间转录组学数据,以研究肿瘤和邻近非肿瘤组织中细胞组织的差异。我们进一步研究了巨噬细胞群体及其在肿瘤环境中经历的分子变化,其中一些变化类似于人类胎儿发育期间的巨噬细胞。
结果¶
NSCLC 样本的 scRNA-seq 和空间图谱¶
为了确定 LUAD 和 LUSC 中免疫和非免疫细胞状态的异质性及其空间分布,我们收集了 25 名未接受治疗的 LUAD(n=13)、LUSC(n=8)或未确定肺癌(LC,n=4)患者的肺组织切除样本,以及两名健康的已故捐赠者的样本(图 1A,B 和补充数据 1)。我们收集了肿瘤和匹配的正常非肿瘤组织(即背景),分离了 CD45+ 免疫细胞(补充图 1A)以及肿瘤和其他非免疫群体(使用 CD235a 柱去除红细胞),并进行了 scRNA-seq。此外,来自上述 25 名患者中的 8 名患者的肿瘤和背景组织切片被用于使用 10x Genomics Visium 平台进行空间转录组学处理(总共 n=36 个切片)(图 1A 和补充数据 1)。
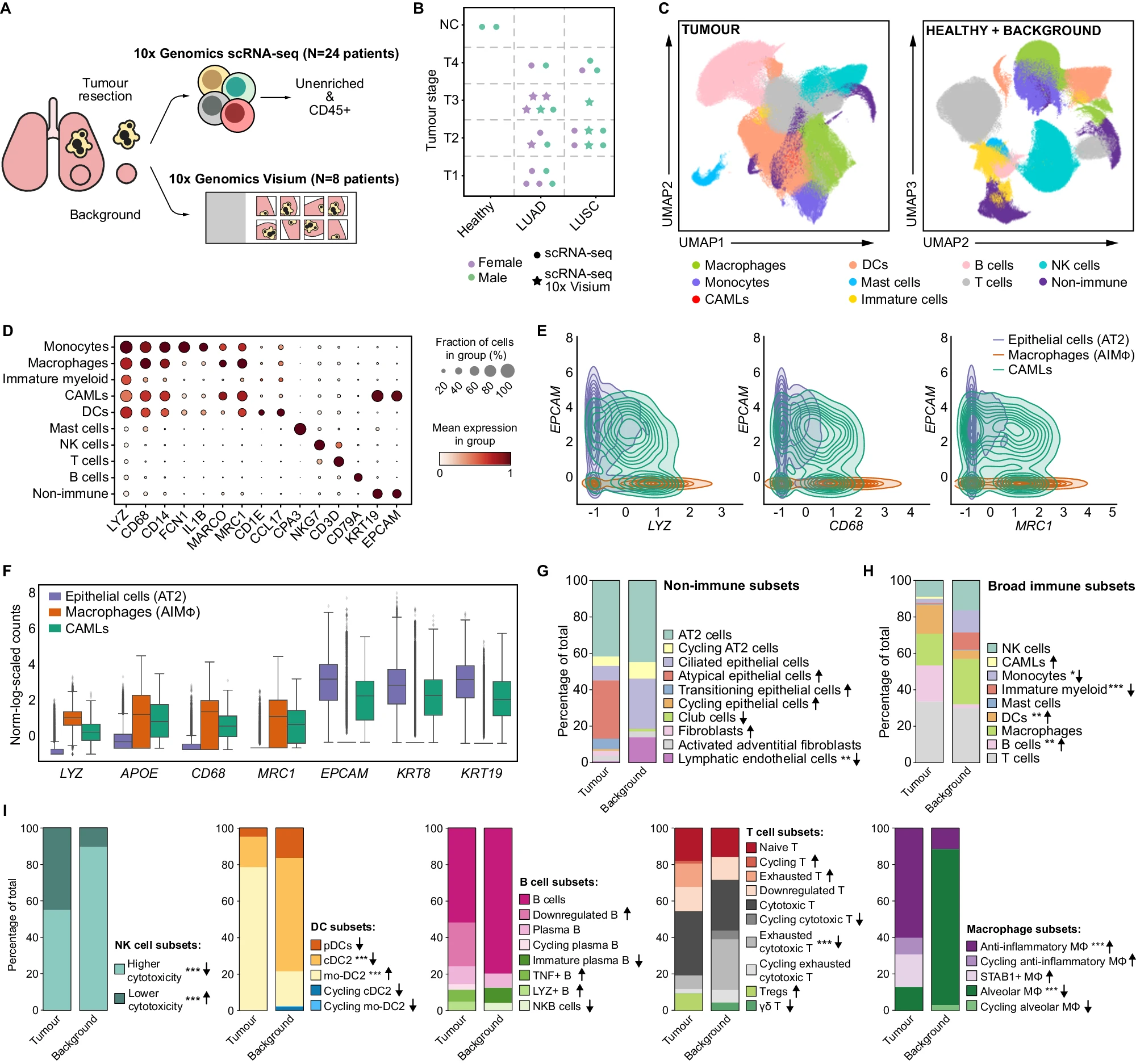
研究概述¶
A. 研究概述。对切除的肿瘤组织、邻近未受影响的组织(背景)和已故捐赠者的健康肺进行单细胞悬浮液处理,富集 CD45+ 或 CD235− 细胞并进行单细胞 RNA 测序(scRNA-seq)。将新鲜、快速冷冻的肿瘤、背景和健康组织的冷冻切片用于 10x Visium 空间转录组学分析。 B. 队列概述。符号代表个体患者和进行的分析 C. 肿瘤和组合的背景 + 健康数据集的 UMAP 投影。 D. 用于肿瘤样本中广泛细胞类型注释的代表性基因的点图。 E. 等高线图显示 AT2 细胞(44,399 个细胞)、CAMLs(2520 个细胞)和 AIMɸ(16,120 个细胞)中髓系(LYZ, CD68, MRC1)和上皮(EPCAM)基因的共表达。基因表达已归一化、缩放和对数转换。 F. 箱线图显示 AT2 细胞、CAMLs 和 AIMɸ 中髓系(LYZ, APOE, CD68, MRC1)和上皮(EPCAM, KRT8, KRT19)基因的归一化、缩放和对数转换后的基因表达。箱子表示四分位数,须线表示 1.5 倍的四分位距。 G. 计算在 CD235− 富集中的肿瘤和背景中的非免疫细胞亚群的相对比例。箭头表示肿瘤相对于背景的增加(↑)或减少(↓)。通过双侧 Wilcoxon 秩和检验和 Bonferroni 校正进行成对比较。**P < 0.01。没有星号的箭头表示该细胞类型仅在肿瘤或背景中发现 H. 计算在 CD235− 富集中鉴定的所有免疫细胞中的肿瘤和背景中的广泛免疫细胞的相对比例。箭头表示肿瘤相对于背景的增加(↑)或减少(↓)。通过双侧 Wilcoxon 秩和检验和 Bonferroni 校正进行成对比较。*P < 0.05,**P < 0.01,**P < 0.001。没有星号的箭头表示该细胞类型仅在肿瘤或背景中发现。 I. 计算在 CD235− 富集中的肿瘤和背景中的广泛注释中的 NK、DC、B、T 和巨噬细胞亚群的相对比例。箭头表示肿瘤相对于背景的增加(↑)或减少(↓)。通过双侧 Wilcoxon 秩和检验和 Bonferroni 校正进行成对比较。**P < 0.001。没有星号的箭头表示该细胞类型仅在肿瘤或背景中发现。
肿瘤相比于邻近肺组织表现出更高的免疫和非免疫细胞多样性¶
在对 scRNA-seq 数据集进行质量控制(QC)后,我们总共识别出 895,806 个高质量细胞,其中 503,549 个来自肿瘤,392,257 个来自组合的背景和健康组织(以下简称 B/H)。在进行归一化和 log1p 转换、高度可变基因选择、降维、批次校正和 Leiden 聚类后,分别对来自肿瘤和 B/H 的细胞进行广泛的细胞类型注释,并通过统一流形近似和投影(UMAP)进行可视化(图 1C,补充图 1B,C 和“方法”)。我们识别出了具有单核细胞、巨噬细胞、树突状细胞(DCs)转录特征的髓系细胞簇,以及肥大细胞、自然杀伤细胞(NK 细胞)、T 细胞、B 细胞和非免疫细胞(图 1C,D)。我们没有检测到中性粒细胞,可能是由于它们在采集后以及特别是在冷冻 - 解冻循环中对降解的敏感性。最后,我们识别出了一个特征为髓系(LYZ, CD68, CD14, MRC1)和上皮基因(KRT19, EPCAM)共表达的簇(图 1D-F)。这些细胞存在于肿瘤中,并表现出与先前描述的癌症相关巨噬细胞样细胞(CAMLs)相似的特征。CAMLs 代表一种具有同时表达上皮肿瘤蛋白的大型髓系细胞的独特群体。这些独特的细胞已在包括 NSCLC 在内的各种恶性肿瘤患者的血样中观察到。CAMLs 的丰度与治疗干预的反应直接相关,突显其功能意义。即使在进一步的亚聚类中,CAMLs 仍保持其独特的髓系 - 上皮双重特征(补充图 1D)。值得注意的是,双细胞检测软件 Scrublet 为 CAMLs 分配了较低的双细胞评分,表明其表达特征不太可能是肿瘤细胞和巨噬细胞偶然测序产生的组合特征(补充图 1E)。所有簇均包含来自多个患者的细胞,簇的大小范围从 2520 到 124,459 个细胞(补充图 1F,G)。此外,我们使用 scArches 进行了参考查询映射,以确认我们在肿瘤和 B/H 数据集中的注释一致性(补充图 2A-C 和补充说明)。
免疫和非免疫成分的组成在肿瘤和背景之间显著不同。在肿瘤中,我们检测到成纤维细胞和淋巴内皮细胞(LECs)的比例减少(Padj = 0.0025,图 1G 和补充数据 2)。此外,上皮细胞群体表现出更高的多样性,包括在肿瘤组织中存在的肺泡 II 型(AT2)、下调上皮标志物(KRT19, EPCAM, CDH1)的非典型上皮细胞、上调髓系标志物(LYZ)的过渡性上皮细胞和循环上皮细胞(图 1G,补充说明和补充图 2D,E)。这些差异与肿瘤标本中上皮细胞可能是突变肿瘤细胞和非突变正常细胞的混合物的事实一致,并且表明肿瘤的转化导致了细胞状态的进一步多样性。我们未检测到肺泡 I 型(AT1)或基底细胞,可能是由于在解离过程中它们的丢失,正如其他人之前报道的那样。
如前所述,与背景相比,肿瘤样本中单核细胞和未成熟髓系细胞的比例显著减少(分别为 Padj = 0.022 和 Padj = 0.00001),而 DCs 和 B 细胞整体增加(分别为 Padj = 0.0023 和 Padj = 0.0044;图 1H 和补充数据 3)。为了进一步了解肿瘤与背景组织的细胞组成差异,我们对每个广泛簇进行亚聚类,并识别出 46 种细胞类型/状态(补充图 2D,E,补充数据 4 和 5,补充图 3 和补充说明)。在肿瘤中,我们发现显著更高比例的 NK 细胞具有较低的细胞毒性表型(补充说明),而且大多数 DCs 来自单核细胞(即 mo-DC2),(补充说明)与背景相比(分别为 Padj = 0.00002 和 Padj = 0.00002,图 1I 和补充数据 6)。这与炎症条件下 mo-DC2 的单核细胞起源一致。同样,我们发现表达 LYZ 和 TNF 的 B 细胞扩展,NKB 细胞减少(图 1I 和补充说明)。在 T 细胞中,肿瘤样本显示调节性 T 细胞(Tregs)积累,已知它们会阻碍肿瘤的免疫监视(图 1I)。相反,肿瘤中耗竭的细胞毒性 T 细胞减少(Padj = 0.00002),并且缺乏与 NSCLC 生存相关的 γδT 细胞(图 1I 和补充数据 6)。γδT 细胞能够识别和裂解各种癌细胞,因此被建议在全癌种免疫治疗中发挥作用。最后,我们观察到抗炎 Mɸ(AIMɸ)的异质性和比例增加,循环抗炎 Mɸ、STAB1 + Mɸ(图 1I)和 CAMLs(图 1H)在肿瘤组织中大量存在。有趣的是,我们发现跨患者的 STAB1 + Mɸ/AIMɸ 和 T/NK 细胞频率之间存在强烈的负相关性,突显了 Mɸ 在限制细胞毒性细胞浸润肺肿瘤组织中的关键作用(图 2A)。这与最近的一项研究描述的人类 NSCLC 中单核细胞衍生的 Mɸ 获得免疫抑制表型并限制 NK 细胞浸润的发现一致。
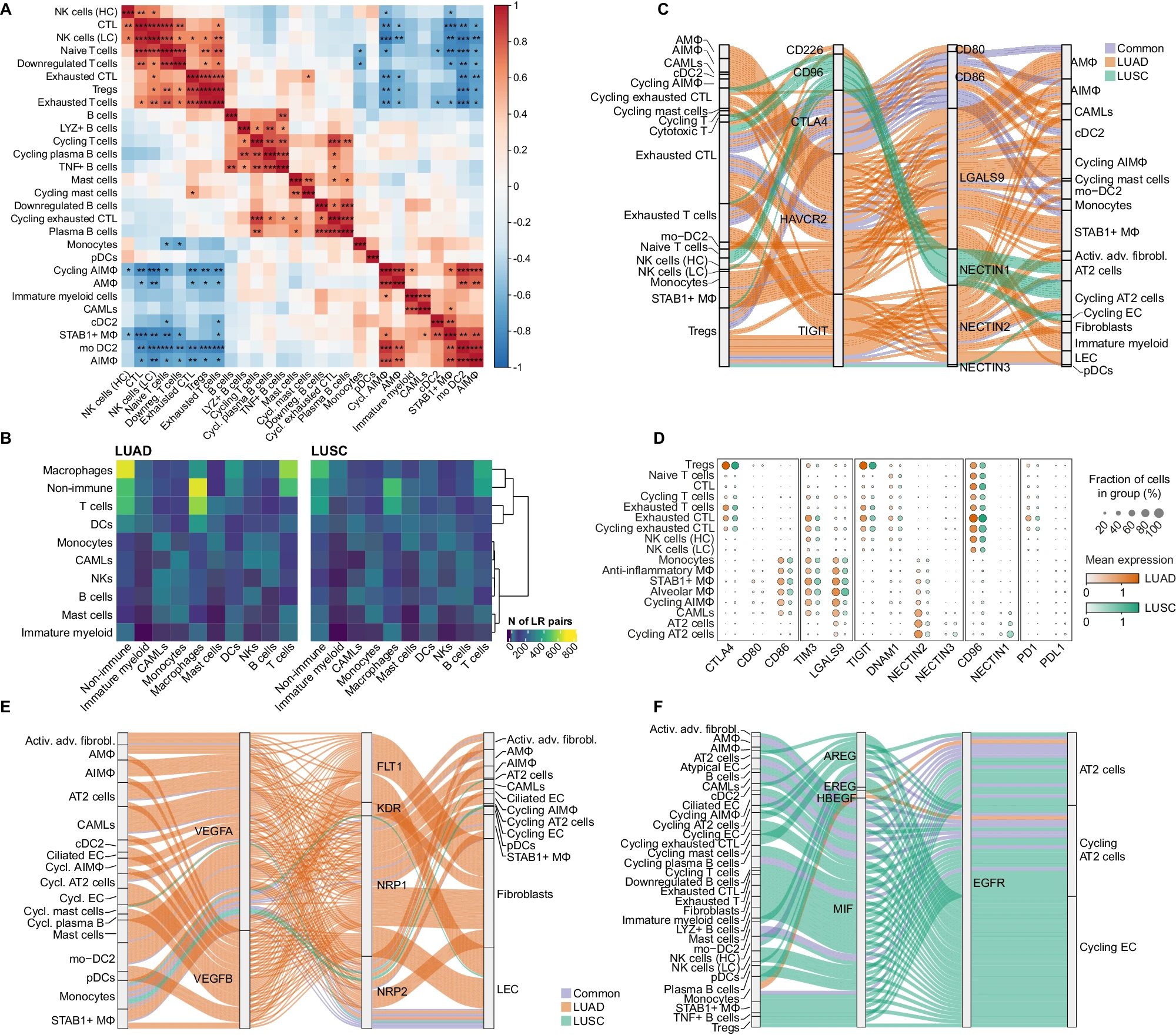
A. 热图显示每种免疫细胞类型的相对细胞类型丰度之间的 Pearson 相关性(在 CD235− 富集中计算)。颜色表示 Pearson 相关值,星号表示基于 Pearson 乘积矩相关系数计算的双侧关联检验的显著性水平(*P < 0.05,P < 0.01,*P < 0.001)。
B. 热图显示在 LUAD(左)和 LUSC(右)中所有细胞类型之间的 LR(配体 - 受体)相互作用数量,按广泛细胞注释总结。行使用欧几里得距离的完全链接法进行层次聚类。
C. Sankey 图显示 CellphoneDB 检测到的 LUAD 和 LUSC 中特定肿瘤的选定 ICIs(免疫检查点抑制剂)相互作用。线条颜色表示 LUAD 中发现的 LR 相互作用(橙色)、LUSC 中发现的 LR 相互作用(绿色)或两种肿瘤类型中均发现的 LR 相互作用(蓝色)。
D. ICI 基因和(C)中突出显示的细胞类型的点图,按肿瘤类型分割。每个点的大小表示簇中表达基因的细胞百分比,颜色表示每组中每个基因的平均归一化、缩放和对数转换表达。
E. Sankey 图显示 CellphoneDB 检测到的 LUAD 和 LUSC 中 VEGFA/B 相互作用的肿瘤特异性相互作用。线条颜色表示 LUAD 中发现的 LR 相互作用(橙色)、LUSC 中发现的 LR 相互作用(绿色)或两种肿瘤类型中均发现的 LR 相互作用(蓝色)。
F. Sankey 图显示 CellphoneDB 检测到的 LUAD 和 LUSC 中 EGFR 相互作用的肿瘤特异性相互作用。线条颜色表示 LUAD 中发现的 LR 相互作用(橙色)、LUSC 中发现的 LR 相互作用(绿色)或两种肿瘤类型中均发现的 LR 相互作用(蓝色)。
LUAD 和 LUSC 具有相似的细胞组成但利用不同的细胞间相互作用网络¶
LUAD 和 LUSC 具有非常不同的预后,通常被认为是不同的临床实体。为了检查临床特征差异是否源于不同的细胞组成,我们比较了 LUAD 与 LUSC 患者 CD235- 样本中免疫细胞和非免疫细胞亚群的频率。我们观察到细胞频率的微小差异,但在 P 值校正后未达到统计显著性(补充图 4A 和补充数据 7 和 8)。此外,患者观察到的免疫细胞和非免疫细胞频率与癌症亚型、癌症分期或性别之间没有明显关联(补充图 4B,C),这表明 LUAD 和 LUSC 的 TME 组成相当相似。尽管 LUAD 和 LUSC 具有相似的细胞组成,但观察到的临床差异可能源于不同的细胞间相互作用。因此,我们检查了 LUAD 与 LUSC 的 TME 中是否使用了不同的细胞间相互作用网络。为此,我们通过推断在背景或健康组织中未检测到的、具有统计显著性的配体 - 受体(L-R)对及其对应的细胞类型,识别出在每种肿瘤类型环境中独有的细胞间相互作用清单。
虽然这两种肿瘤亚型显示了类似的相互作用网络,主要涉及非免疫细胞、AIMɸ 和 T 细胞之间的相互作用,但也有一些显著差异。
首先,我们在 LUAD 数据集中总体上识别出更多的 L-R 对,这并不是由 LUAD(n = 105,749 个细胞)和 LUSC(n = 230,066 个细胞)数据集中细胞数量的差异所驱动的。其次,一些免疫检查点抑制剂(ICI)及其各自的抑制分子在 LUAD 和 LUSC 中的共表达有所不同。例如,LGALS9-HAVCR2(TIM3)、NECTIN2-CD226(DNAM1)和 NECTIN2/NECTIN3-TIGIT 在 LUAD 中经常被识别,而推定的 ICI CD96-NECTIN1 则在 LUSC 中优先发现。相比之下,CD80/CD86-CTLA4 和 HLAF-LILRB1/2 在两种肿瘤亚型中均被发现。LILRBs(白细胞 Ig 样受体)作为下一代免疫治疗的潜在靶点,正在受到关注,因为其阻断可以增强免疫反应。最常用的肺癌免疫疗法阻断 PD1 和 PDL1 之间的相互作用,最近的临床试验表明,抗 CTLA4 和抗 PD1 的组合疗法提高了患者的生存率,而与肿瘤 PD1 表达无关。在我们的数据集中,我们未观察到任一肿瘤亚型中的 PD1-PDL1 相互作用。我们的初步分析表明,其他 ICIs(如 CTLA4、TIGIT、LILRB1/2 和 TIM3)可能是治疗 NSCLC 的有希望的靶点。
在 LUAD 和 LUSC 中检测到的显著 L-Rs 中,我们注意到几对参与血管生成信号传导的髓系细胞群体,例如 VEGFA/B-FLT1、VEGFA-KDR 和 VEGFA-NRP1/2。尽管在 LUAD 和 LUSC 中均发现了 VEGFA 和 VEGFB 的表达,但其受体在 LUAD 中更频繁地被发现,特别是在成纤维细胞中。类似地,我们观察到在 AT2 和循环上皮细胞中,EGFR 配体信号显著表达,例如 EGFR-EREG、EGFR-AREG、EGFR-HBEGF 和 EGFR-MIF,尽管 MIF 在 LUSC 细胞中更频繁地表达。最后,我们观察到支持淋巴细胞激活所需的关键共刺激信号,例如 CD40-CD40LG、CD2-CD58、CD28-CD86、CCL21-CCR7 和 TNFRSF13B/C-TNFSF13B(TACI/BAFFR-BAFF),这些信号通常与主要由 B 细胞、T 细胞和 DCs 组成的异位淋巴器官(即三级淋巴结构)有关。TLS 通常与 NSCLC 中的较长无复发生存期相关。
整合 scRNA-seq 和空间转录组验证原位 L-R 相互作用¶
显著的 L-Rs 及其相互作用细胞类型是基于不同细胞类型簇中基因共表达通过 CellPhoneDB 计算得出的。然而,为了辨别生物学上显著的相互作用,必须确定被识别为相互作用的细胞类型确实在空间上共定位。为此,我们考虑了 scRNA-seq 识别的细胞类型在组织切片上的空间排列。我们采用了一种综合方法,将肿瘤和背景样本的 scRNA-seq 与新鲜冷冻肿瘤和背景组织切片的空间转录组(STx)配置文件结合起来。我们对来自 8 名患者的两张连续的 10 微米切片进行了 10× Visium 处理,其中 7 名患者的样本与用于 scRNA-seq 的样本匹配。我们总共分析了 36 张切片(肿瘤 n = 20,背景 n = 16),肿瘤中的平均 UMI 计数为 6894/spot,背景为 3350/spot。接下来,我们使用 cell2location 和来自我们的 scRNA-seq 数据集的细胞类型特异性表达谱对组织上的细胞类型丰度进行解卷积(图 3A,参见“方法”)。
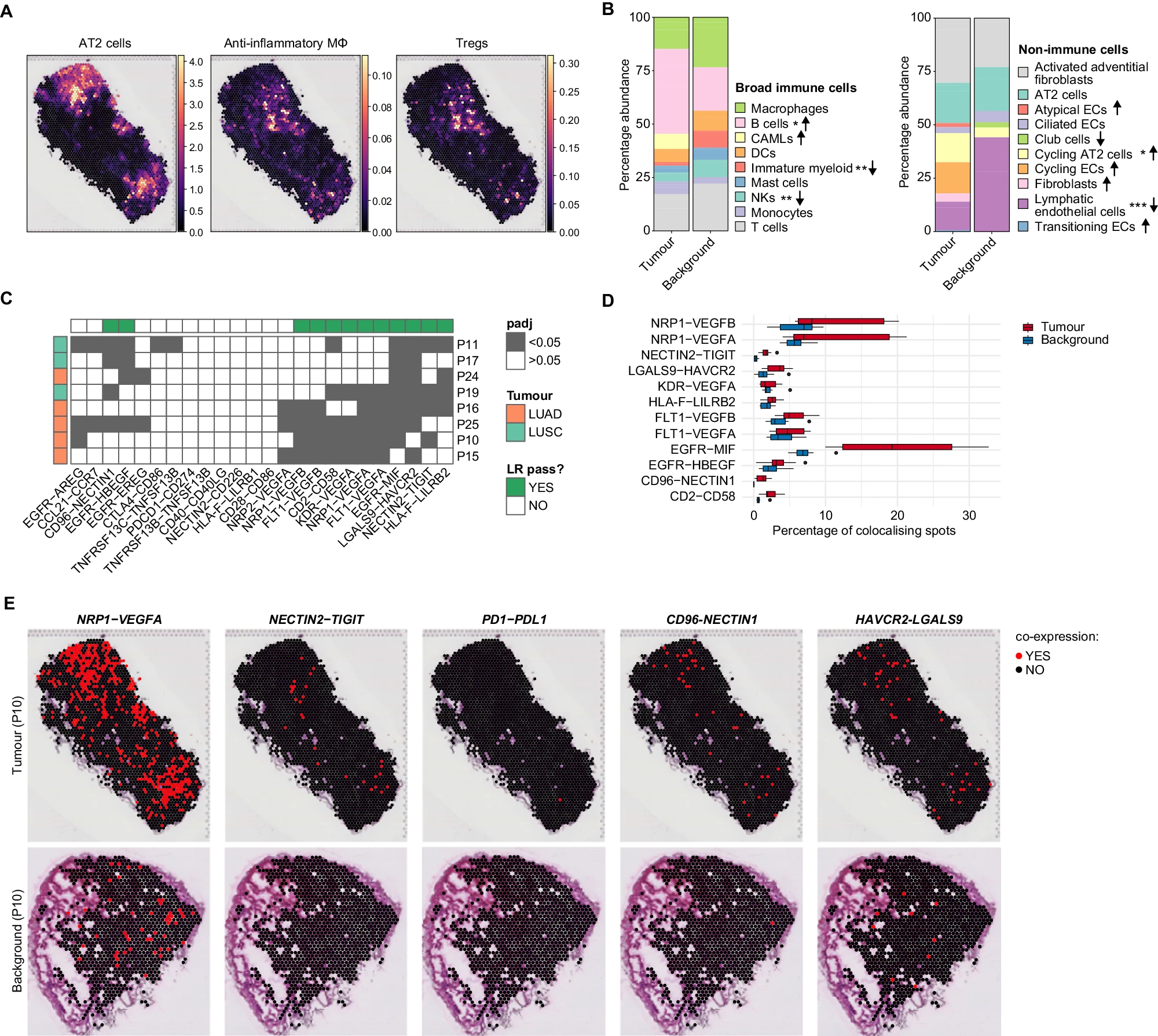
A. 空间图像显示通过 cell2location 估算的 AT2 细胞、AIMɸ 和 Tregs 在代表性肿瘤切片上的细胞丰度。 B. 基于 cell2location 对肿瘤和背景切片中细胞丰度估算的免疫(左)和非免疫(右)细胞类型的相对比例。免疫细胞根据其广泛注释进行分组。箭头表示肿瘤中相对于背景的增加(↑)或减少(↓)。使用双侧 Wilcoxon 秩和检验和 Bonferroni 多重比较校正进行配对比较。*P < 0.05,**P < 0.01,***P < 0.001。没有星号的箭头表示该细胞类型仅在肿瘤或背景中发现。请参阅补充数据 13 和 14 以获取确切的 P 值。 C. 空间 LR 共定位的热图。在所有切片中,每个点估计 LR 基因对的共表达,并使用 χ² 检验比较肿瘤和背景中共定位与非共定位点的频率,随后进行 Bonferroni 多重比较校正。深灰色方块表示肿瘤和背景切片中共定位基因对频率显著不同。绿色列注释表示在至少八名患者中四名中显著的 LR 对。行注释表示肿瘤类型。 D. 箱线图显示每个分析切片中肿瘤与背景中显著不同的共定位 LR 对的频率。N = 8 名患者。箱线图使用 Python Seaborn 包中的默认设置绘制,即,箱线显示四分位数,须长为四分位距的 1.5 倍。源数据作为源数据文件提供。 E. 空间图像显示在肿瘤(上)和背景(下)中找到的共表达 LR 对的位置,对于 NRP1-VEGFA、NECTIN2-TIGIT、PD1-PDL1、CD96-NECTIN1 和 HAVCR2-LGALS9。来自一名患者的代表性切片。
肿瘤和背景组织中不同细胞类型的分布¶
确定组织切片上的细胞类型后,我们检查了肿瘤和背景组织中所有切片上不同细胞类型的频率。通过汇总通过 cell2location 估算的细胞丰度的后验 5% 分位数(q05)值并在通过 QC 的点上进行汇总,我们计算了肿瘤和背景中的细胞类型丰度(“方法”)。我们的分析证实了肿瘤与背景中所有切片上细胞类型频率的差异与 scRNA-seq 数据中获得的结果一致(图 3B)。例如,在肿瘤中,我们发现 B 细胞(Padj = 0.0372)和循环 AT2 细胞(Padj = 0.0147)的比例增加,而未成熟细胞(Padj = 0.0012)、NK 细胞(Padj = 0.0012)和 LECs(Padj = 0.00077,补充数据 13 和 14)的比例减少。然而,从 scRNA-seq 数据或 STx 数据中估算出的肿瘤或背景中的其他细胞类型的比例显示出一些差异(补充图 4H,I)。这种差异在非免疫群体中特别明显,其中 STx 估算的 LECs、活化外膜成纤维细胞和循环亚群的比例较高,而 scRNA-seq 则较低。之前的研究也表明,不同方法之间的细胞比例差异可能是由于 scRNA-seq 和 STx 技术(如 Visium)的固有取样偏差所致 35,36。在 scRNA-seq 中,细胞消化敏感性的变化可能导致细胞类型的差异表示。而在 Visium 中,差异可能源于肿瘤切除位置的变化以及与 scRNA-seq 研究相比的样本量差异。然而,通过 scRNA-seq 和 Visium 获得的结果的总体一致性表明,我们的不同细胞类型的空间“地图”忠实地反映了它们在组织中的分布。
接下来,我们检查了通过 cellphoneDB 识别的 L-Rs 的空间共定位。如果两个基因在同一个点上表达并且高于该基因在切片点上的中值,则认为 L-Rs 共定位。然后,我们使用χ²检验比较了在匹配的肿瘤和背景切片中 L-R 基因共定位与非共定位点的频率(“方法”)。由于从 LUSC 和 LUAD 患者收集的组织块数量较少(NLUSC = 3,NLUAD = 5),统计功效不足以在 LUAD/LUSC 特异性 L-Rs 的空间定位之间进行比较分析。然而,我们证实了一些上述的肿瘤特异性 L-Rs 在肿瘤切片中比在背景切片中显著更多地共定位,包括 NRP1-VEGFA 和 ICI NECTIN2-TIGIT、LGALS9-HAVCR2 和 CD96-NECTIN1(图 3C-E 和补充数据 15)。与 cellphoneDB 结果一致,我们在肿瘤切片中没有发现 PD1-PDL1 的显著共定位。
CAMLs 与肿瘤细胞共享相似的拷贝数异常(CNAs)¶
从手术切除的肿瘤样本中可以获得恶性和残余正常上皮细胞。人类肿瘤的 scRNA-seq 中的一个重大挑战在于区分癌细胞和非恶性细胞。因此,我们应用肿瘤拷贝数核型分析(CopyKAT37)来识别单个细胞中的全基因组非整倍性。从 scRNA-seq 数据中计算 DNA 拷贝数事件的原理是基于相邻基因的表达水平可以提供有价值的信息,用于推断该特定基因组段内的基因组拷贝数。由于非整倍性在人类癌症中很常见,具有全基因组 CNA 的细胞被视为肿瘤细胞。
使用 CopyKAT 分析显示,肿瘤组织中存在广泛的、患者特异性的 CNA(图 4A 和补充图 5A),但在背景中没有。在个体肿瘤样本中,CNA 在 AT2 和循环 AT2 细胞中检测到,并且在一些患者中,这些基因改变在 AT2/循环 AT2 细胞和非典型上皮细胞之间共享,表明不同上皮亚群之间的紧密谱系关系(图 4A 和补充图 5A)。我们通过推断肿瘤中非血细胞群体的轨迹来证实这一发现,使用基于划分的图抽象(PAGA)38。PAGA 显示了 AT2 细胞、循环 AT2/上皮细胞和非典型上皮细胞之间的一侧分化连续性,而另一侧是纤毛上皮细胞和过渡上皮细胞(图 4B)。此外,盲法组织学评估证实了病理学家定义的肿瘤部位与 cell2location 预测的 AT2 和循环 AT2 细胞之间的重叠,表明它们的肿瘤细胞状态(图 4C)。非典型上皮细胞的重叠较少(图 4C)。AT2 细胞与背景相比的差异表达分析(DEA)显示,肿瘤中的 AT2 细胞在肿瘤中上调了与缺氧、TP53 途径和代谢重构相关的基因。肿瘤中的 AT2 细胞上调了参与糖酵解和氧化磷酸化的基因(图 4D 和补充数据 16)。虽然糖酵解在肿瘤细胞中的重要性已经得到充分证实 39,但最近有报告称,人类 NSCLC 使用葡萄糖和乳酸来燃料三羧酸循环(TCA)40。此外,与背景 AT2 细胞相比,肿瘤 AT2 细胞被注意到更多地表达 LYPD3(log2FC = 2.04,Padj = 0.039,补充数据 16),一种以前与 NSCLC 预后不良相关的粘附蛋白,目前正在进行临床前和临床研究 41,42。
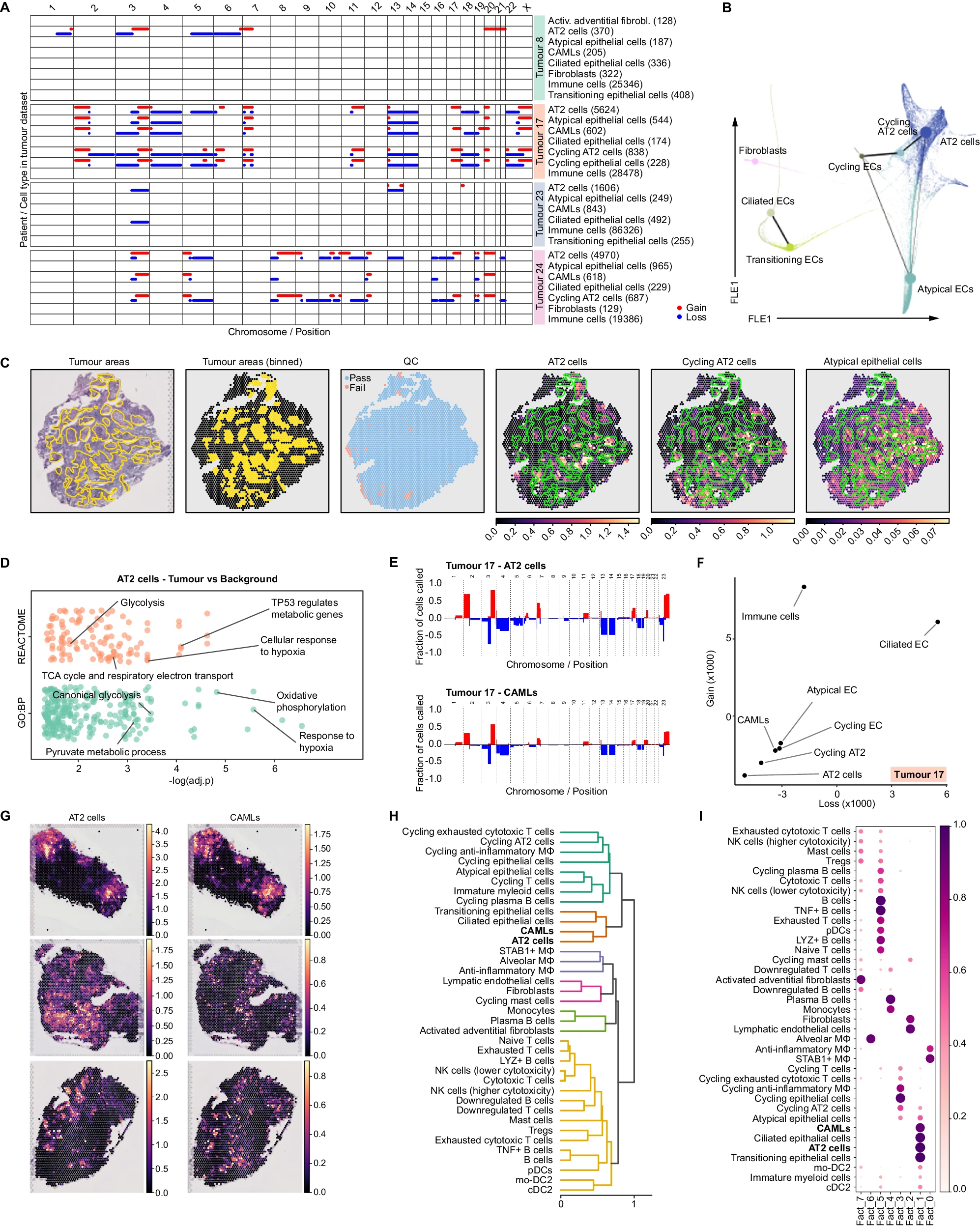
A CNA 分析。该图显示了通过 CopyKat 估算的每条染色体臂中不同细胞类型和肿瘤数据集中患者的染色体增益(红线)和损失(蓝线)。为了绘图目的,所有免疫细胞类型被归为一组。
B PAGA 图覆盖在肿瘤中非免疫细胞类型计算的扩散图(力导向布局—FLE 嵌入)上。
C 前三个面板——合格病理学家的代表性盲注释,显示肿瘤浸润区域(左),Visium 点上的肿瘤区域分区(中)和通过 QC 的点(右)。后三个面板——在同一切片上,cell2location 对 AT2 细胞(左)、循环 AT2 细胞(中)和非典型上皮细胞(右)的估计,覆盖了病理学家对肿瘤浸润的注释(绿色轮廓)。
D 使用 clusterProfiler R 包对基因本体论—生物过程(GO:BP)和 REACTOME 数据库进行过度表现分析,使用肿瘤 vs 背景中 AT2 细胞上调的 DEGs。源数据作为源数据文件提供。
E 来自一名代表性患者肿瘤的 AT2 和 CAMLs 的 CNA 详细概述。条形图显示了在特定染色体区域中具有染色体增益(红条)或损失(蓝条)的细胞频率。
F 散点图显示了肿瘤数据集中每种细胞类型的损失(x 轴)和增益(y 轴)的 KL 散度,使用它们的增益和损失分布计算。为了绘图目的,所有免疫细胞类型被归为一组。
G 空间图像显示了 cell2location 估计的三个代表性肿瘤切片上的 AT2 细胞和 CAMLs 的细胞丰度。
H 在所有肿瘤切片中,通过 QC 的点上(cell2location 估计的)细胞类型组成计算的相关距离的层次聚类。
I 在所有肿瘤切片中,通过 QC 的点上(cell2location 估计的)细胞类型丰度的 q05 估计上建立的非负矩阵分解。
有趣的是,CAMLs 群体也表现出与同一患者的 AT2 细胞和循环 AT2 细胞相似的显著 CNAs(图 4A、E 和补充图 5A、B)。为了以统计上稳健的方式测量不同细胞类型之间基因组增益和损失分布的差异,我们计算了 Kullback–Leibler(KL)散度(图 4F 和补充图 5C)。CAMLs 的 KL 散度值与携带 CNA 的肿瘤细胞相当,从而确认了它们的 CNA 特征的相似性(图 4F 和补充图 5C)。由于 CAMLs 共同表达广泛的髓系基因以及典型的上皮基因(图 1D–F 和补充图 1D),双细胞评分低且与肿瘤细胞共享相同的 CNA 特征,我们假设这些细胞可能代表紧密附着于癌细胞的一部分 Mɸ。这些 Mɸ 可能正在经历吞噬作用或融合。
CAMLs 以前已从癌症患者的外周血中分离出来,并被描述为促进循环肿瘤细胞远端转移 16。我们的分析表明,CAMLs 也可以从肿瘤组织中分离出来。为了验证 CAMLs 在原位与肿瘤细胞的物理接近,我们检查了我们的 STx 切片。我们计算了所有切片(8 名患者,nsections = 20)中居住在同一位置并因此共同定位的细胞类型的相对丰度之间的皮尔逊相关性。我们的分析表明,CAMLs 确实与 AT2 细胞共同定位(图 4G、H)。我们使用 cell2location 估计的绝对细胞类型丰度上的非负矩阵分解(NMF)确认了这一发现,该方法定义了共同出现的细胞状态的因素(图 4I)。
为了确定 CAMLs 可能来源的特定 Mɸ 群体,我们使用 PAGA 来阐明肿瘤数据集中髓系细胞群体的分化路径(补充图 5D)。分析揭示了不同髓系细胞群体之间分化过渡的连续性 43。在 PAGA 轨迹中,肺泡 Mɸ(AMɸ)和 AIMɸ 显示出较高的 PAGA 连接性,表明它们具有高度的转录相似性。AIMɸ 和 AMɸ 在 PAGA 轨迹上显示出与 STAB1 + Mɸ 的最强连接性,后者又与 CAMLs 相关联。与轨迹分析一致,CAMLs 共同表达许多特定于 STAB1 + Mɸ 的基因(补充图 2A),支持 CAMLs 可能是在与肿瘤细胞紧密接触后由 STAB1 + Mɸ 衍生的假设。最后,LUSC 与 LUAD 患者的 CAMLs 之间的 DEA 分析显示,在 LUSC 样本中 KRT17、KRT5 和 KRT6A 上调(补充数据 17)。这些 KRT 基因以前在多项研究中被确定为 LUSC 的标志 44,45,这支持了 CAMLs 来源于 Mɸ 与肿瘤细胞之间相互作用的假设。
TAMs 在肿瘤中促进胆固醇和铁的外流¶
传统上被分类为 M1(经典激活)和 M2(替代激活)表型的 Mɸ,现在被认为存在于一个功能状态的动态光谱中 46。Mɸ 可塑性的概念强调了它们能够根据来自微环境的复杂信号在促炎和抗炎角色之间无缝转换的能力(补充图 5D)。为了更好地了解不同 Mɸ 群体在 TME 中经历的转录变化,我们进行了 DEA。在肿瘤中,AMɸ 和 AIMɸ 上调了参与胆固醇和脂质运输及代谢的基因(如 ABCA1、APOC1、APOE、FABP3 和 FABP5),与背景组织相比(图 5A、B 和补充数据 18 和 19)。由于癌细胞增殖期间对新合成细胞膜的高需求,胆固醇在肿瘤生长中起着重要作用。与背景相比,肿瘤中的 AT2 细胞上调了与缺氧相关的基因(图 4D),这可能通过抑制胆固醇合成促进肿瘤细胞的胆固醇嗜好性,从而迫使它们依赖外源胆固醇的摄取 47。在我们的数据集中,我们检测到 AMɸ 和 AIMɸ 中胆固醇外运蛋白 ABCA1 的高表达,而低密度脂蛋白受体(LDLR)没有表达,后者负责将携带胆固醇的脂蛋白颗粒摄入细胞,这表明 TAMs 优先将胆固醇从 TAMs 排出到 TME 中(图 5A)。有趣的是,我们还注意到 AMɸ 和 AIMɸ 中高表达 TREM2(图 5A),它在小胶质细胞中的胆固醇外流中起着重要作用 48,49,50。为了验证 TME 中胆固醇水平的增加,我们用 BODIPY™ 493/503(染色胆固醇和其他中性脂质)对匹配的肿瘤和背景组织切片进行了染色。我们发现肿瘤切片中的 BODIPY 信号显著增加,相比于背景组织(图 5C、D),这证实了中性脂质在肿瘤中的可用性增加,可能是由于 TAMs 增加了外流。
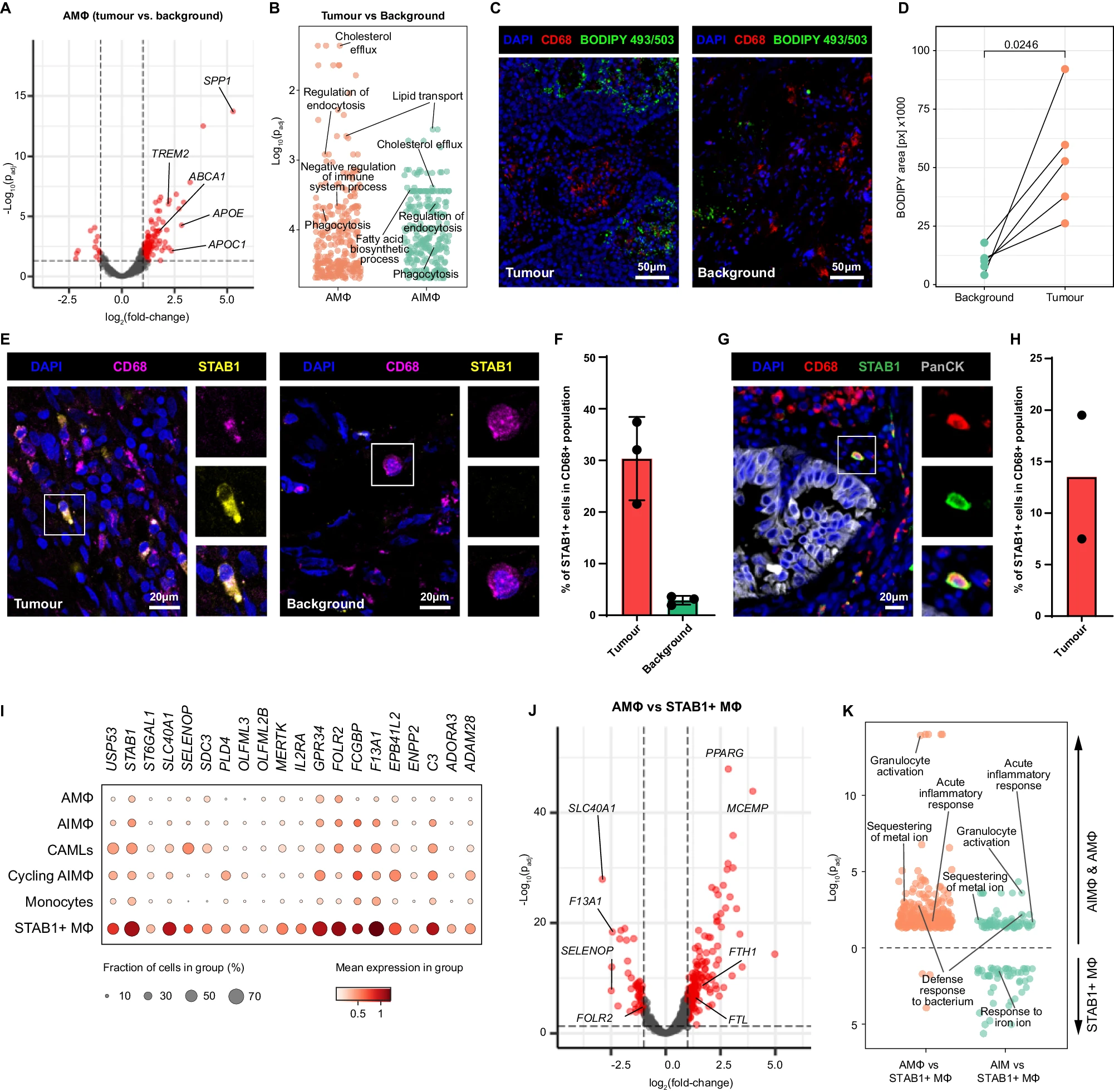
A 火山图展示了肿瘤 vs 背景中 AIMɸ 的差异表达基因(DEGs,红色),使用 py_DESeq2 包提取。
B 对基因本体——生物过程数据库的过度代表性分析,使用 clusterProfiler R 包,对在肿瘤 vs 背景中上调的肺泡 Mɸ 和 AIMɸ 的 DEGs 进行分析。数据来源提供于 Source Data 文件中。
C 肿瘤和背景组织切片上的 CD68 和中性脂质(BODIPY 493/503)的免疫组织化学染色。Z 轴堆叠的最大强度投影。比例尺 50 µm。
D 肿瘤和背景切片中 BODIPY 信号覆盖的面积。BODIPY 面积覆盖差异由配对、双侧 t 检验确定,匹配来自同一患者的肿瘤和背景切片。N = 5 名患者。数据来源提供于 Source Data 文件中。
E 肿瘤(左)和背景(右)组织切片上的 CD68 和 STAB1 的免疫组织化学染色。Z 轴堆叠的最大强度投影。插图显示了单个细胞的详细放大图。比例尺 20 µm。
F CD68+ 巨噬细胞群体内 STAB1+ 细胞的定量。显示为总 CD68+ 区域的百分比的 STAB1 + CD68+ 区域的比例。数据以平均值和标准差表示(n = 3 个生物复制)。数据来源提供于 Source Data 文件中。
G 肿瘤组织切片上的 CD68、STAB1 和 PanCK 的染色。Z 轴堆叠的最大强度投影。插图显示了单个细胞的详细放大图。比例尺 20 µm。
H NSCLC 中 CD68+ 巨噬细胞群体内 STAB1 + CD68+ 细胞的定量。数据以平均值和单个数据点表示(n = 2 个生物复制)。数据来源提供于 Source Data 文件中。
I 点图显示了肿瘤中所有巨噬细胞亚群和 CAMLs 的“STAB1 特征基因”的表达。
J 火山图展示了肿瘤中肺泡 Mɸ vs STAB1 Mɸ 的差异表达基因(DEGs,红色),使用 py_DESeq2 包识别。
K 对基因本体——生物过程数据库的过度代表性分析,使用 clusterProfiler R 包,对肿瘤中肺泡 Mɸ vs STAB1 Mɸ(顶部)和 AIMɸ vs STAB1 Mɸ(底部)的 DEGs 进行分析(左——STAB1 Mɸ 上调;右——肺泡 Mɸ 或 AIMɸ 上调)。数据来源提供于 Source Data 文件中。
STAB1 + Mɸ 在肿瘤切除组织中被识别出来(图 5E–H,补充图 2 和补充说明),因此我们使用 DEA(差异表达分析)来识别与肿瘤 AIMɸ 或 AMɸ 相比,特异性表达于 STAB1 + Mɸ 的基因集。我们识别了 20 个基因,从现在开始称为“STAB1 特征基因”(图 5I)。有趣的是,STAB1 + Mɸ 独特地表达了编码铁转运蛋白的 SLC40A1,这是唯一已知的将二价铁从细胞质跨过细胞膜输出的蛋白质,对巨噬细胞的铁释放活动至关重要(图 5I,J 和补充数据 20 和 21)51。研究表明,M2 型巨噬细胞通过铁转运蛋白介导的自由铁释放可以在体外促进肾细胞癌细胞的增殖,这可能是由于支持了由于 DNA 合成增加而导致的高铁需求 52。此外,与 AMɸ 相比,STAB1 + Mɸ 表达较低水平的编码铁储存蛋白铁蛋白的 FTH1 和 FTL(图 5J 和补充数据 20)。与持续向细胞外基质输出自由铁的假设一致,STAB1 + Mɸ 下调了参与铁隔离的基因(图 5K)。总之,我们的分析表明,巨噬细胞在肿瘤微环境(TME)内经历“重编程”,并采用了一种转录特征,促进胆固醇流出和铁输出,从而支持肿瘤进展。
肿瘤组织中的 STAB1 + Mɸ 进行癌胎重编程 胚胎发育与肿瘤组织共享许多特征,包括快速细胞分裂、细胞灵活性和高度血管化的微环境。最近的研究报告表明,在肿瘤发生过程中,巨噬细胞可以经历癌胎重编程 53,并获得支持肿瘤生长和转移的胎儿样转录身份 53。考虑到一些 STAB1 特征基因通常由胎儿巨噬细胞表达(如 STAB1、FOLR2、SLC40A1、MERTK、GPR34 和 F13A1)54,我们想探讨肿瘤来源的 STAB1 + Mɸ 和从人类胎儿肺中分离的巨噬细胞之间是否存在进一步的转录相似性。为此,我们将我们的数据集中来源于肿瘤和背景的髓系细胞(n = 347,364 个细胞)与一个公开可用的胎儿肺 scRNA-seq 数据集中的髓系和祖细胞(n = 6,947 个细胞)结合在一起,使用 Harmony 进行分析。接下来,我们在邻域图上执行 Leiden 聚类,并检查细胞类型在簇内的分布情况(补充图 6A,B)。为了检查它们的基因表达谱的相似性,我们应用层次聚类,并通过估算和谐 PC 嵌入空间中的细胞类型之间的相关距离来构建一个树状图,在层次聚类的完全链接标准下进行(图 6A)。
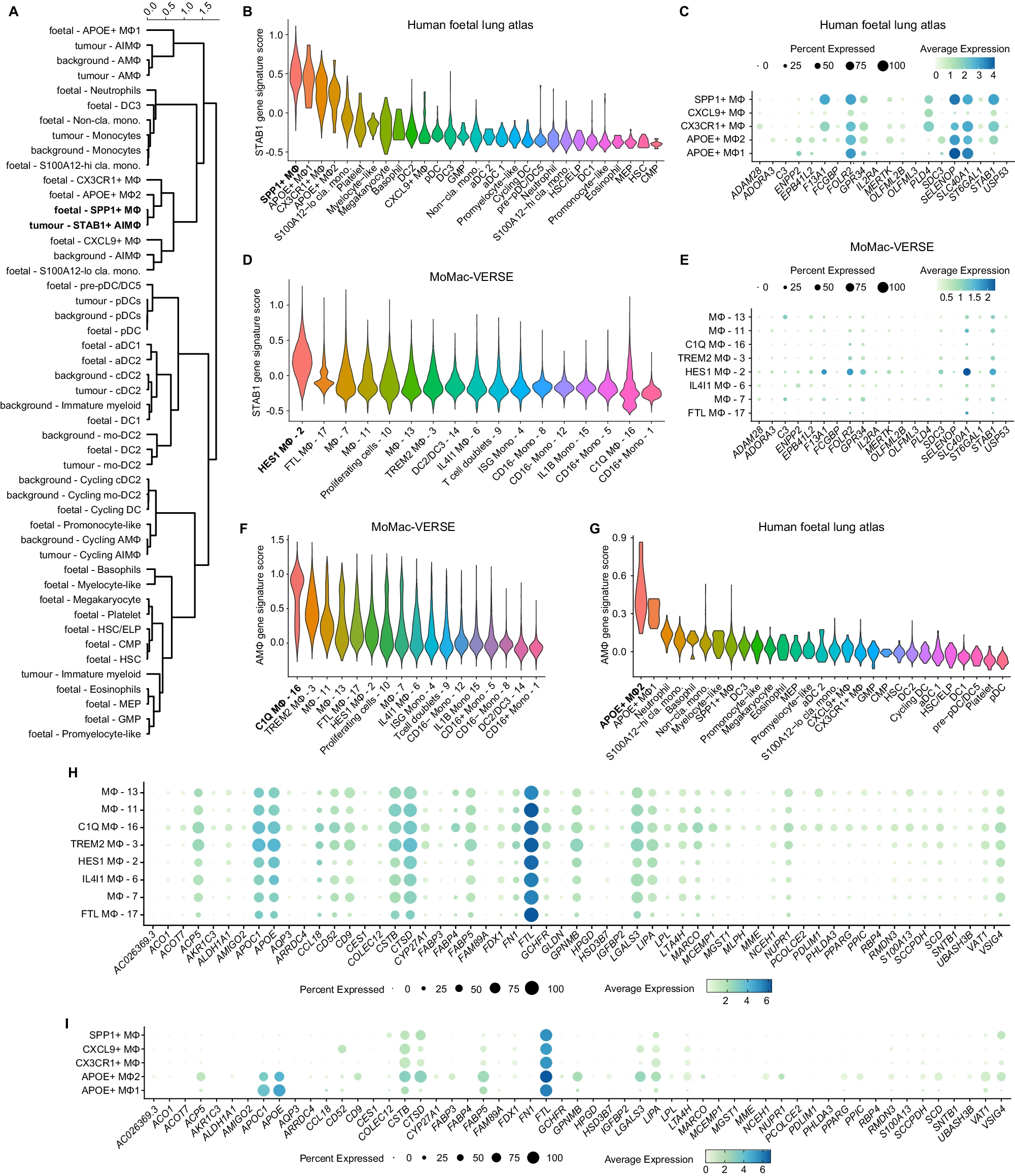
A 在和谐(肿瘤髓系 + 背景髓系 + 胎儿肺髓系)PC 空间中计算每个细胞的相关距离的层次聚类。
B 小提琴图显示在公开可用的人类胎儿肺图谱中识别的髓系细胞和祖细胞群体中“STAB1 基因特征”的表达水平。
C 点图显示在选定的胎儿肺巨噬细胞群体中每个“STAB1 基因特征”基因的表达。每个点的大小代表簇中表达该基因的细胞百分比,颜色代表每个簇中每个基因的平均表达水平。
D 小提琴图显示在公开可用的 MoMac-VERSE 数据集中识别的簇中“STAB1 基因特征”的表达水平。
E 点图显示在 MoMac-VERSE 数据集中选定的巨噬细胞群体中每个“STAB1 基因特征”基因的表达。每个点的大小代表簇中表达该基因的细胞百分比,颜色代表每个簇中每个基因的平均表达水平。
F 小提琴图显示在公开可用的 MoMac-VERSE 数据集中识别的髓系细胞和祖细胞群体中“AMɸ 基因特征”的表达水平。
G 小提琴图显示在公开可用的人类胎儿肺图谱中识别的髓系细胞和祖细胞群体中“AMɸ 基因特征”的表达水平。
H 点图显示在 MoMac-VERSE 数据集中识别的选定巨噬细胞群体中每个“AMɸ 基因特征”基因的表达。每个点的大小代表簇中表达该基因的细胞百分比,颜色代表每个簇中每个基因的平均表达水平。
I 点图显示在胎儿肺巨噬细胞群体中每个“AMɸ 基因特征”基因的表达。每个点的大小代表簇中表达该基因的细胞百分比,颜色代表每个簇中每个基因的平均表达水平。
我们观察到,肿瘤 cDC2 与背景 cDC2 表现出最强的相关性,而肿瘤 mo-DC2 则与胎儿 DC2 以及更广泛的背景 mo-DC2 具有最高的相关性。来自肿瘤、背景和胎儿肺的 pDC 群体密切相关。同样,肿瘤单核细胞与胎儿经典单核细胞和背景单核细胞相关性较高。相比之下,肿瘤中的巨噬细胞群体,特别是 STAB1 + Mɸ,与胎儿巨噬细胞相关。STAB1 + Mɸ主要聚集在胎儿 SPP1 + Mɸ中(图 6A),这类细胞占参考文献 55 中报告的所有胎儿肺巨噬细胞的 80% 以上。与这一发现一致,SPP1 + Mɸ相比其他造血群体,高度表达“STAB1 基因特征”(图 6B,C)。我们的分析证实了肿瘤环境中的单核细胞在分化为抗炎巨噬细胞的过程中,获得了类似于胎儿巨噬细胞的转录特征。这种独特的转录特征在周围正常组织中的巨噬细胞中未观察到。
为了进一步研究 STAB1 + Mɸ在其他病理学中的普遍性,包括其他癌症,我们使用一个名为 MoMac-VERSE 的已发表的人类单核细胞和巨噬细胞图谱,分析了来自 12 种不同健康和病理组织(n = 140,327 个细胞)的多样化髓系细胞群体中的“STAB1 基因特征”基因表达。MoMac-VERSE 中鉴定的“HES1+ 巨噬细胞”簇显示出最高的“STAB1 基因特征”基因表达(图 6D,E)。与 STAB1 + Mɸ相似,HES1+ 巨噬细胞在肺癌患者的肿瘤中积累,但也存在于肝癌患者中 57,并被认为代表了具有胎儿样转录特征的“长期驻留样”巨噬细胞簇 56。相比之下,MoMac-VERSE 中的“C1Q”巨噬细胞,被描述为肺泡巨噬细胞,显示出独特的肿瘤肺泡 AMɸ基因高表达(从这里开始称为“AMɸ基因特征”,图 6F,H)。在胎儿肺中,一种罕见的 APOE + 巨噬细胞群体,占参考文献 55 中报告的所有胎儿肺巨噬细胞的不到 1%,具有高 AMɸ基因特征评分(补充说明和图 6G,I,参见“方法”)。
综上所述,我们的分析表明,肿瘤巨噬细胞,特别是 STAB1 + Mɸ,展示了类似于胎儿肺发育期间巨噬细胞的转录特征,表明它们在 NSCLC 肿瘤环境中经历了癌胎重新编程。
讨论¶
我们的研究代表了对来自未接受治疗的 NSCLC 患者样本进行的大规模单细胞多组学分析。我们整合了来自 25 名未接受治疗的患者的肿瘤切除和相邻非恶性组织中的近 90 万个细胞的 scRNA-seq 数据,并结合空间转录组学,构建了肺癌中免疫和非免疫成分的图谱。
LUAD 和 LUSC 是最常见的两种 NSCLC 亚型,它们表现出显著不同的预后结果,并显示出针对亚型的治疗潜力【28】。尽管细胞类型组成相似,但我们观察到 LUAD 和 LUSC 在若干 ICIs 和抑制分子的共表达方面存在显著差异,突显了治疗机会。LUAD 样本中频繁表达 TIGIT 和 TIM3(HAVCR2),而在 LUSC 中,我们发现了假定的 ICI CD96-NECTIN1。目前,针对 TIGIT 的多个高级临床试验,包括在 NSCLC 患者中的试验正在进行中【58】;然而,针对 TIM3 和 CD96 的进展较为有限【59】。一项评估抗 CD96 单克隆抗体 GSK6097608 作为单药治疗或与抗 PD1(dostarlimab)联合使用的一期人体研究最近才开始招募患者【60】。总的来说,我们的数据表明,LUAD 和 LUSC 患者可能受益于特异性免疫治疗,针对 ICIs 如 TIM3、TIGIT 和 CD96。
TME 在调节 Mɸ的群体和行为方面起着关键作用【4】。我们发现,与相邻的非肿瘤组织相比,肿瘤切除物中单核细胞比例较低,但单核细胞来源的细胞比例较高,如 mo-DC2 和抗炎 Mɸ,表明 TME 中单核细胞分化增强【7,9】。抗炎 Mɸ,包括 STAB1 + Mɸ的普遍性,与肿瘤环境中 NK 细胞和 T 细胞的丰度呈负相关;肿瘤中的 NK 细胞表现出较低的细胞毒性活动。我们的结果与最近的研究一致,该研究发现肺 Mɸ清除肿瘤细胞碎片会导致其转化为免疫抑制表型,从而阻碍 NK 细胞向 TME 的浸润【27】。报告显示,具有高水平肿瘤碎片的 Mɸ上调了涉及胆固醇运输和脂质代谢的基因,这一特征与我们数据集中抗炎 Mɸ共享。因此,它们下调了共刺激分子、细胞因子和趋化因子【27】,这些分子对于招募 CD8 + T 细胞至关重要,因此变得更具免疫抑制性。
在肿瘤中的 Mɸ群体中,我们还鉴定了表现出最高免疫抑制标记物水平的 STAB1 + Mɸ。这些 STAB1 + Mɸ展示了类似于胎儿肺 Mɸ的基因表达模式,并表现出铁代谢的改变,标志着在 TME 中与铁释放相关的基因表达增加。因此,我们假设 STAB1 + Mɸ可能在通过维持高循环肿瘤细胞的铁需求来支持肿瘤进展中起关键作用【52,61】。在皮下 LLC1 刘易斯肺腺癌模型中,缺乏 Stab1 表达的 Mɸ的小鼠,肿瘤生长减少。这一结果归因于 TAM 向促炎表型的转变和 TME 中 CD8 + T 细胞的强烈浸润【62】。STAB1 + Mɸ在转录上与 CAMLs 相似,后者同时表达与 Mɸ和上皮细胞相关的基因,并表现出与肿瘤细胞相似的拷贝数变异(CNAs)。STAB1+ 在通过特异性与磷脂酰丝氨酸相互作用促进凋亡细胞的粘附和吞噬中起关键作用,这支持了 CAMLs 中 Mɸ与肿瘤细胞强相互作用的假设【63】。在先前的研究中,通过免疫荧光在患有各种实体肿瘤的个体的外周血中鉴定了 CAMLs,并被提出促进循环肿瘤细胞在远处转移部位的传播和建立【16】。在这里,我们根据复合基因表达特征、肿瘤特异性拷贝数变异以及 Visium 切片中肿瘤细胞的物理接近性,报告了它们在多个肿瘤切除物中的存在。综上所述,我们的综合数据集帮助识别了肺肿瘤微环境中 Mɸ群体的多种分子变化,这将有助于开发针对 NSCLC 的治疗策略。
数据可用性¶
本研究中生成的 scRNA-seq 和 Visium 数据集可在 BioStudies 公开获取(https://www.ebi.ac.uk/biostudies/),登录号分别为 E-MTAB-13526 和 E-MTAB-13530。其余数据可在本文、补充信息或源数据文件中获取,源数据随本文提供。
代码可用性¶
用于所有分析和生成本文所有图表的脚本可在以下网址获取: - https://gitlab.com/cvejic-group/lung - https://github.com/sdentro/copykat_pipeline