Single cell characterization of macrophages in uveal melanoma uncovers transcriptionally heterogeneous subsets conferring poor prognosis and aggressive behavior(范例 1)
摘要¶
葡萄膜黑色素瘤(UM)是最常见的原发性眼内恶性肿瘤,具有高转移潜力和不良预后。巨噬细胞是一种最丰富的浸润性免疫细胞,在癌症中具有多种功能。然而,在 UM 中,巨噬细胞的细胞异质性和功能多样性仍然未被充分探索。在这项研究中,我们分析了来自 11 例 UM 患者的 63,264 个单细胞转录组,并鉴定了四个转录上不同的巨噬细胞亚群(命名为 MΦ-C1 至 MΦ-C4)。其中,我们发现 MΦ-C4 表现出相对较低的 M1 和 M2 标志基因表达,失去了炎症途径和抗原呈递,而表现出增强的增殖信号、线粒体功能和代谢1。我们从单细胞和大量转录组跨越五个队列量化了 MΦ-C4 的浸润丰度,并发现增加的 MΦ-C4 浸润与侵袭行为相关2,并可能作为独立的预后指标,预示不良结果3。我们提出了一种基于巨噬细胞的新型亚型方案,通过整合 MΦ-C4 的转录特征和机器学习将患者分层为富含 MΦ-C4 或贫含 MΦ-C4 的亚型。这两个亚型显示出显著不同的临床结果,并通过大量 RNA 测序和免疫荧光分析在公共多中心队列和我们的内部队列中得到验证。在进一步的转化研究之后,我们的发现突显了通过靶向巨噬细胞亚群来控制转移性疾病,并持续改善 UM 患者预后的潜在治疗策略。
引言¶
葡萄膜黑色素瘤(UM)源于葡萄膜道的黑色素细胞,在成人中是最常见的原发性眼内恶性肿瘤,占所有眼内黑色素瘤的 85% 以上。局部治疗如保守放疗和手术切除已被广泛用于控制原发性 UM,改善患者预后,并提高生存率和生活质量。然而,UM 是一种高度转移性疾病,约 50% 的患者发展为远处转移,导致致命后果,中位总生存期(OS)为 12 个月。因此,迫切需要改进的治疗选择和目标以有效治疗 UM 转移并改善长期生存率。
关于 UM 遗传学和发病机制的研究揭示了该疾病的异质性特质,导致了分子和临床上的不同亚型的识别。肿瘤是由癌细胞、非恶性基质细胞和免疫细胞组成的复杂生态系统,统称为肿瘤微环境(TME)。尽管眼睛是一个重要的免疫特权区域,但证据表明,TME 中的免疫异质性显著影响 UM 生物学的各个方面,包括发展、进展、转移、预后和治疗反应。以往的研究已经从大量组织数据中表征了 UM 中的免疫浸润,并将 M2 系列的单核细胞或巨噬细胞识别为主要的肿瘤浸润免疫细胞群体;这些细胞与肿瘤生长和不良预后相关4。单细胞 RNA 测序的最新进展突显了巨噬细胞的异质性和功能多样性,与 M1/M2 模型无关。例如,Chen 等人鉴定了一种新的巨噬细胞亚群作为胶质瘤中的间质促瘤标记。在肾脏损伤急性阶段进行的一项时间序列分析确认了 S100A8/A9+ 巨噬细胞浸润在组织损伤中的相关性。最近的全癌单细胞分析也揭示了不同癌症类型中巨噬细胞亚群的复杂性和异质性水平,强调了需要单独研究 TME 中巨噬细胞的复杂表型。然而,巨噬细胞的细胞异质性和功能多样性尚未在 UM 的单细胞水平上得到充分阐明。
在本研究中,我们对 UM 患者的巨噬细胞进行了单细胞 RNA 测序分析,以表征巨噬细胞的异质性,并鉴定了四个巨噬细胞亚群。然后,我们利用多中心的大量和单细胞队列探讨了四个巨噬细胞亚群对肿瘤表型和预后的临床影响,并通过 RNA-seq 和免疫荧光成像验证了它们对分子亚型的贡献。我们的研究为 UM 中巨噬细胞的多样性提供了重要见解,并展示了靶向巨噬细胞亚群以改善日常临床实践的潜力。
材料与方法¶
单细胞 RNA 测序数据及分析¶
葡萄膜黑色素瘤(UM)患者的单细胞 RNA 测序数据(10x Genomics)和临床病理信息从 Durante 等人的研究中回顾性地收集自基因表达 Omnibus(GEO)数据库,登记号为 GSE139829。
单细胞 RNA 测序数据的处理和分析采用了 Seurat v4 R 软件包。除非另有说明,所有功能均采用默认参数执行。首先使用 merge 函数将所有 UM 样本的单细胞 RNA 测序数据进行整合,并移除低质量细胞(<200 个基因/细胞或>8000 个基因/细胞和>10% 线粒体基因)。通过质量控制后,使用 SCTransform 方法对单细胞 RNA 测序数据进行归一化,采用回归方法确定线粒体百分比。使用 Harmony 方法纠正样本间的潜在批次效应。进行主成分分析以降维,选取前 20 个主成分,采用基于图的聚类方法(使用 FindClusters 函数,resolution = 2)来识别不同细胞群。利用已知的细胞类型标记基因对主要细胞群进行注释,并使用 t 分布随机邻域嵌入(tSNE)散点图进行可视化。使用 Seurat 中的 FindAllMarker 函数鉴定每个细胞类型与所有其他细胞类型之间的差异表达基因(DEGs),并使用 Wilcoxon 秩和检验进行显著性检验,并使用 Bonferroni 校正。基因根据|logFC| > 0.5 和调整后的 P 值<0.05 的阈值选择为 DEGs。
批量转录组数据及分析¶
从 UCSC Xena(GDC The Cancer Genome Atlas (TCGA)-UVM 队列; https://xenabrowser.net/datapages/)获取了 80 个 UM 样本的批量 RNA 测序数据和临床病理信息。使用 HiSeq Illumina 平台测量的基因表达水平以 FPKM-UQ 进行量化,并使用 log2 转换进行归一化。
从 GEO 获取了 120 例 UM 患者的批量微阵列数据和临床病理信息,其中包括来自 Laurent 等人的研究的 63 例患者(登记号 GSE22138)、Gangemi 等人的研究的 29 例患者(登记号 GSE27831)以及 van Essen 等人的研究的 28 例患者(登记号 GSE84976)。
使用鲁棒多芯片平均(RMA)算法对来自 Human Genome U133 Plus 2.0 阵列的原始微阵列数据进行了预处理和归一化,进行了背景校正、分位数归一化和 log2 转换。使用作者提供的 Illumina HumanHT-12 V4.0 表达微珠芯片的处理过的表达数据进行了分析。
特征分数的定义¶
TEX 分数由 TEX 相关基因集(PDCD1、CTLA4、LAG3、TIGIT、TOX 和 TCF7)的平均表达值定义。 M1 和 M2-MΦ签名富集分数使用 Seurat 中的“AddModuleScore”函数以规范的 M1 和 M2-MΦ签名进行计算(见附表 1)。
轨迹分析¶
使用 Monocle2(v2.16.0)和 CytoTRACE(https://cytotrace.stanford.edu/)确定细胞类型和簇之间的巨大转化关系。对于 Monocle2,使用参数“expressionFamily = negbinomial”创建了 CellDataSet 对象。在降维后,使用默认参数构建了分化轨迹。轨迹推断的基因使用 dispersionTable 函数确定。仅选择平均表达量>1 的基因进行分析。使用差异基因测试函数识别了沿伪时间动态差异表达的基因,其中 q <0.001。对于 CytoTRACE,我们将巨噬细胞的表达计数矩阵输入在线工具,然后获得结果。
SCENIC 分析¶
SCENIC 分析用于探究转录因子(TF)调控,默认参数 37。使用 Seurat 对象中的表达计数矩阵对基因进行筛选,使用 Seurat 中的 geneFiltering 函数进行筛选,并使用默认阈值(minCountsPerGene,0.03 × ncells; minSamples,0.01 × ncells)进行规范化。使用 GRNboost 推断 TF 靶基因共表达模块,并使用 RcisTarget 识别调控元,使用默认参数和以下 cisTarget 数据库:hg19-500 bp-upstream-7species.mc9nr.feather 和 hg19-tss-centered-10 kb-7species.mc9nr.feather。使用 AUCell 方法对单个细胞的每个调控元的活性进行评分。
富集分析¶
使用 R 软件包“GSVA”进行单样本基因集富集分析(ssGSEA)计算特定基因集或通路活性的富集分数。为了进行巨噬细胞亚型的功能富集分析,我们确定了每个巨噬细胞亚型的排序差异表达基因(DEGs),并使用 clusterProfiler(v3.18.0)软件包进行基因集富集分析(GSEA)。
患者生物标本采集和 RNA 提取¶
本研究按照《赫尔辛基宣言》进行,并获得了温州医科大学眼科医院伦理委员会的批准(伦理批准号 2022-043-K-28-02)。所有患者均提供了书面知情同意。所有数据均进行了匿名分析。从 5 名被诊断为 UM 的患者在温州医科大学眼科医院进行外科切除期间收集了人类 UM 组织。患者在收集前未接受术前治疗。
使用 TRIzol®试剂(Invitrogen, Thermo Fisher Scientific, Inc.)从 UM 组织中提取总 RNA。 OD260 / OD280 比值被用作 RNA 纯度的指标;纯 RNA 的比值需要接近 2.0(可接受范围为 1.8-2.1)。使用 NanoDrop 2000 分光光度计(Invitrogen, Thermo Fisher Scientific, Inc.)测量 RNA 浓度和纯度。
免疫荧光分析¶
肿瘤组织在 4°C 下用 4%戊二醛(PFA)固定过夜,用 10%、20%和 30%蔗糖溶液脱水,转移至 OCT 中,并在 -80°C 冷冻保存以备后续使用。制备 10μm 厚度的组织,并使用磷酸盐缓冲液(PBS)进行洗涤。应用 CD68(稀释 1:200; cat. no. sc-20060, Santa Cruz Biotechnology, Inc.)和铁蛋白轻链(稀释 1:100; cat no. ab69090; Abcam)的一抗。将组织切片在 4°C 过夜与一抗孵育,然后与二抗,Alexa Fluor 594 标记的驴抗兔 IgG [(H+L); cat no. 34212ES60; Shanghai Yeasen Biotechnology Co., Ltd.] 和 Alexa Fluor 488 标记的驴抗鼠 IgG [(H+L); cat. no. 34106ES60; Shanghai Yeasen Biotechnology Co., Ltd.]。用 DAPI 染色核,信号使用 DM4B(Leica Microsystems GmbH)可视化。
统计分析¶
所有统计分析均在 R 软件(v4.0.3)中进行,使用 R studio 界面(v1.3.959)。使用 Wilcoxon 秩和检验或 Student's t 检验比较两组之间的差异。使用 R 软件包“ConsensusClusterPlus”进行一致性聚类分析,参数为:pItem = 0.8,pFeature = 1,reps = 1000,以及“Pam”聚类方法。利用单变量和多变量 Cox 比例危险回归模型评估变量与生存时间之间的关联。使用 Kaplan-Meier 方法和 log-rank 检验比较不同患者组之间的生存差异。计算了风险比(HRs)、95% 置信区间(CIs)和相应的 P 值,并使用 R 软件包“forestplot”进行协变量效应的可视化。斯皮尔曼秩相关系数用于相关分析。统计显著性水平设为 P <0.05。
结果¶
单细胞转录组分析揭示了 UM 中四种转录上不同的巨噬细胞亚群¶
为了探索 UM TME 中的细胞异质性,我们对 11 名 UM 患者的 scRNA-seq 数据进行了回顾性分析。经过质量控制和批次效应校正后,保留了总共 63,264 个合格细胞,平均表达了 1555 个基因,用于降维和无监督基于图的细胞聚类分析。该分析确定了八个主要的细胞簇,通过 t-SNE 可视化,并根据 Cellmarker 数据库中列出的经典基因标记和先前发表的数据进行手动注释。这些细胞簇包括肿瘤细胞(MLANA+、MITF+ 和 PRAME+)、T 细胞(CCL5+、CXCR4+、CD8A+)、NK 细胞(NKG7+、GNLY+、GZMB+)、B/浆细胞(IGL3-1+、IGLV2-14+、IGLV1-40+)、髓系细胞(C1QB+、C1QC+ 和 CD74+)、癌相关成纤维细胞(CAF,MGP+、RGS5+、COL1A1+)以及其他细胞类型的极小比例:内皮细胞(PECAM1+、RAMP2+ 和 CCL21+)和光感受细胞(RCVRN+)(见图 1a-c)。
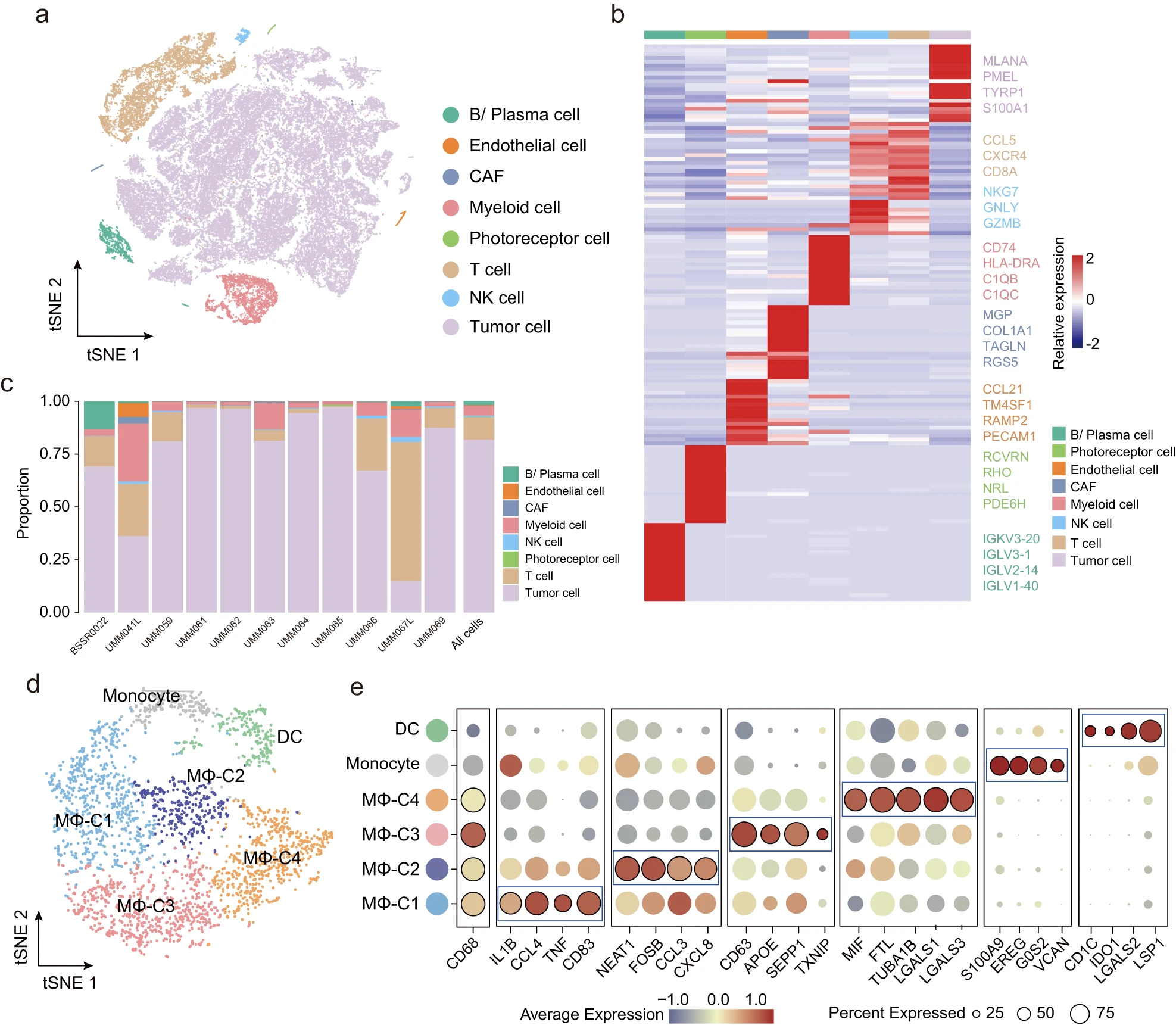
a. 所有单个细胞的 t-SNE 图,根据其分配的主要细胞类型进行着色。 b. 热图显示指定细胞簇中顶部已知标记的平均表达。 c. 在不同患者中显示的八个主要细胞类型的比例,用条形图表示。 d. t-SNE 图显示了 2847 个髓系细胞的六个亚群。每个亚群根据细胞亚群进行着色。 e. 泡泡热图可视化了每个髓系亚群的标记基因表达,其中颜色和点大小分别表示每个髓系亚群的平均标度表达水平和基因表达的百分比。
与 GEP 第 1 类肿瘤和原发性肿瘤相比,髓系细胞在 GEP 第 2 类肿瘤或==转移性肿瘤==中显示出相对较高的浸润丰度(附图 1a),表明髓系细胞浸润与肿瘤的==侵袭性行为有关==。我们进一步基于 3000 个高变异基因(HVGs)对 2847 个合格髓系细胞进行基于 SNN 的聚类,以解剖浸润的髓系细胞组分,并鉴定了六个聚类:单核细胞、树突状细胞(DCs)和==四个巨噬细胞亚群(称为 MΦ-C1 到 MΦ-C4)==(图 1d)。单核细胞表达高水平的单核细胞相关基因,如 S100A9、EREG、G0S2 和 VCAN,而 DCs 表达 CD1C、IDO1、LGALS2 和 LSP1(图 1e)。对于巨噬细胞,MΦ-C1 和 MΦ-C2 表达高水平的促炎介质(如 IL1B、CCL3、CCL4、FOSB 和 CD83)和趋化因子(CXCL8),而 MΦ-C3 高表达 M2-MΦ标志物(如 APOE、SEPP1 和 TXNIP)。相比之下,MΦ-C4 并未高表达 M1/M2 特征基因,而是表现出高水平的微管相关基因(TUBA1B)、铁蛋白轻链(FTL)和几种免疫调节分子的表达(迁移抑制因子(MIF)、半乳糖凝集素 -1(GAL1)和半乳糖凝集素 -3(GAL3))。对四个巨噬细胞聚类的 M1 和 M2-MΦ经典特征的进一步富集分析也表明,高 M1-MΦ特征富集分数表征了 MΦ-C2 聚类;MΦ-C3 聚类显示出高 M2-MΦ特征富集分数,而 MΦ-C1 则以 M1/M2-MΦ特征富集分数为特征。值得注意的是,MΦ-C4 聚类的 M1 和 M2-MΦ特征富集分数较低(附图 1b)。根据每个髓系亚群的平均基因表达值,我们根据其转录模式计算了所有亚群的 Spearman 秩相关系数,并发现了 MΦ-C4 聚类的独特转录谱,使其与其他三个巨噬细胞聚类有所区别(附图 1c)。
在 UM 中不同巨噬细胞亚群的功能特征化¶
为了调查巨噬细胞亚群的功能异质性,我们对一个巨噬细胞亚群和其他三个巨噬细胞亚群之间进行了单细胞差异表达分析,以识别特定亚群中显著上调的基因(图 2a 和附表 2)。对癌症标志途径的功能富集分析显示,MΦ-C1 的上调基因富集于 IL-6/JAK/STAT3 信号传导、IL-2/STAT5 信号传导和炎症应答,MΦ-C2 中富集于 TNF-α通过 NF-κB 信号传导、缺氧和炎症应答、IL-6/JAK/STAT3 信号传导等途径,MΦ-C3 中富集于干扰素α应答。而 MΦ-C4 则富集于氧化磷酸化、脂肪酸代谢和 MYC 靶基因 V1(图 2b)。此外,还应用基于 Reactome 数据库的基因集变异分析来计算生物学功能的活性。结果显示,MΦ-C2 富集于‘IL-6 型细胞因子受体 - 配体相互作用’、‘NF-kB 激活并信号传导生存’、‘WNT 在癌症中的信号传导’、‘NOTCH3 胞内结构域调节转录’和‘p75NTR 通过 NF-kB 信号传导’。MΦ-C3 富集于‘PD-1 信号传导’、‘IRF3 介导的 I 型干扰素诱导’、‘补体级联’和‘MHC-II 抗原呈递’。MΦ-C1 不仅与 MΦ-C2 和 MΦ-C3 共享类似的途径,还富集于白细胞介素 -35 信号传导和免疫系统中的细胞因子信号传导。相反,代谢相关途径和 G2 期只在 MΦ-C4 中富集,表明这个亚群可能具有独特的代谢和增殖特性(图 2c)。因此,我们利用 Seurat 根据 S 和 G2/M 期标记基因的表达水平计算了每个巨噬细胞亚群的细胞周期阶段得分。如图 2d 所示,MΦ-C4 的 S 和 G2/M 期得分高于其他三个巨噬细胞亚群5(图 2d)。
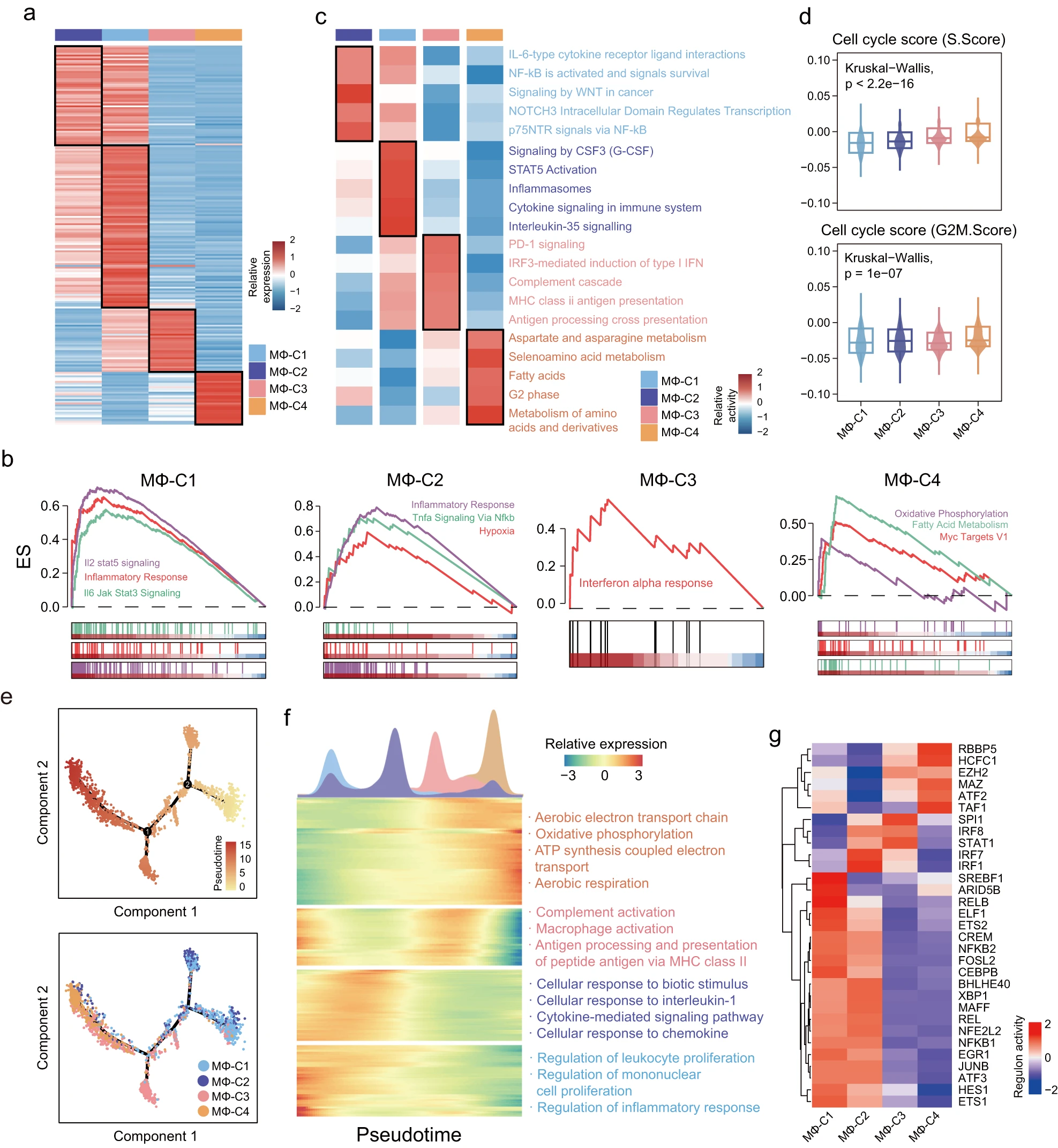
a. 四个巨噬细胞亚群的差异表达基因(DEGs)热图。 b. 每个巨噬细胞亚群中上调基因集的 GSEA 富集图。 c. 显示不同巨噬细胞亚群中统计富集的 Reactome 通路活性的热图。 d. 不同巨噬细胞亚群的细胞周期得分小提琴图。使用 Kruskal-Wallis 检验确定统计学差异。 e. 由 Monocle 2 推断的不同巨噬细胞亚群的潜在极化轨迹。顶部面板显示极化方向,底部面板显示发育轨迹中每个巨噬细胞亚群的分布。 f. 沿着伪时间的差异表达基因被分层聚类成四个轮廓。显示代表性基因功能和通路。 g. 由 SCENIC 估计的不同巨噬细胞亚群的统计富集转录因子的调控子活性的热图。
此外,对这四个巨噬细胞亚群的先天免疫功能进行了研究,结果显示 MΦ-C4 的内在免疫基因特征最低,表明其可能存在内在免疫功能失调(附图 2a)。进一步检查 Toll 样受体(TLR)家族的情况显示,与其他三个巨噬细胞亚群相比,MΦ-C4 的表达水平较低6(附图 2b)。TLRs 是巨噬细胞中启动先天免疫应答的重要模式识别受体。这表明 TLR 缺陷可能在一定程度上导致 MΦ-C4 内在免疫功能失调。
随后使用 Monocle 进行轨迹分析,以研究 UM 中巨噬细胞的极化轨迹。该轨迹分析显示了一个分叉的结构,从 MΦ-C1 和 MΦ-C2 开始,并分成多个巨噬细胞极化状态(图 2e)。如图 2e 所示,MΦ-C1、MΦ-C2 和 MΦ-C3 在轨迹的早期和中期更为冗余,而 MΦ-C4 主要位于终端分支。为了验证,我们使用另一个轨迹重构计算工具 cytoTRACE 来再现巨噬细胞的分化轨迹,结果显示 MΦ-C1 和 MΦ-C2 具有最高的发育潜力,而 MΦ-C4 处于最终分化状态(显示为低发育潜力,附图 2c,d)。在伪时间上探索了转录和功能上的变化。伪时间上的差异表达基因富集分析显示炎症应答、细胞因子介导的信号通路和补体激活在早期和中期被激活,但在极化的后期被统一下调(图 2f)。相反,代谢途径和参与细胞能量学的 ATP 生物合成过程在极化的后期显示出高度富集(图 2f)。
为了确定决定每个巨噬细胞亚群状态的关键转录因子(TF),我们应用了 SCENIC 方法,以确定不同巨噬细胞亚群之间 TF 与不同转录程序之间的相关性,并识别了多个 TF 活性模式(图 2g)。炎症相关的 TF(NFKB1、NFKB2、JUNB 和 FOSB)在 MΦ-C1 和 MΦ-C2 中被激活,而经典的干扰素调节 TF(IRF 和 STAT)在 MΦ-C3 中被鉴定。共有六个 TF(EZH2、RBBP5、ATF2、TAF1、HCFC1 和 MAZ)在 MΦ-C4 中高度升高,可能介导 MΦ-C4 的表型和特征。7
单细胞转录上与 UM 的侵袭性行为相关的差异巨噬细胞亚群¶
为了研究转录上不同巨噬细胞亚群的临床相关性,我们检查了单细胞和患者水平上的浸润丰度差异。根据单细胞数据确定了 GEP 类 1 和 GEP 类 2 肿瘤以及原发性和转移性肿瘤中不同巨噬细胞亚群的相对比例。MΦ-C4 在 GEP 类 2 肿瘤(29.39%)和转移性肿瘤(32.84%)中的相对比例高于 GEP 类 1 肿瘤(12.47%)和原发性肿瘤(22.98%)(图 3a)。图 3b 显示了 MΦ-C4 普遍存在于每个 UM 样本中,尽管样本间的水平不同,范围从 4.7% 到 58.3% 不等。此外,在分析浸润丰度与临床特征之间的相关性时,发现 MΦ-C4 的浸润密度在 GEP 类 2 肿瘤(P = 0.024)中高于 GEP 类 1 肿瘤(图 3c,附图 3a),MΦ-C4 在转移性肿瘤中的浸润更多(附图 3b)。此外,与其他巨噬细胞亚群相比,肿瘤直径与 MΦ-C4 的浸润密度呈显著正相关(r = 0.71,P = 0.047;图 3d,附图 3c)。这些结果表明,MΦ-C4 的增加浸润与侵袭性行为相关。
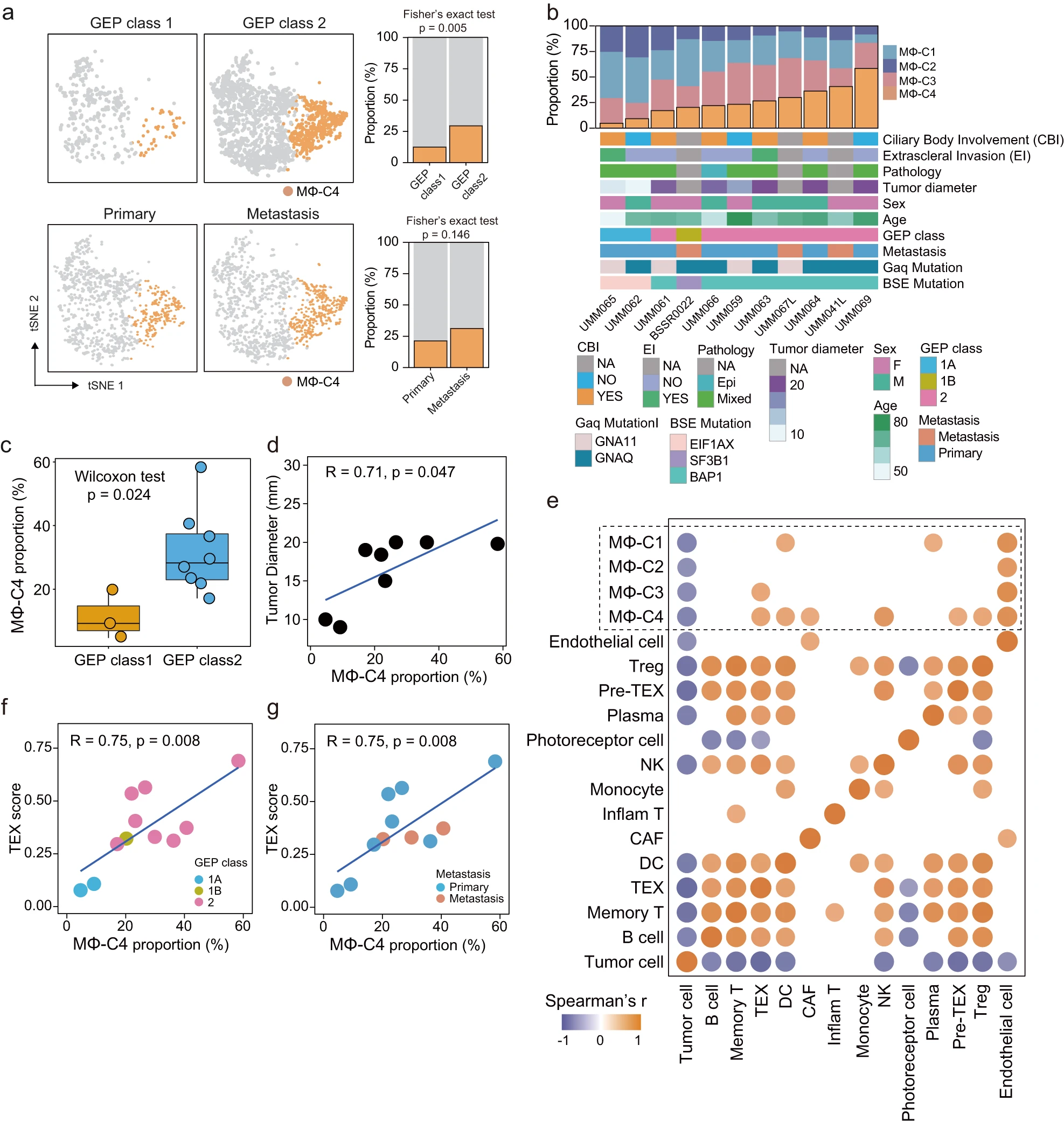 a 单细胞数据中比较了 GEP 类 1 和 GEP 类 2 肿瘤以及原发性和转移性肿瘤中 MΦ-C4 的比例。统计学差异由 Fisher 确切检验确定。
b 每个患者中 MΦ-C4 的比例和临床特征的景观。
c 盒图显示了 GEP 类 1 和 GEP 类 2 肿瘤中 MΦ-C4 比例的差异。统计学差异由 Wilcoxon 秩和检验确定。
d 散点图显示了 MΦ-C4 比例与肿瘤直径的相关性。相关性由 Spearman 相关系数评估。
e 热图显示了不同细胞亚群比例之间的 Spearman 相关系数。P < 0.05 被视为截断值。
f 散点图显示了 MΦ-C4 比例与 T 细胞耗竭(TEX)分数的相关性。相关性由 Spearman 相关系数评估,点颜色表示 GEP 分类。
g 散点图显示了 MΦ-C4 比例与 T 细胞耗竭(TEX)分数的相关性。相关性由 Spearman 相关系数评估,点颜色表示原发性肿瘤或转移灶。
a 单细胞数据中比较了 GEP 类 1 和 GEP 类 2 肿瘤以及原发性和转移性肿瘤中 MΦ-C4 的比例。统计学差异由 Fisher 确切检验确定。
b 每个患者中 MΦ-C4 的比例和临床特征的景观。
c 盒图显示了 GEP 类 1 和 GEP 类 2 肿瘤中 MΦ-C4 比例的差异。统计学差异由 Wilcoxon 秩和检验确定。
d 散点图显示了 MΦ-C4 比例与肿瘤直径的相关性。相关性由 Spearman 相关系数评估。
e 热图显示了不同细胞亚群比例之间的 Spearman 相关系数。P < 0.05 被视为截断值。
f 散点图显示了 MΦ-C4 比例与 T 细胞耗竭(TEX)分数的相关性。相关性由 Spearman 相关系数评估,点颜色表示 GEP 分类。
g 散点图显示了 MΦ-C4 比例与 T 细胞耗竭(TEX)分数的相关性。相关性由 Spearman 相关系数评估,点颜色表示原发性肿瘤或转移灶。
为了探究各种细胞组分之间的相互作用,我们进一步将 T 细胞分为八个亚群,基于已建立的标记:记忆 T 细胞、调节性 T 细胞(Tregs)、表达 TNF 和 IFNG 的与炎症相关的 T 细胞(Inflam T),以及表达中度和高度耗竭标记的前耗竭(Pre-TEX)和耗竭(TEX)T 细胞(补充图 4a,b)8。B 细胞和浆细胞也被区分开来(补充图 4c,d)。对于每个亚群,计算了 Spearman 等级相关系数(图 3e)。与其他巨噬细胞亚群相比,MΦ-C4 与肿瘤来源的癌相关成纤维细胞(CAFs)和免疫抑制性 T 淋巴细胞亚群(包括 Tregs、前耗竭 T 细胞(Pre-TEX)和耗竭 T 细胞(TEX))之间存在高度相关性(图 3e)。还观察到 MΦ-C4 与 T 细胞耗竭(TEX)之间存在显著的正相关关系(r = 0.75,P = 0.008,图 3f 和 g)。
-
从表达基因,代谢路径,代谢情况来分群 ↩
-
对感兴趣细胞的浸润程度判别以及生物学行为探究 ↩
-
归因至临床意义上 ↩
-
研究动机 ↩
-
在条件干预的情况下,部分细胞处于非稳定状态,如增殖类细胞出现由于细胞周期相关基因的不同导致细胞聚类发生一定的偏移。基于 G2/M 和 S 期经典 marker 基因的表达,计算每个细胞可能处于某一细胞分裂时期的分数,那些都不表达 marker 基因的细胞可能处于 G1 期 ↩
-
从免疫角度说明该细胞在肿瘤发展中的重要性 ↩
-
从转录因子角度 ↩
-
T 细胞耗竭是一个概念宽泛的术语,用于描述长期刺激造成的 T 细胞功能障碍。耗竭性t细胞可表现出独特的表型,包括抑制性标志物(例如 PD-1、LAG-3 和 TIM-3)过度表达,以及 T 细胞释放促炎性细胞因子(IFNγ 和 TNFα)的能力减弱。t细胞耗竭通常发生在肿瘤微环境中,耗竭 t 细胞将丧失细胞毒性作用,无法杀伤癌细胞 ↩