Single cell spatial multi omics and deep learning dissect enhancer driven gene regulatory networks in liver zonation
摘要¶
在哺乳动物的肝脏中,肝细胞根据其在肝小叶中的位置显示出多样的代谢和功能特性。然而,这种称为分区的空间变化是否受到明确定义的基因调控代码的管理尚不清楚。在这里,我们结合了单细胞多组学、空间组学、大规模平行报告基因分析和深度学习技术,绘制了小鼠肝脏细胞类型的增强子 - 基因调控网络。我们发现,分区影响肝细胞中的==基因表达和**==染色质可及性,以及其他细胞类型。这些状态由抑制因子 TCF7L1 和 TBX3 以及其他核心肝细胞转录因子如 HNF4A、CEBPA、FOXA1 和 ONECUT1 共同驱动**。为了检查驱动这些细胞状态的增强子的架构,我们训练了一个名为 DeepLiver 的分层深度学习模型。我们的研究提供了一个多模态理解,涉及肝细胞身份及其分区状态下的调控代码,这可以用于设计具有特定活性水平和分区模式的增强子。
正文¶
细胞身份由基因调控网络(GRN)编码,转录因子(TF)通过结合增强子和启动子来调节基因表达。单细胞技术的进步使得我们能够同时测量单个细胞内的基因表达情况(单细胞 RNA 测序(scRNA-seq))和可及染色质1(带测序的单核转座酶可接近染色质测定(snATAC-seq)),提供了生成组织完整细胞状态空间无偏视图的机会,并探索特定细胞类型 GRN 的机制。
近期研究将细胞类型定义为一组(连续的)细胞状态,这些状态与细胞类型及其微环境之间的相互作用产生的细胞表型范围相对应。某些细胞类型可能会受到其在组织中的空间位置的影响,如在哺乳动物肝脏中。在每个肝小叶中,血液从门静脉流向中心静脉,形成营养物、氧气、激素和形态素的梯度,导致高度变化的环境轴(图 1a)。肝细胞的功能和代谢根据其在门静 - 中心轴上的位置而变化,因为它们暴露于不同的微环境中,这一现象被称为分区。以前的单细胞和空间转录组学研究已显示,不仅肝细胞功能,而且它们的转录组沿此轴线变化。然而,这些变化状态如何被基因组调控程序和增强子逻辑编码,以及分区状态如何与核心肝细胞 GRN 相互作用,目前尚不十分清楚。
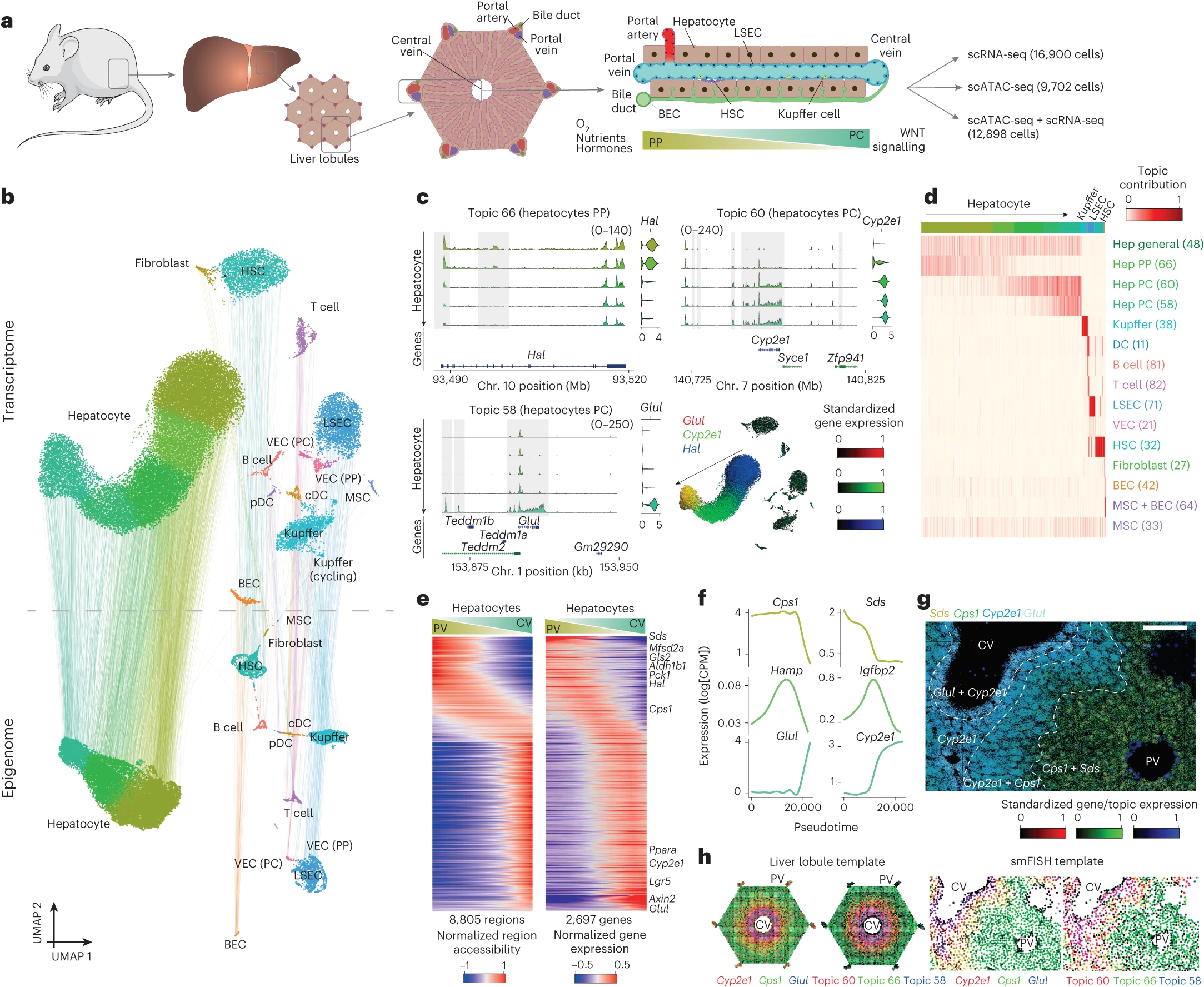
a,小鼠肝脏及实验设置概览。肝脏由称为肝小叶的六边形结构组成,血液从门静脉和肝动脉流入,并在中心静脉排出,形成氧气、营养物、激素和形态素(如 WNT)的梯度。b,基于转录组和表观基因组的均匀流形近似和投影(UMAP)投影(分别为 29,798 和 22,600 个细胞)。连接 UMAP 投影的线条连接来自同一细胞的转录组和表观基因组 UMAP 位置(通过单细胞多组学分析)。c,不同基因位点的假体染色质配置文件,以及显示每一类中相关基因的标准化基因表达的小提琴图。UMAP 投影显示了相关基因的基因表达,使用 RGB 编码。d,细胞话题贡献热图。e,标准化区域可及性和基因表达分区热图。细胞按伪时间排序(从门静脉(PP)到中心(PC)),显示受分区影响的区域和基因(8,805 个区域和 2,697 个基因)。基因表达热图右侧突出显示的基因位于以下排名位置(从上到下):3, 155, 201, 207, 233, 239, 866, 2,112, 2,535, 2,544, 2,556, 2,579。CV,中心静脉;PV,门静脉。f,沿着分区伪时间选择基因的 GAM 拟合基因表达轮廓。CPM,每百万计数。g,肝切片图像显示 Glul、Cyp2e1、Cps1 和 Sds 的 smFISH 轮廓。进行了三次独立实验,结果相似。刻度尺,100 微米。h,ScoMAP 肝小叶(4,498 个细胞)和 smFISH,按基因表达和话题贡献使用 RGB 编码上色。对于转录组和表观基因组数据,分别合并了来自五个和四个生物重复的细胞。cDCs,常规树突状细胞;pDCs,浆细胞样树突状细胞。
分区驱动肝细胞状态的异质性¶
为了表征肝脏细胞类型和状态在转录组和染色质水平上的特性,我们在小鼠肝脏中进行了两次 10x 单细胞多组学(ATAC + RNA)实验,共得到 12,898 个高质量细胞(方法)。为了提高分辨率,我们还进行了四次 10x 单核 RNA 测序(snRNA-seq)和两次 10x 单核 ATAC 测序(snATAC-seq)实验,分别提供了额外的 16,900 个单细胞转录组和 9,702 个单细胞 ATAC 配置文件(图 1a)。从 snRNA-seq 数据中,我们平均每个细胞获得了 5,863 个独特分子标识符(UMI)和 2,377 个表达基因。snATAC-seq 数据产生了 486,888 个可接近区域,使用 pycisTopic7 分组成 82 个调控话题(方法),平均每个细胞有 12,083 个独特片段和 7,241 个可接近区域,中位转录起始位点(TSS)富集为 16.1,峰值读数占比(FRIP)为 66%。我们观察到多组学实验和独立实验之间的整体质量相似(扩展数据图 1)。snRNA-seq 和 snATAC-seq 数据都区分了相同的细胞群体,对应于 14 种不同的细胞类型(图 1b),包括肝细胞、肝星状细胞(HSCs)、肝窦状内皮细胞(LSECs)、胆管上皮细胞(BECs)、库普弗细胞、门静脉和中心静脉血管内皮细胞(VECs)、间皮细胞(MSCs)、成纤维细胞和其他免疫细胞(例如 T 细胞、B 细胞和浆细胞样/常规树突状细胞)。此外,我们发现基因平均染色质可接近性和基因表达之间存在显著相关性(P < 2.2 × 10^-16;扩展数据图 2a-e)。
在所有动物中,我们发现肝细胞内存在单向梯度,通过基因表达和区域可接近性得到证实(图 1c-f)。为了确定这种梯度是否代表沿门静 - 中心轴的空间变化,我们用 100 个选定基因的单分子荧光原位杂交(smFISH)2进行了跨细胞类型和细胞状态的肝脏分析(图 1g,扩展数据图 2f-l 和补充表 1)。在肝细胞中,我们识别了三个主要区域,与肝细胞中门静 - 中心轴发现的三个调控话题一致;一个包含围绕中心静脉的肝细胞的 Glul+ 区域(话题 58),一个包括中心和中叶肝细胞的 Cyp2e1+ 区域(话题 60),以及一个包含中叶至门静肝细胞的 Cps1+ 区域3(话题 66)。这些区域与早期的空间转录组学研究一致。我们还识别了一个中叶区域,其中 Cyp2e1 和 Cps1 共表达,这也反映了话题 60(中心间接)和话题 66(门静)细胞贡献的重叠。对于每个基因和区域,我们通过广义加法模型(GAM)拟合了伪空间排序的细胞(从门静到中心肝细胞;方法),并发现 2,697 个基因(在至少有 3 个 UMI 计数的 6,823 个肝细胞基因中)和 8,805 个肝细胞区域(在 14,005 个共享肝细胞区域中)的相应表达和可接近性沿门静 - 中心轴显著变化(图 1e,f 和方法;调整后 P < 0.01)。此外,LSEC 和 HSC 簇也以梯度形式呈现,反映出不同的染色质可接近性话题和基因表达(扩展数据图 3)。这种集成的空间和单细胞分析也证实了 Ntn4+ LSECs 和 Ngfr+ HSCs 位于门静端,而 Kit+ LSECs 和 Spon2+ HSCs 位于中心端,这与先前的报道一致。我们识别了在 LSECs 和 HSCs 中沿门静 - 中心轴变化的 220 和 275 个基因以及 281 和 475 个区域(调整后 P < 0.01;方法)。在 220 个 LSEC 分区基因中,69% 与之前描述的分区肝内皮标记重叠。此外,BEC 簇可以清楚地定位在胆管中;中心和门静 VECs 围绕相应的血管,连同成纤维细胞;库普弗细胞主要位于门静和中区,没有强烈的分区模式,这与最近的研究一致。其他免疫细胞类型(如 B 细胞和 T 细胞)分布在所有区域(扩展数据图 2k)。
为了将整个转录组和表观基因组映射到 smFISH 空间图中,我们实现了 R 包单细胞组学映射到空间轴使用伪时间排序(ScoMAP)的新版本,结果是肝小叶的简化模板,其中可以可视化基因表达和区域可接近性(图 1h 和方法)。我们还在肝细胞中识别了一个有趣的批次效应,与老鼠之间的生理状态差异有关(补充说明和补充图 1-5)。总之,我们的小鼠肝脏空间单细胞多组学图谱显示,细胞类型身份和细胞状态在转录组和染色质可接近性水平上是一致反映的。
肝细胞核心 GRN 受分区抑制性转录因子调控¶
为了识别在肝脏不同细胞类型和分区状态下的候选转录因子(TF),我们使用 pycisTarget7(方法)对不同调控话题和细胞类型间的差异性可接近区域(DARs)进行了基序富集分析。由于基序往往可以与多个 TF 关联(经常是同一家族的几个成员),我们通过要求 TF 表达与基序富集的相关性来修剪注释的 TF 列表。这导致识别了在所有肝细胞可接近区域的 HNF1A、PPARA、NFIA、NFIB、HNF4A、CEBPA、FOXA1 和 ONECUT1 的基序,以及在门静脉和中心静脉可接近区域的 TBX3 和 TCF7L1/2 的基序(图 2a)。值得注意的是,Tbx3 仅在中心静脉肝细胞中表达,而其候选靶向区域仅在门静脉可接近。相反,Tcf7l1 在门静脉表达,其候选靶向区域在中心静脉可接近,而 Tcf7l2 在所有肝细胞中表达(图 2a)。TCF7L1 是 TCF7L2 的同源物,TCF7L2 是中心静脉活跃的 WNT 效应转录因子;这可能表明 TCF7L1 和 TCF7L2 绑定相同的基序—TCF7L2 在中心静脉用于激活,而 TCF7L1 在门静脉用于抑制。4
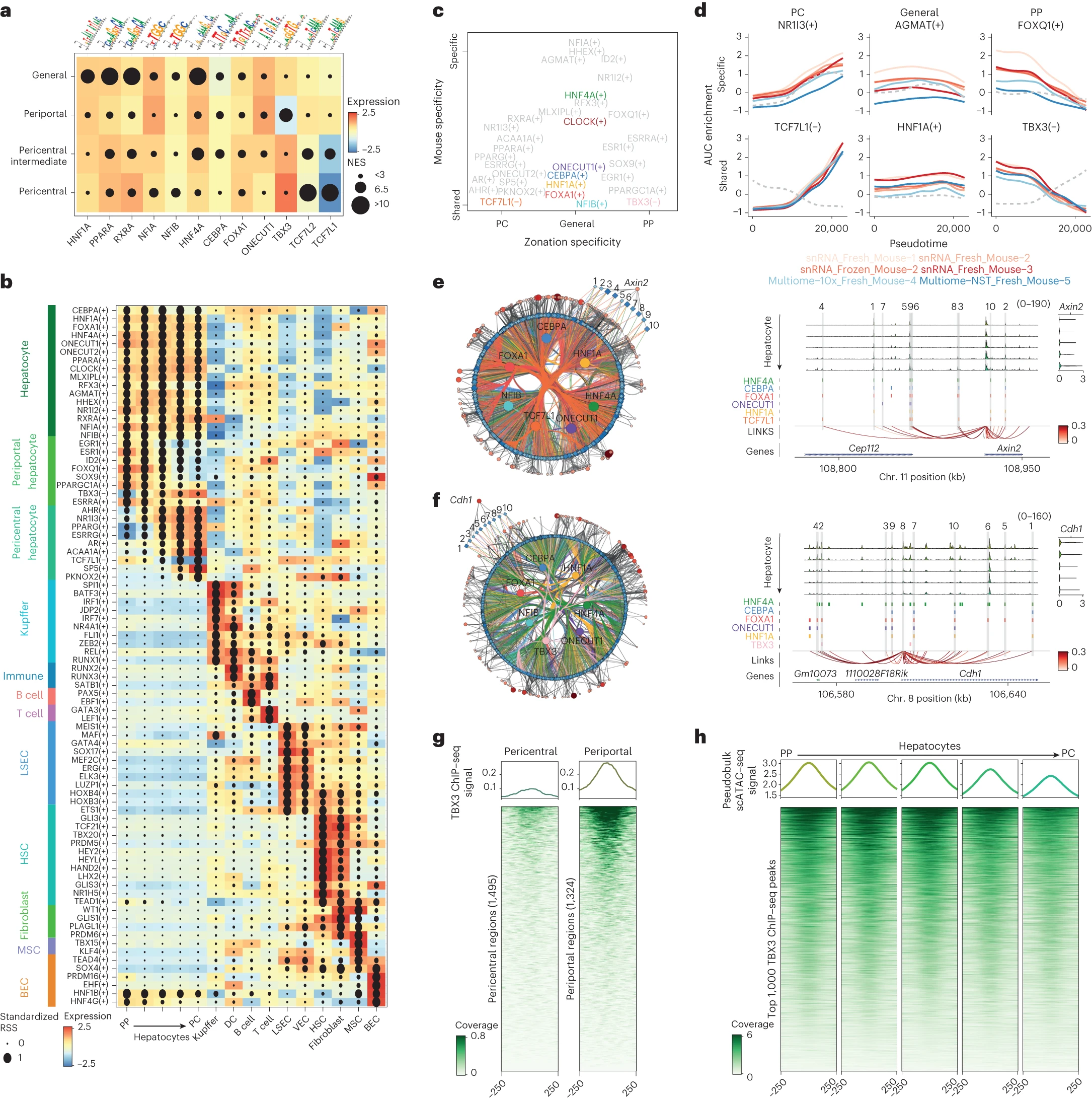
a,不同肝细胞类别中特定可接近区域内选定转录因子(TF)基序的最高标准化富集得分(NES;圆圈大小)。batch 按该肝细胞类别中相应 TF 的表达颜色显示。b,SCENIC+ eGRN 富集点图。基于基因的 eRegulon 特异性分数(RSS)由圆圈大小表示,颜色代表相应细胞类型中 TF 的表达。括号中的符号表示 TF 是激活(+)还是抑制(-)其靶基因。c,基于 eGRN AUC 的 PCA 图,其中第一主成分代表分区特异性,第二主成分代表 eRegulons 是在小鼠间共享还是特定于某些小鼠。d,沿分区伪时间为选定的 eRegulons 拟合的 GAM-fitted eGRN AUC 轮廓。灰色虚线表示 TF 表达轮廓的 GAM 拟合。snRNA-seq 样本用红色表示,单细胞多组学样本用蓝色表示。e,中心静脉核心肝细胞 eGRN,包含 265 个中心静脉标记基因和 1,439 个由选定核心 TF(CRM 得分>3)靶向且在小鼠间保守的区域。作为示例显示的是肝细胞假体染色质可接近性轮廓(从门静脉到中心静脉)在 Axin2 基因位点;预测的 TF 结合位点,区域到基因链接按 SCENIC+ 相关性得分着色,以及跨分区肝细胞类别的基因表达(从门静脉到中心静脉)。核心 eGRN 中的区域以灰色突出显示并编号。Chr.,染色体。f,门静脉核心肝细胞 eGRN,包含 175 个门静脉标记基因和 972 个由选定核心 TF(CRM 得分>3)靶向且在小鼠间保守的区域。作为示例显示的是肝细胞假体染色质可接近性轮廓(从门静脉到中心静脉)在 Cdh1 基因位点;预测的 TF 结合位点,区域到基因链接按 SCENIC+ 相关性得分着色,以及跨分区肝细胞类别的基因表达(从门静脉到中心静脉)。核心 eGRN 中的区域以灰色突出显示并编号。g,覆盖图显示 TBX3 ChIP-seq 在中心静脉和门静脉肝细胞区域的覆盖情况。h,肝细胞在前 1,000 个 TBX3 ChIP-seq 区域的覆盖情况。对于转录组和表观基因组数据,分别将来自五个和四个生物重复的细胞合并。
接下来,我们研究了预测的转录因子结合位点和增强子如何与候选靶基因相连。通过遵循 SCENIC+ 流程(方法),我们使用 pycisTopic 预测的染色质可接近性、pycisTarget 的 TF 顺式作用区(即 TF 及其潜在靶区域)和基因表达矩阵作为输入,编制了增强子 - 基因调控网络(eGRN)。通过机器学习方法,推断了区域 - 基因链接(在每个基因上下游 150kb 的空间内)和 TF- 基因关系。接下来,我们采用富集分析方法评估 TF 共表达模块是否与从基序/区域基链接中恢复的基因显著重叠,并随后保留了每个 TF 的最佳靶基因和区域集。一个 TF 及其预测的靶增强子和区域集被称为==增强子调节子(eRegulon)==。
此分析揭示了 180 个 eRegulons,包括库普弗细胞中的 SPI1、JDP2、RUNX1;B 细胞中的 EBF1 和 PAX5;T 细胞中的 GATA3 和 LEF1;LSECs 中的 GATA4、MEIS1 和 MAF;HSCs 中的 LHX2 和 TEAD1;成纤维细胞中的 WT1;以及 BECs 中的 TEAD4、DOX4 和 HNF1B 等,并且这些发现与文献相符。我们发现了一般性肝细胞特有的 eRegulons,例如 CEBPA、HNF1A、HNF4A、ONECUT1、FOXA1 和 NFIB,以及与分区相关的 eRegulons,如门静脉端的 ESR1 和 SOX9,中心静脉端的 PPARG 和 AR。重要的是,SCENIC+ 识别了 TBX3 和 TCF7L1 作为中心静脉和门静脉肝细胞中的转录抑制因子,分别靶向 193 和 520 个区域以及 77 和 119 个基因(图 2b)。换句话说,与 Tbx3 表达负相关的染色质区域位于与 Tbx3 反表达的基因附近(Tcf7l1 亦然)。此外,我们使用公开的 Hi-C 数据和转录因子染色质免疫沉淀后的测序(ChIP-seq)数据验证了 SCENIC+ eGRNs(扩展数据图 4a-e)。
由于我们之前观察到的转录组和表观基因组在不同生理状态的小鼠之间的差异,我们进行了 eGRN 富集分数的主成分分析(PCA),以依据它们的分区状态和小鼠特异性对肝细胞 eRegulons 进行分类(图 2c)。这使我们能够识别依赖于营养状态的 eRegulons,如 AGMAT 和 MLXIPL;依赖于激素水平的,如 NR1I2、NR1I3 和 RXRA;以及依赖于昼夜节律的,如 CLOCK。一些 eRegulons 受到小鼠生理状态和分区的双重影响,包括门静脉端的 ESRRA 和 FOXQ1 以及中心静脉端的 PPARA。在所有小鼠共有的核心 eRegulons 中,我们识别了 ONECUT1、CEBPA、HNF1A、FOXA1 和 NFIB,而 TBX3 和 TCF7L1 分别是核心中心静脉和门静脉(抑制性)eRegulons(图 2d)。这些生理状态并非独立于肝细胞核心 eGRN。例如,具有 3,442 个靶基因和 26,127 个靶区域的 HNF4A eRegulon,既有助于核心程序,也有助于依赖于生理状态的程序(扩展数据图 4f-i)。肝细胞 GRN 与控制细胞状态的 TFs 的合作,或者说混合,在分区方面表现得更为强烈:TBX3 和 TCF7L1 的靶区分别与至少一个一般核心 TF(HNF4A、HNF1A、CEBPA、ONECUT1、FOXA1、NFIB)的靶区有 94.8% 和 89.6% 的重叠。这表明肝细胞分区 eGRN 是一般肝细胞 eGRN 的一个子集(或层)。例如,预测在中心静脉肝细胞基因 Axin2 附近的候选增强子中有六个 TCF7L1 靶区。在门静脉基因 Cdh1 附近的候选增强子中预测有两个 TBX3 靶区。在这两种情况下,这些区域都被额外的一般核心 TFs 结合(图 2e,f)。使用公开的雄性和雌性小鼠的 scRNA-seq 数据,我们确认了肝细胞核心 eGRN 不受性别二态性的影响(补充说明和补充图 6)。综上所述,eGRN 推断显示,门静脉和中心静脉基因的表达直接在染色质水平上受到调节,与最近聚焦于它们启动子可接近性的研究形成对比(扩展数据图 5a,b)。
增强子序列决定肝细胞中的活性¶
到目前为止,我们所做的增强子和基因调控网络(GRN)预测基本上是基于基因表达、染色质可接近性和统计基序富集。然而,染色质可接近性并不总是与增强子活性相关联。为了评估预测的增强子是否活跃,我们使用之前发布的增强子条形码策略进行了大规模平行报告基因分析(MPRA)(图 3a)。我们选择了 10,845 个基因区域,这些区域仅基于它们在肝细胞中的可接近性,以混合方式克隆它们(方法),并将这个库转染到小鼠肝脏(7 个重复)和人类 HepG2 细胞(2 个重复)中(图 3a、方法、扩展数据图 6a-d 和补充表 2)。我们选择 HepG2 细胞作为体外模型,这是基于核心肝细胞 eGRN 转录因子的表达水平和库增强子的可接近性(在转换后),与其他小鼠肝细胞模型如 AML12 和 Hepa1-6 进行比较(补充说明和补充图 7 和 8)。
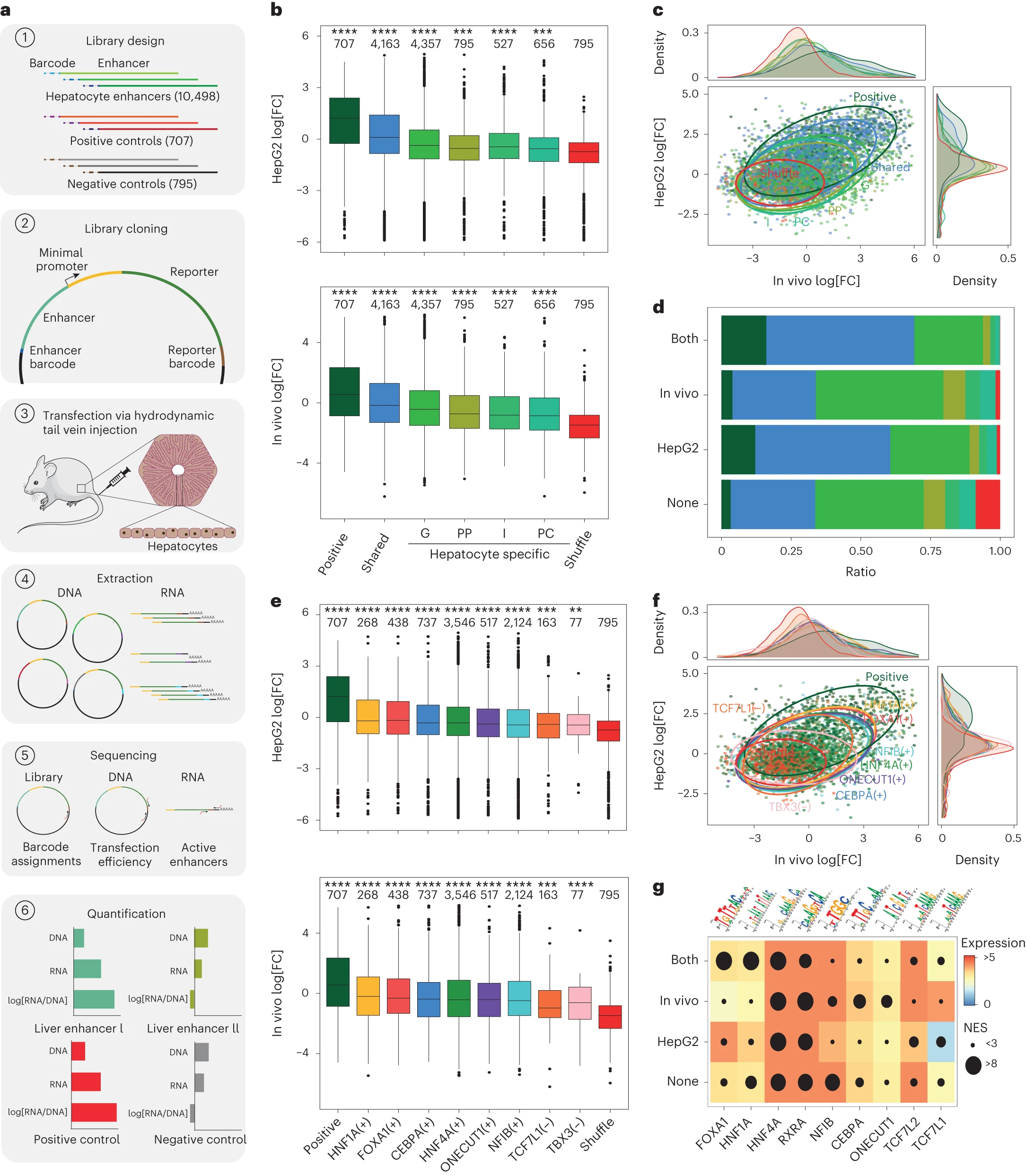
a, 小鼠肝脏的 MPRA 示意图。b, 每个增强子类别的 MPRA 对数变换后的倍数变化(log2[FC])。n = 9 个生物样本。每类增强子的数量在顶部标明。G, 通用;I, 中间。c, HepG2 细胞中高置信度增强子(n = 7,198)的 log2[FC] 值与体内的相关性,按增强子类型着色,数据椭圆表示各组。d, 高置信度活性类别中增强子类别的比例。None, 无活性(n = 4,285);in vivo, 仅体内活性(n = 806);HepG2, 仅在 HepG2 细胞中活性(n = 921);both, 在 HepG2 细胞和体内均有活性(n = 1,186)。e, 每个 eRegulon 的 MPRA 对数变换后的倍数变化(log2[FC])。n = 9 个生物样本。每个 eRegulon 测试的增强子数量在顶部标明。f, HepG2 细胞和体内高质量增强子(n = 7,198)的 log2[FC] 值的相关性,按 eRegulon 着色,数据椭圆表示各组。g, 在不同增强子(MPRA)活性类别中与选定转录因子(TF)相关的基序的最高标准化富集得分(圆圈大小),按 HepG2 细胞、体内或平均(both 和 none)中相应 TF 的表达着色。对于 b 和 e 中的箱形图,中心线显示中位数,顶部和底部铰链代表上四分位数和下四分位数,须从铰链向外延伸到最大值和最小值,不超过铰链的 1.5 倍四分位距。进行单侧秩和 Wilcoxon 测试以评估每组的 log2[FC] 值是否大于随机区域的值。星号表示比较的 Bonferroni 调整后 P 值;*, P ≤ 0.0001;*, P ≤ 0.001;**, P ≤ 0.01;, P ≤ 0.05;NS, P > 0.05。体内和 HepG2 细胞实验分别使用了七个和两个生物重复。源数值数据提供为源数据。
在肝细胞中可接近的区域表现出比随机序列显著更高的增强子活性(图 3b、c),并且多次重复的 MPRA 分析显示出很强的相关性(相关系数在 0.82 到 1 之间;扩展数据图 6a)。我们使用随机序列作为背景来确定一个最佳活性截断值(方法),这将 2,913 个增强子分类为至少在其中一个系统中活跃(806 个仅在体内,921 个仅在 HepG2 细胞中,1,186 个在两者中均活跃,调整后的 P < 0.1;方法)和 4,285 个区域为非活跃。换句话说,40.5% 的 ATAC 峰通过 MPRA 显示为活跃,这与其他研究一致。在小鼠肝脏中体内活跃的小鼠区域中,64% 是远端增强子,27% 是启动子。相比之下,在人类 HepG2 细胞中活跃的区域,54% 是启动子(图 3d 及扩展数据图 6c)。
SCENIC+ 预测的 HNF1A、HNF4A、FOXA1、CEBPA、ONECUT1、NFIB 和 TCF7L1 的靶区域的活性均显著高于随机区域,它们预测的靶区域分别有 45%、39%、43%、39%、35%、32% 和 26% 是活跃的(图 3e、f)。TBX3 的靶区域在体内更活跃(20% 的区域在体内活跃,而在 HepG2 细胞中只有 5% 的区域活跃)。与此一致,活跃与非活跃区域的基序富集分析,随后使用随机森林模型的基于分类器的特征选择识别了 HNF1A、HNF4A、FOXA1、CREB 和 AP-1 基序作为活跃增强子的决定性特征(图 3g、方法及扩展数据图 6d、e)。这种基于基序的分类器预测增强子活性的接收者操作特征曲线(AUROC)下面积为 0.71(随机 AUROC 为 0.54),精确度 - 召回曲线(AUPR)下面积为 0.44(随机 AUPR 为 0.27;扩展数据图 6e)。
DeepLiver 解码肝细胞增强子语法¶
为了进一步审查增强子逻辑如何支撑增强子活性和分区,我们训练了一个名为 DeepLiver 的分层深度学习模型。我们首先训练了一个卷积神经网络(CNN)来分类 DNA 序列到肝脏调控话题(称为话题 -CNN),使用 219,823 个标注区域作为输入。然后,使用话题 -CNN 模型学习的权重来初始化另外两个 CNN,一个用于预测体内 MPRA 活性(MPRA-CNN),另一个用于预测分区(分区 -CNN,使用分区可接近性类别作为输出变量)。这种迁移学习策略克服了我们可用于活性(4,215 个区域)和分区(4,181 个中心静脉,1,372 个门静脉,12,122 个通用)的区域数量有限的问题(方法)。对于每个模型,最佳训练周期是基于其在测试数据(输入数据的 10%;扩展数据图 7a)上的准确性和损失选择的。这三个模型的 AUROC 和 AUPR 都比随机对照分类器高,与细胞类型相关的话题的性能比低贡献话题更高(扩展数据图 7b-d)。为了验证 DeepLiver 的预测,我们使用之前发布的在体内对合成序列进行的 MPRA 数据集进行验证,发现 DeepLiver 的预测与实验测量结果相关性很好(R = 0.68)(扩展数据图 7e)。总之,DeepLiver 将给定的 DNA 序列分配给肝脏中的细胞类型(由话题表示),并预测肝细胞增强子的活性和分区模式(图 4a)。
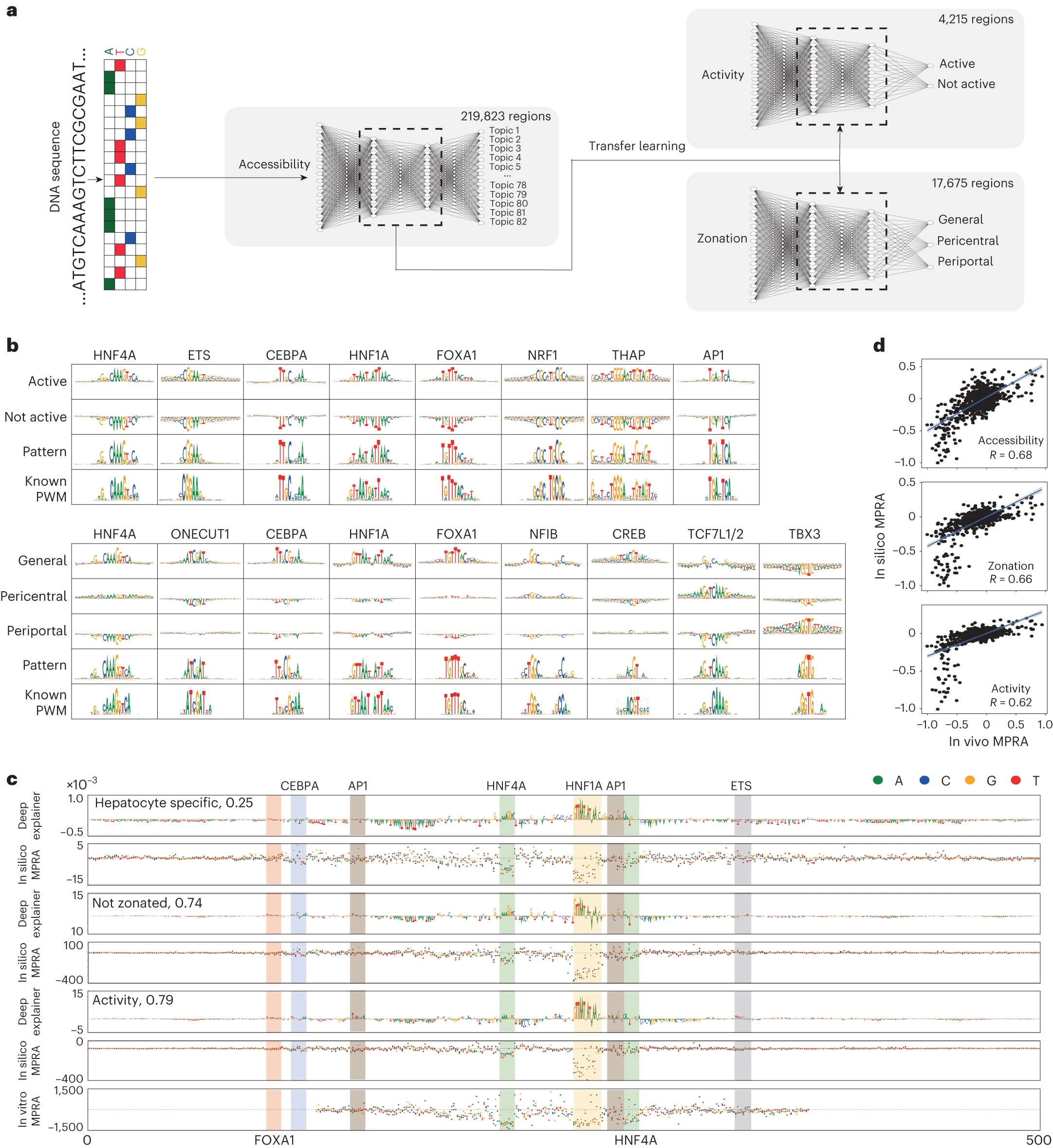
a, DeepLiver 概览。首先,训练一个 CNN 来将 DNA 序列分类到相应的调控话题中(219,823 个序列)。在第一个模型中学到的权重被用来初始化活性和分区模型。活性模型基于 DNA 序列在体内的 MPRA 活性进行分类(使用 4,215 个高置信度区域),而分区模型则基于序列在肝细胞上的分区模式进行分类(中心静脉、门静脉或非分区/通用,使用 17,675 个区域进行训练)。b, 在活性和分区模型中识别的 TF-MoDISco 模式,展示了每个类别的贡献分数以及与 cisTarget 基序集合中最相似的 PWM。c, 使用 DeepExplainer 和饱和突变分析图对 Aldob 增强子的可接近性、分区和活性模型进行分析(hg19:染色体 9:104195449–104195449),并突出显示了基序。下方显示的饱和突变之前在此增强子上执行过。d, DeepLiver 体内突变分析与 Aldob 增强子中的实验饱和突变之间的相关性。蓝线代表拟合的线性回归,灰色带代表 95% 置信区间带。
接下来,我们使用 DeepExplainer 来评估增强子分类中每个核苷酸的贡献,并使用 TF-MoDISco 来从贡献分数中的重复模式识别基序(图 4b)。对于 MPRA-CNN,我们识别了促进增强子活性的模式,这些模式对应于 HNF4A、CEBPA、HNF1A、FOXA1 和 AP-1 的基序,以及几个与启动子相关的基序,如 ETS、NRF1 和 THAP。在分区模型中,识别出的 HNF4A、ONECUT1、CEBPA、HNF1A、FOXA1、CREB 和 NFIB 基序被确定为调节所有肝细胞可接近性的调控因子;而 TCF7L1/2 和 TBX3 基序分别与中心静脉和门静脉的可接近性相关,这与我们的 GRN 级别分析一致。
我们进一步使用 DeepLiver 来研究序列变异对增强子特异性、活性和分区的影响。为了验证 DeepLiver 的体内突变分析(方法),我们将突变的预测效果与早期研究中六个增强子的实验饱和突变数据进行了比较(分别来自体内和 HepG2 细胞的三个;图 4c,d 及扩展数据图 7g,h)。DeepLiver 对增强子突变效果的预测与实验结果相关性良好(相关系数在 0.36 到 0.75 之间;扩展数据图 7f)。
接下来,我们使用 TF-MoDISco 模式和 SCENIC+ 位置权重矩阵(PWMs)来识别肝细胞序列中的 TF 结合位点(方法)。我们为 TBX3、TCF7L1、FOXA1、HNF1A、HNF4A、NFIB、ONECUT1 和 CEBPA 确定了 1,235 至 6,991 个靶区域,与 SCENIC+ 预测的靶区域有良好的重叠(17-70%;扩展数据图 8a-c)。为了验证预测的结合位点,我们将我们的预测与之前发布的 HNF4A、CEBPA、FOXA1 和 ONECUT1 的 ChIP–seq 数据进行了比较,发现当区域集中在预测的结合位点上时,对应的 TF 有特定的信号(扩展数据图 8d)。最后,我们评估了重叠区域中基序实例之间的距离。这表明 TCF7L1 和 HNF4A 经常重叠,这可能是由于基序之间的相似性(分别是 GATCAAAG 和 CAAAGTCA,其中共同的基底以粗体突出显示)。另一方面,FOXA1、HNF1A、CEBPA、NFIB 和 TBX3 通常位于 HNF4A 基序附近(扩展数据图 8e,f)。
接下来,我们使用 DeepLiver 来解释 SCENIC+ 中核心中心静脉和门静脉 eGRN 的增强子,现在是以碱基对分辨率进行(图 5 和 6;https://doi.org/10.6084/m9.figshare.24115986)。例如,在 Cdh1 增强子上,DeepLiver 发现 FOXA1、HNF4A 和 HNF1A 位点是增强子可接近性和活性的驱动因子,而 TBX3 位点(一个二聚体基序和两个单体)被预测将使增强子变为门静脉(图 5a)。一致地,我们在这个区域发现了 HNF4A 和 FOXA1 的 ChIP–seq 信号,但没有发现 CEBPA 或 ONECUT1 的 ChIP–seq 信号(图 5e)。这个增强子的可接近性和 Cdh1 基因表达都与 TBX3 表达呈负相关(分别为 -0.44 和 -0.17;图 5b-e)。
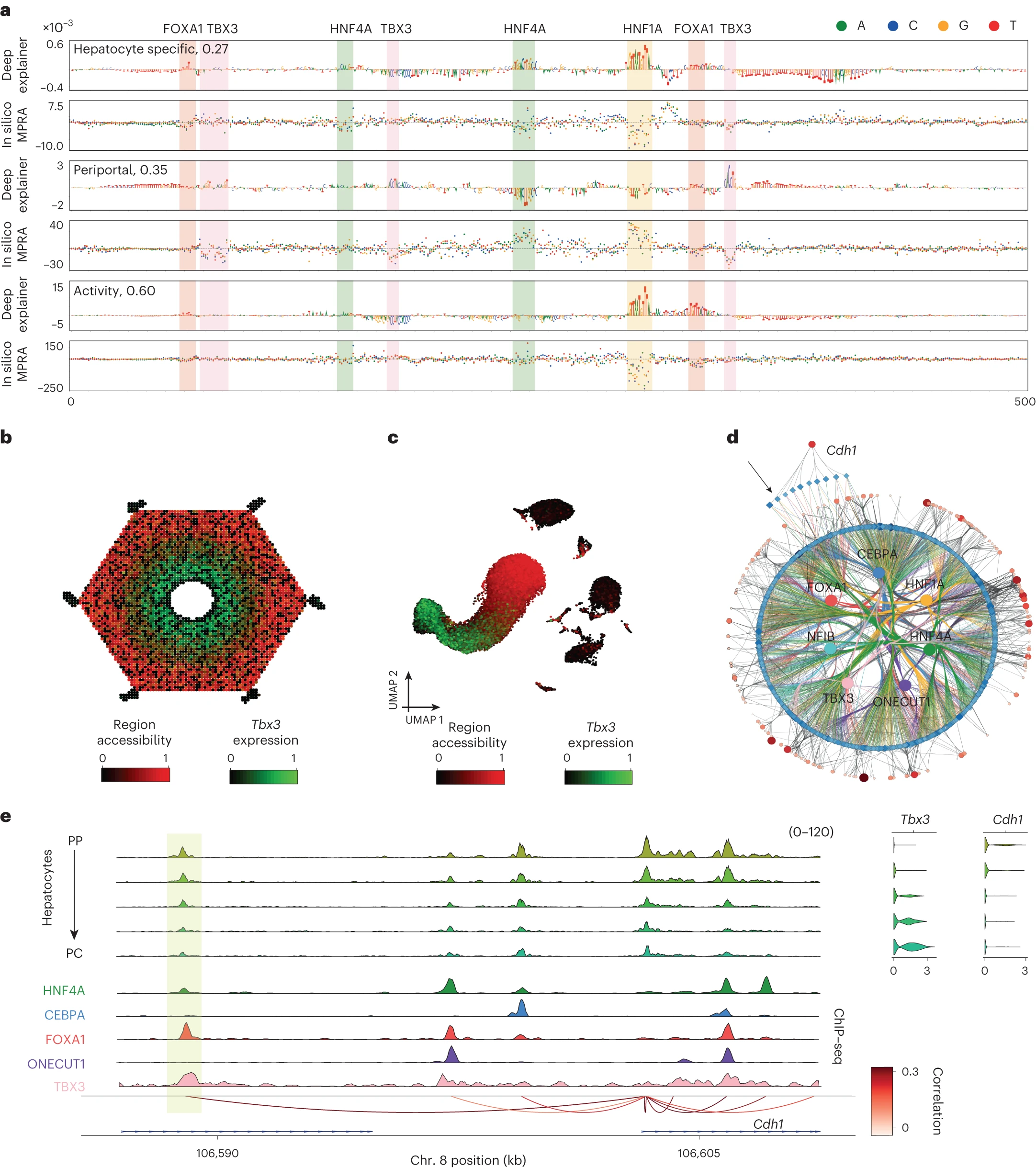
a, 使用 DeepExplainer 和饱和突变分析图展示 Cdh1 门静脉增强子(染色体 8: 106588720–106589220)上的可接近性、分区和活性模型,其中突出显示了基序。可接近性模型突出显示了使增强子在肝细胞中(与肝脏中其他细胞类型相比)可接近的核苷酸;分区模型突出显示了促使增强子成为门静脉的核苷酸;活性模型突出显示了在其活性中起作用的核苷酸。b, ScoMAP 肝小叶模板(4,498 个细胞)按区域可接近性和 TBX3 表达着色。c, 基于转录组的 UMAPs(29,798 个细胞)按区域可接近性和 TBX3 表达着色。d, 门静脉核心肝细胞 eGRN,包含 175 个门静脉标记基因和 972 个由选定核心转录因子(CRM 得分>3)靶向且在小鼠间保守的区域。Cdh1 增强子区域由箭头指示。e, 覆盖图显示伪批量可接近性轮廓、ChIP-seq 覆盖(针对 HNF4A、CEBPA、FOXA1、ONECUT1 和 TBX3)、SCENIC+ 区域到基因链接按相关性得分着色,以及 Cdh1 和 Tbx3 在分区肝细胞类别中的表达(从门静脉到中心静脉)。Cdh1 增强子区域以黄色突出显示。对于转录组和表观基因组数据,分别合并了来自五个和四个生物重复的细胞。
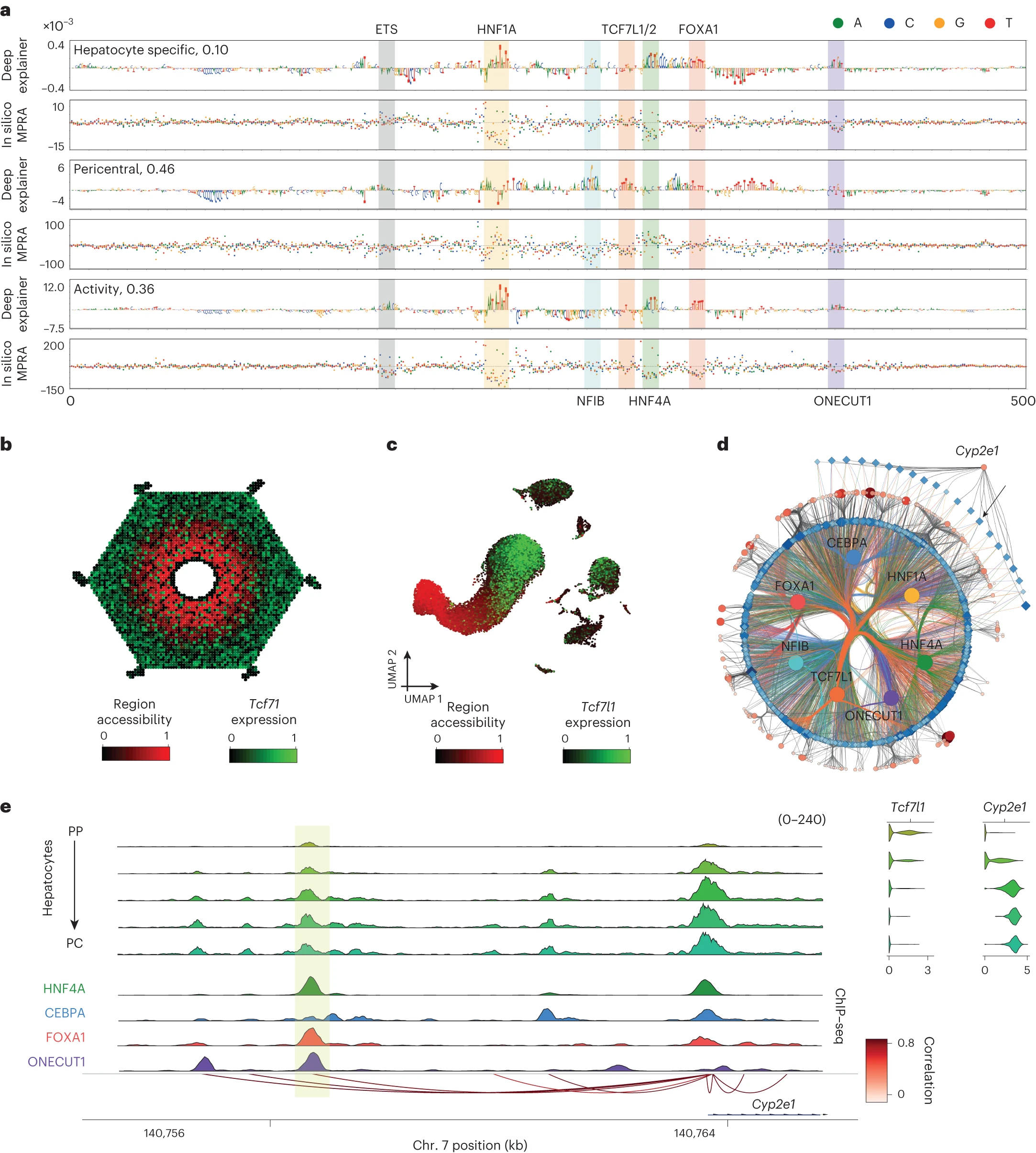
a, 使用 DeepExplainer 和饱和突变分析图对 Cyp2e1 中心静脉增强子(染色体 7: 140756424–140756924)上的可接近性、分区和活性模型进行分析。可接近性模型突出显示了在肝细胞上使增强子可接近的核苷酸(与肝脏中其他细胞类型相比);分区模型突出显示了促使增强子成为中心静脉的核苷酸;活性模型突出显示了在其活性中起作用的核苷酸。b, ScoMAP 肝小叶模板(4,498 个细胞)按区域可接近性和 Tcf7l1 表达着色。c, 基于转录组的 UMAPs(29,798 个细胞)按区域可接近性和 Tcf7l1 表达着色。d, 中心静脉核心肝细胞 eGRN,包括 265 个中心静脉标记基因和 1,439 个由选定核心转录因子(CRM 得分>3)靶向且在小鼠间保守的区域。Cyp2e1 增强子区域由箭头指示。e, 覆盖图显示伪批量可接近性轮廓、ChIP–seq 覆盖(针对 HNF4A、CEBPA、FOXA1 和 ONECUT1),SCENIC+ 区域到基因链接按相关性得分着色,以及 Cyp2e1 和 Tcf7l1 在分区肝细胞类别中的表达(从门静脉到中心静脉)。Cyp2e1 增强子区域以黄色突出显示。对于转录组和表观基因组数据,分别合并了来自五个和四个生物重复的细胞。
在中心静脉 Cyp2e1 增强子上,DeepLiver 识别出 HNF1A、FOXA1 和 ONECUT1 位点,这些位点有助于增强子的可接近性和活性,以及一个 ETS 位点,它有助于活性但不影响可接近性,这在其他增强子中也有观察到(图 6a;https://doi.org/10.6084/m9.figshare.24115986)。另一方面,NFIB 位点有助于可接近性(但不影响活性),而 TCF7L1/2 位点则独特地出现在分区模型中,有助于使增强子成为中心静脉。与此一致,我们在这一区域观察到 HNF4A、FOXA1 和 ONECUT1 的 ChIP-seq 信号(图 6e)。TCF7L1 表达与区域可接近性和基因表达呈负相关(分别为 -0.32 和 -0.40;图 6b-e)。这些观察结果表明 TBX3 和 TCF7L1 可能抑制这些区域。总之,DeepLiver 在碱基对分辨率上解码增强子的可接近性、活性和分区,并能预测调节肝细胞增强子活性和分区的变异。
验证分区抑制性转录因子和增强子¶
DeepLiver 模型为肝细胞增强子提供了有意义的解释,并预测这些增强子由核心肝细胞代码组成,混合了分区抑制性转录因子 TBX3 和 TCF7L1 的结合位点,分别使增强子活性偏向中心静脉区或门静脉区。为了进一步测试这些预测,我们首先在 SCENIC+ 网络上进行了模拟实验,遵循我们之前发布的扰动模拟策略。在肝细胞中模拟 Tbx3 或 Tcf7l1 的敲低和过表达(方法)表明,Tbx3 的过表达和 Tcf7l1 的敲低可以将门静脉肝细胞转变为中心静脉状态,而 Tbx3 的敲低或 Tcf7l1 的过表达可以将中心静脉肝细胞转变为门静脉状态(图 7a-c)。SCENIC+ eGRN 预测 TBX3 和 TCF7L1 直接相互抑制。因此,敲低或过表达其中一个转录因子会引起另一个的上调或下调(同时也会导致另一个的靶基因下调和上调;图 7c)。
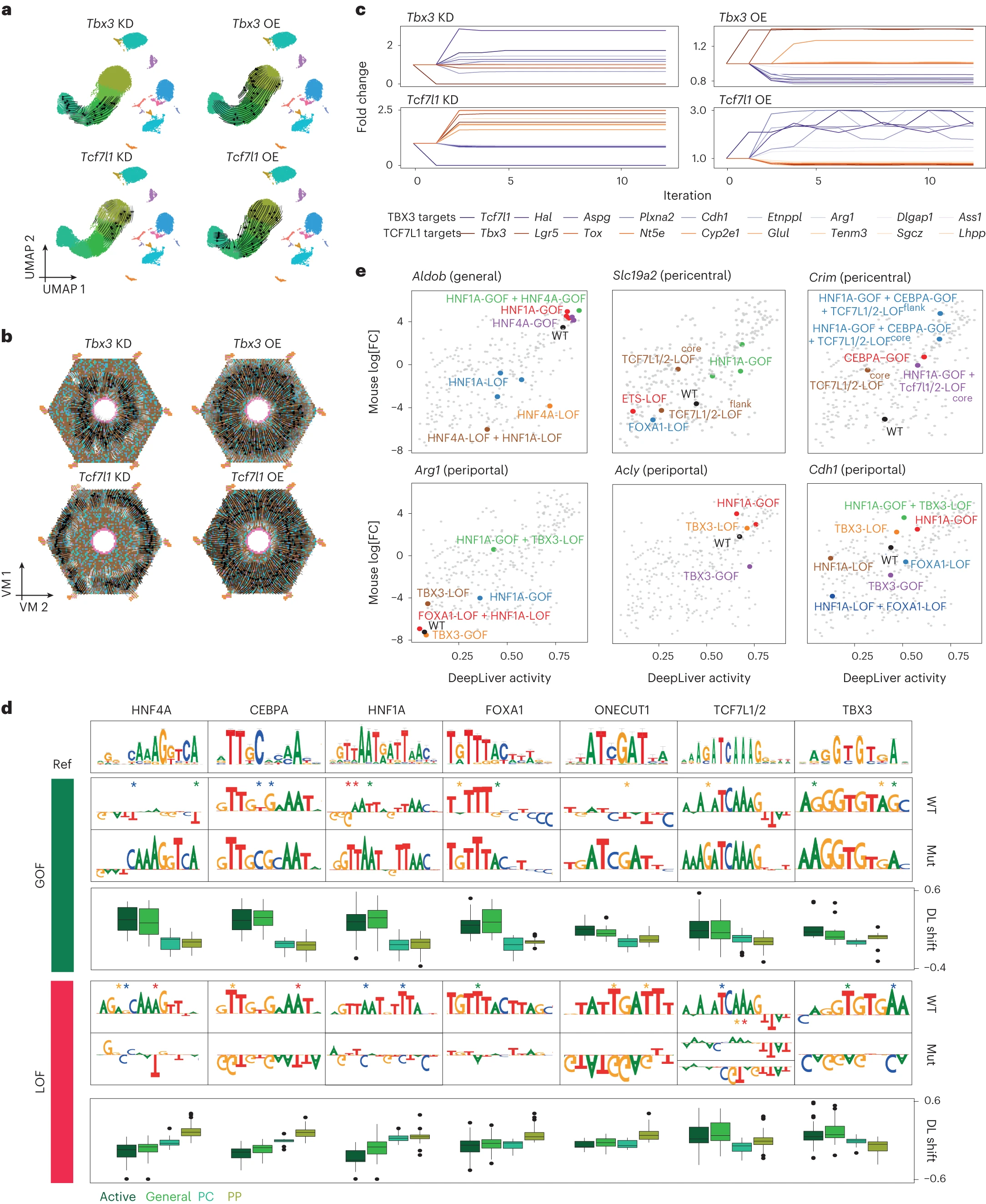
a, 在 Tbx3 或 Tcf7l1 敲低(KD)或过表达(OE)后,通过单核 RNA 测序(snRNA-seq)UMAP(29,798 个细胞)显示的模拟细胞转移,用箭头表示。箭头的阴影基于模拟后每个细胞移动的距离。对于 UMAP,综合了来自四个 snRNA-seq 和两个单细胞多组学实验的细胞数据。b, 在 Tbx3 或 Tcf7l1 敲低(KD)或过表达(OE)后,ScoMAP 肝小叶虚拟图(VM;4,498 个元细胞)上的模拟细胞转移,用箭头表示。箭头的阴影基于模拟后每个细胞移动的距离。c, 在中心静脉肝细胞中模拟 Tbx3 敲低和 Tcf7l1 过表达,以及在门静脉肝细胞中模拟 Tcf7l1 敲低和 Tbx3 过表达后,选定基因(TBX3 靶基因以紫色显示,TCF7L1 靶基因以橙色显示)的预测倍数变化。d, 基于 DeepLiver 的序列突变(mut)概览,这些突变引入到野生型增强子中,以改变活性和分区模式。这些变异引起了改善的基序(GOF)的出现或它们的破坏(LOF)。顶部显示来自 cisTarget 数据库的参考转录因子基序(Ref)。每个变异下方的箱形图显示了 DeepLiver 预测的活性(活跃)或分区(通用、中心静脉或门静脉)得分的转变。在箱形图中,顶部/下部铰链代表上/下四分位数,须从铰链向外延伸至最大/最小值,不超过 1.5 倍四分位距。中位数作为中心。DL 表示深度学习。e, 体内 MPRA 对数变换后的倍数变化(log2[FC])与 DeepLiver 活性得分的比较,突出显示了每个增强子的序列变异。对于 MPRA 实验,在 HepG2 细胞和体内分别进行了三次和八次生物重复实验。
在第二个实验中,我们在 DeepLiver 模型的指导下,对一组肝细胞增强子引入了特定的突变,然后使用体内 MPRA 测量它们的活性。我们选定了 13 个门静脉和 21 个中心静脉增强子,这些增强子被预测会分别由 TBX3(在中心静脉)和 TCF7L1(在门静脉)抑制,这一预测由 SCENIC+ 和 DeepLiver 共同支持。我们引入了影响 HNF4A、CEBPA、HNF1A 和 FOXA1 结合位点的功能增益(GOF)和功能丧失(LOF)突变,以及 TBX3 和 TCF7L1/2 基序的突变(图 7d 和扩展数据图 9a-c),共产生了 455 个序列。首先在小鼠肝脏和人类 HepG2 细胞上使用批量 MPRA 测试了这些增强子变异体的活性(方法、扩展数据图 9d-g 和补充表 3),其中 HNF4A、HNF1A、CEBPA 和 FOXA1 的 GOF 变异确实如 DeepLiver 预测的那样导致了更高的活性(图 7e 和扩展数据图 9c,f,g)。预测的分区转录因子 TBX3 和 TCF7L1 结合位点的变异也显示了变化,但这些变化在批量实验中更难评估,因为门静脉和中心静脉肝细胞是混合的(图 7e)。
为了解决这个问题,我们对通过荧光活细胞分选(FACS)按区域分选的肝细胞进行了 MPRA 实验,使用中心静脉和门静脉表面蛋白 CD73(由 Nt5e 编码)和 ECAD(由 Cdh1 编码)分别进行分选,按照之前发布的协议(图 8a、方法和扩展数据图 10a)。分选的细胞组确实代表了中心静脉和门静脉肝细胞,如单独分数的批量 ATAC-seq 轮廓所示,这与 snATAC-seq 的分区轮廓一致(图 8b)。接下来,我们使用 MPRA 分析了分选分数上的增强子活性(方法和补充表 3)。正如预期的那样,中心静脉增强子在中心静脉分数中显示出更高的活性,反之亦然(图 8c 和扩展数据图 10b,c)。HNF1A 和 HNF4A 的 GOF 变异在两个分数中都导致活性增加,CEBPA 和 FOXA1 变异的效果较轻。这些基序的破坏与它们的野生型相比降低了增强子活性(图 8c,d 和扩展数据图 10d)。TBX3 的 LOF 和 TCF7L1 的 GOF 导致中心静脉分数中活性增加,TCF7L1 的 LOF 导致门静脉群体中活性增加(图 8c,d 和扩展数据图 10d)。
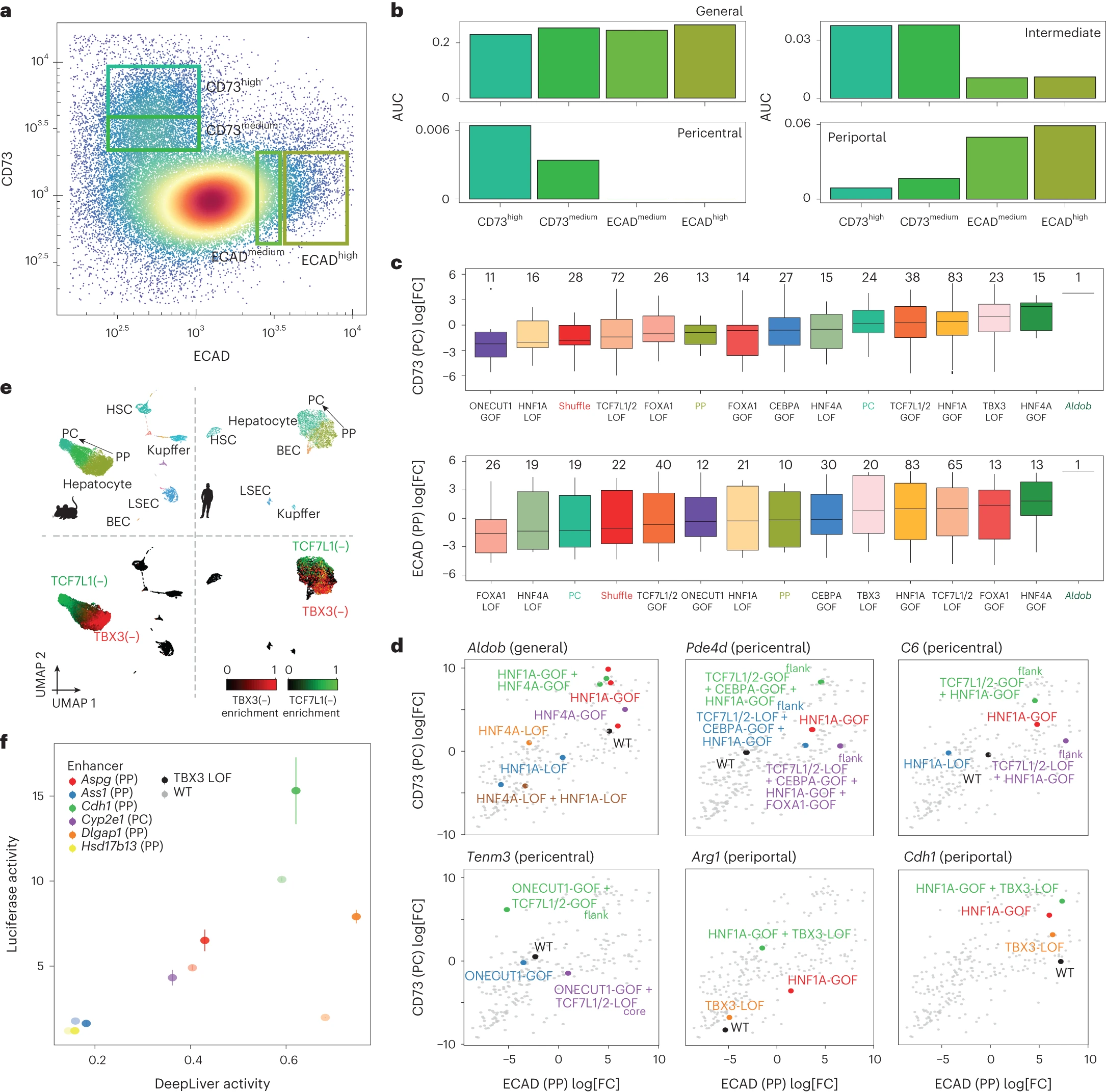
a, 根据 CD73 和 ECAD 的强度进行的选定细胞的 FACS 分析。矩形框标示了沿门静 - 中心轴选定的区域。b, 在分选的细胞群体上,核心一般区域、中心静脉区域、中心静脉 - 中间区域和门静脉区域的 AUCell 富集情况。c, 每个变异类别下,聚合的 CD73 和 ECAD 群体的体内 MPRA 对数变换后的倍数变化(log2[FC])值。每组增强子的数量在顶部标示。Aldob 增强子的活性由一条线指示。中心线显示中位数,顶部和底部铰链代表上下四分位数,须从铰链向外延伸到最大和最小值,不超过铰链的 1.5 倍四分位距。对照组和野生型序列被突出显示。使用了四个生物重复。d, CD73 MPRA 对数变换后的倍数变化(log2[FC])与 ECAD MPRA 对数变换后的倍数变化(log2[FC])值的比较,突出显示了每个增强子的序列变异。使用了四个生物重复。e, 小鼠和人类肝脏 snATAC–seq UMAP(分别为 22,600 和 6,366 个细胞)按细胞类型(顶部)和 eRegulon 富集(底部)着色。对于小鼠 UMAP,综合了来自四个生物重复的细胞。f, 在 HepG2 细胞中的荧光素活性与 DeepLiver 活性得分对选定增强子及其变异体的比较。每个增强子进行了 4 次生物独立的荧光素实验。数据表示为均值±标准误。WT 表示野生型。
-
细胞核的内部是一个高频动态的体系,大多数染色质以直径为30 nm染色质纤维的形式存在。这是在核小体串珠结构的基础上螺旋化而成的结构形式。核小体在整个基因组上的分布是不均匀的。核小体结构致密的区域,其与基因表达相关的结构区域相对封闭,不利于转录因子等调控元件与之结合,从而导致基因沉默;而核小体分布较少的区域,允许转录调控元件与这些区域的启动子、增强子相互接近并结合,进而调控基因表达。因此,染色质DNA在行使转录以及复制合成的功能过程中,蛋白质需要脱离DNA,使其处于裸露状态,即折叠结构变成松散状态时,这种裸露的DNA便于启动转录以调控基因的表达。这种允许启动子、增强子、绝缘子、沉默子等顺式调控元件和反式作用因子能够与染色质DNA物理接触的程度就称为染色质可及性(Chromatin Accessibility),由核小体的占有率和拓扑结构以及其他阻碍DNA结合的染色质结合因子决定。简而言之,染色质可及性就是指染色质的物理压缩程度。这个过程类似于将一份压缩文件(封闭染色质)进行解压(开放染色质)后才能看到具体的文件内容 ↩
-
单分子荧光原位杂交(single-molecule fluorescence in situ hybridization,smFISH)技术是一种通过用偶联荧光基团的寡核苷酸探针,对固定细胞或组织中单个mRNA分子进行成像的方法。smFISH可对RNA进行定位、定量,以此对目标转录本进行实时研究 ↩
-
FISH证实拟时序分析(多组学分析结果)结果正确 ↩
-
富集分析显示在肝脏的不同区域,肝细胞的染色质可及位点有不同的改变(不同空间位置中,DNA 层次的表达情况) ↩