Single cell transcriptomic analyses reveal distinct immune cell contributions to epithelial barrier dysfunction in checkpoint inhibitor colitis
摘要¶
免疫检查点抑制剂(ICI)疗法已经革新了肿瘤学领域,但其治疗受到免疫相关不良事件(irAEs)的限制,包括检查点抑制剂结肠炎(irColitis)。对于驱动 irColitis 的致病机制知之甚少,因为这种情况在小鼠等模型生物中并不常见。为了确定 irColitis 的分子驱动因素,我们使用单细胞多组学技术,对 13 名患有癌症并发展为 irColitis 的患者(9 人接受抗 PD-1 或抗 CTLA-4 单药治疗,4 人接受双重 ICI 治疗;大多数患者患有皮肤癌或肺癌)、8 名接受 ICI 治疗的对照患者和 8 名健康对照的结肠黏膜和血液中的约 300,000 个细胞进行了分析。患有 irColitis 的患者显示出黏膜调节性 T 细胞(Tregs)的扩展,表达 CXCL13 和 Th17 基因程序的 ITGAEHi CD8 组织驻留记忆 T 细胞和循环的 ITGB2Hi CD8 T 细胞。与抗 PD-1 治疗相比,抗 PD-1/CTLA-4 治疗相关的结肠炎中细胞毒性 GNLYHi CD4 T 细胞、循环的 ITGB2Hi CD8 T 细胞和表达缺氧基因程序的内皮细胞进一步扩展。患有 irColitis 的患者的腔内上皮细胞表达与细胞凋亡、细胞更新增加和吸收不良相关的 PCSK9、PD-L1 和干扰素诱导的特征。总体来看,这些数据表明循环 T 细胞和上皮 - 免疫细胞互作在 PD-1/CTLA-4 依赖的耐受性和屏障功能中的关键作用,并确定了 irColitis 的潜在治疗靶点。
正文¶
免疫检查点抑制剂(ICIs)靶向 PD-1 和 CTLA-4,已经革新了免疫肿瘤学领域。然而,这些治疗受到免疫相关不良事件(IRAEs)的限制,这些炎症副作用的严重程度从轻微到潜在致命不等。在接受免疫治疗的患者中,超过 85% 会出现 IRAEs,导致 10-40% 的患者中断治疗【1】。免疫检查点抑制剂相关的结肠炎(irColitis)是最常见和严重的 IRAEs,发生在多达 25% 的患者中【2】。irColitis 表现为上皮内淋巴细胞增加、中性粒细胞浸润、上皮细胞凋亡和/或孤立的淋巴细胞性结肠炎【3,4】。由于部分原因是接受 ICI 阻断抗体治疗的小鼠不会出现结肠炎【5,6】,目前对导致 irColitis 的机制了解甚少。相关研究建议 T 调节细胞(Treg 细胞)、CD21 低 B 细胞、TNF-α、IL-17A、MAIT 细胞、菌群失调和早期 T 细胞受体(TCR)多样化在其中起作用【7-13】。先前对结肠黏膜免疫细胞的单细胞 RNA 测序(scRNA-seq)研究显示,组织 CD8 T 细胞扩展【14,15】,然而,由于这些研究未检查组织中的上皮细胞或间质细胞或循环免疫细胞,因此尚不清楚这些附加群体和免疫细胞与非免疫细胞之间的相互作用如何促进疾病的发病。irColitis 通常用类固醇作为一线治疗,对于类固醇难治性疾病,使用靶向 TNF-α或整合素α4β7 的生物制剂【16,17】。多项研究表明,用大剂量类固醇治疗 IRAEs 会削弱 ICI 的抗肿瘤功效【18,19】。急需对 IRAEs 发病机制的改进理解,以便于早期诊断和指导新治疗靶点的开发,同时保留抗肿瘤反应。
在本研究中,我们调查了 irColitis 的五个尚不明确的方面:(1)CD8 T 细胞与维持炎症的其他免疫和非免疫细胞之间的相互作用;(2)循环细胞对疾病的贡献;(3)定义结肠功能障碍的上皮和间质缺陷;(4)抗 PD-1 与抗 PD-1/CTLA-4 相关的 irColitis 特征;(5)新潜在治疗靶点的识别。我们的发现揭示了 irColitis 背后巨大的细胞转录异质性,并确定了临床诊断和治疗干预的路径。
结果¶
研究设计¶
为了定义与 irColitis 相关的细胞群体,我们从患有 irColitis 的患者、没有结肠炎的 ICI 治疗患者和健康对照的结肠免疫细胞、上皮和间质细胞核以及循环外周血单个核细胞(PBMCs)中生成了 scRNA-seq 数据(图 1a–c,补充表 1–4,扩展数据图 1 和补充图 1)。大多数接受 ICI 治疗的患者患有皮肤癌,并接受了 PD-1 阻断治疗。生物样本在症状发作后 2 周内获取。由于样本有限,并非每位患者都进行了所有测试(补充表 2)。我们观察到两组对照群体之间的细胞谱系差异相对较少(补充图 1 和 2,扩展数据图 2 和补充表 5–7);因此,这些对照群体被合并用于后续分析。病例与对照之间差异显著的簇和差异表达(DE)基因的总数总结在图 1d 中,并突出显示了在结肠黏膜中而不是在血液中的疾病特异性变化。
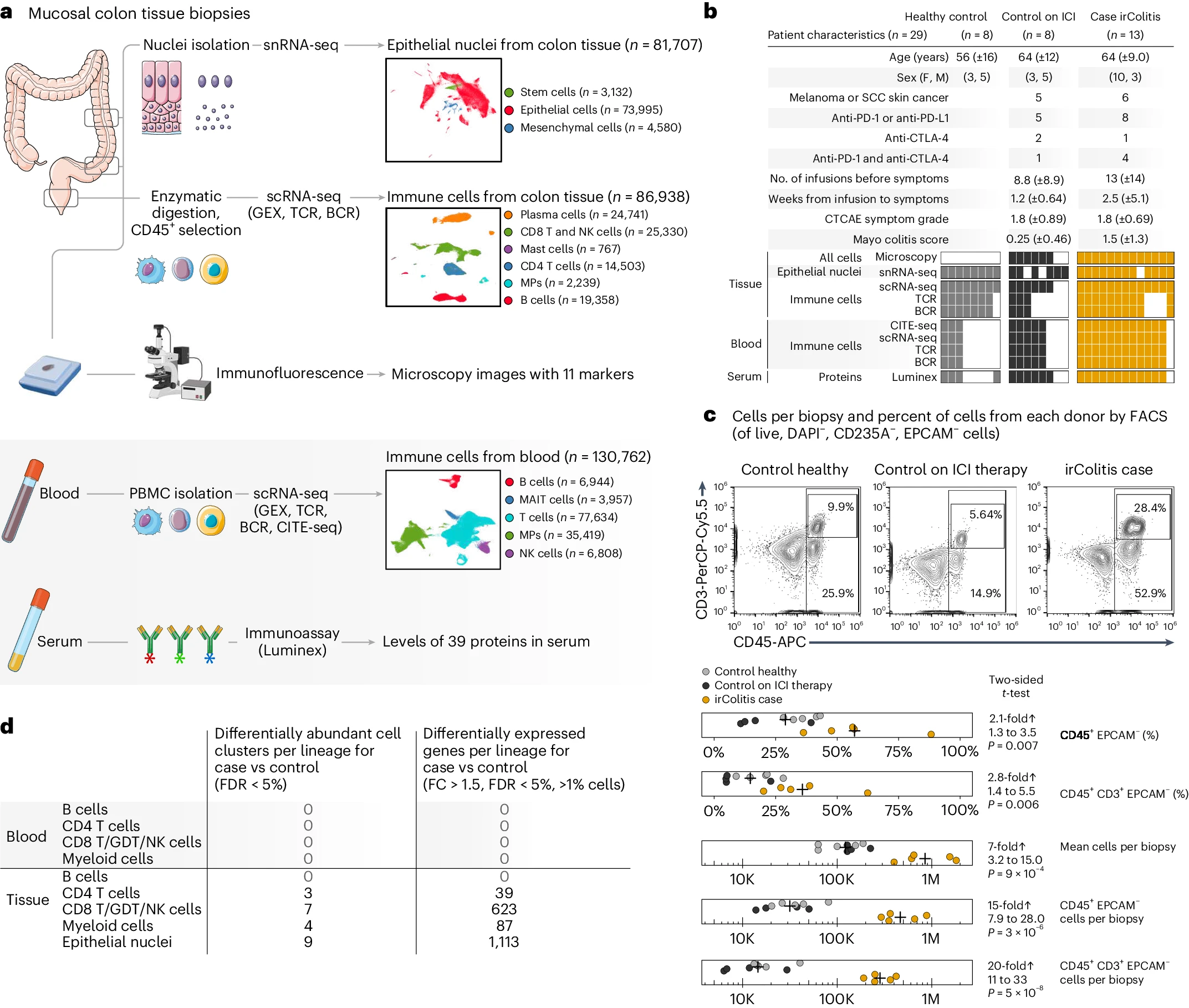
a. 样本处理流程概述。患者在进行内窥镜检查时收集结肠黏膜活检样本,以及配对的血清和 PBMC 标本。对活检样本进行 snRNA-seq 和 scRNA-seq,并进行配对的 TCR 和 BCR 测序。右侧的 UMAP 嵌入面板显示了来自组织和血液 scRNA-seq/snRNA-seq 数据集的指定细胞谱系的聚类解决方案。对于每次采集,活检标本也固定用于多光谱荧光显微镜分析多达 11 个标志物。从血清中使用 Luminex 测量分泌因子,并在 PBMCs 上进行 scRNA-seq 和配对的 TCR、BCR 和 CITE-seq。
b. 患者队列概述,报告来自 29 名患者的样本按临床元数据和实验方法的分布。CTCAE,常见不良事件术语标准;SCC,鳞状细胞癌。
c. 内窥镜结肠黏膜活检中 CD45+ 免疫细胞和 CD45+CD3+ T 细胞的代表性流式细胞术图(上面板),以及这些相应细胞群体在三个不同患者队列中的频率(从顶部算起的第二和第三个面板)。显示了每次活检的所有细胞、每次活检的 CD45+ 免疫细胞和每次活检的 CD45+CD3+ T 细胞的绝对数量(三个底行)。通过对所有患者样本使用相同大小的内窥镜活检钳并对每位患者的 6-16 次活检取平均值来规范化每次活检的细胞数量(补充表 5 和方法)。然后,使用 FACS 定义的每个相应群体的免疫和 T 细胞频率从总细胞数估计每次活检的绝对 CD45+ 和 CD45+CD3+ 细胞百分比。显示了 95% 置信区间和双侧 t 检验的 P 值。
d. 表格显示了 scRNA-seq/snRNA-seq 血液和结肠组织数据集中差异显著的细胞簇(左)和差异表达基因(右)的数量。部分图形使用 Servier Medical Art 的图片绘制。Servier Medical Art 由 Servier 提供,并根据知识共享署名 3.0 非本地化许可进行许可。
组织中的 ITGAE Hi 和 ITGB2 Hi CD8 T 细胞在 irColitis 中扩展¶
如报道所述【3】,患有 irColitis 的患者的结肠黏膜免疫细胞显著增加,包括 T 细胞(图 1c 和补充表 5)。为了表征扩展的群体,我们对代表 CD4 T 细胞、CD8 T 细胞/γδT(GDT)细胞/自然杀伤(NK)细胞、单核吞噬细胞(MPs)和 B 细胞的谱系进行亚聚类(方法,扩展数据图 1,补充图 1 和补充表 1-4)。与对照组相比,患有 irColitis 的患者的所有组织 CD8 T/GDT/NK 亚群的绝对数量增加(扩展数据图 1a),其中主要是表达不同 ITGAE 或 ITGB2 的 CD8 T 细胞,表现出 TRM 表型(图 2a-d),如之前描述的那样【20】。ITGAE(即 CD103)和 ITGB7 形成异二聚体,介导上皮归巢,而 ITGB2 与 ITGAD/ITGAL(LFA-1)/ITGAM/ITGAX 二聚化,诱导炎症组织中 ICAM/VCAM 的归巢,并调节 T 细胞分化和细胞毒性【21】。ITGAEHi CD8 T 细胞表现出从表达 IL7R 和 TCF7 的初始/静止细胞(簇 1 和 6)到扩展的 GZMBHi 效应细胞(簇 7)的表型梯度(图 2c 和补充表 6)。CD8 TRM 细胞具有不同的转录调控因子 EOMES、ZNF683 和 KZF2 的表达,这些在簇 1 和 5 中富集,并在衰竭 T 细胞中显著上调【22】。实际上,CD8 T 细胞簇 5 上调了 KIR2DL4、HAVCR2 和 TIGIT,这些构成了衰竭 T 细胞的转录程序【22,23】。病例显示扩展的 ITGB2Hi CD8 T 细胞群体,包括 GZMKHi 簇 3 和 CX3CR1Hi 簇 11 细胞,它们表达循环标志物 S1PR1 和 SELL,表明最近从血液中迁出或再循环(图 2b-d)。此外,CX3CR1Hi、GZMBHi 簇 11 细胞显示出细胞毒性表型,类似于描述的血液限制的 CX3CR1Hi 效应记忆 CD8 T 细胞【24】(图 2b-d)。

a. CD8 T/GDT/NK 细胞的组织 UMAP 图,以身份着色,并列出主要亚群:ITGB2Hi、ITGAEHi 和混合谱系。箭头指示关联方向。 b. 各细胞簇(行)中基因表达(列)的标准化。 c. 病例(橙色;n=13)和对照(灰色;n=14)之间的细胞丰度差异。 e. 对照(灰色;n=12)、接受抗 PD-1 治疗的病例(橙色;n=8)或抗 PD-1/CTLA-4 治疗的病例(蓝色;n=4)之间的细胞丰度差异。 d,f,k. UMAP 嵌入中的基因表达(log2CPM)、检测到表达的细胞数和百分比。 f. 抗 PD-1 治疗(左)和抗 PD-1/CTLA-4 治疗(右)之间 DE 基因的基因表达。 g. 对照(黑点)和接受抗 PD-1 治疗(橙色)或抗 PD-1/CTLA-4 治疗(蓝色)病例中 CD8 T/GDT/NK 伪批量表达水平。FC 和未调整的双侧 P 值。 h. 热图颜色表示簇对之间共享 TCR 的相对富集或减少,标准化为洗牌数据的均值。白点:置换 P < 8 × 10^-4。 i. 颜色表示簇对之间共享 TCR 克隆的百分比。 j. 具有 TCR 克隆的细胞的累积百分比,按降序丰度排列。 k. 血液 CD8 T/GDT/NK 细胞 UMAP,以身份着色,并跨四个亚群组织,带有标记基因 UMAP 图(底部)。 l,m. 左,组织(l)和血液(m)CD8 T/GDT/NK UMAP;箭头指示血液 TCR 在组织中的富集或减少(l)和组织 TCR 在血液中的富集或减少(m)。中,差异丰度的 TCR 克隆仅在组织中或在血液和组织中均观察到的逻辑回归 OR(l),仅在血液中或在血液和组织中均观察到的逻辑回归 OR(m);红色字体表示相关簇。右,每个组织簇中细胞在组织和血液之间共享或不共享 TCR 的百分比。显示 FDR < 5% 的簇的似然比检验 P 值。 c,e,j,l,m. 箱线图显示中位数和四分位范围。每个点代表一名患者。误差条表示每个细胞簇中病例细胞差异丰度的逻辑回归 OR 和 95% CI。未调整的似然比检验 P 值。
由于联合治疗相比单一治疗使 irColitis 风险增加 5-10 倍【1】,我们调查了接受 PD-1/CTLA-4 阻断治疗与抗 PD-1 治疗的 irColitis 患者的 CD8 T 细胞的转录差异(图 2e–g,补充图 1m 和补充表 6、7)。两个 ITGB2Hi CD8 T 细胞群(簇 3 和 11)在接受双重治疗的患者中更富集(图 2e 和补充表 6)。此外,双重治疗患者的 CD8 T 细胞上调了反映 TCR 信号增加的基因(CD28 和 TNFRSF4)、向炎症组织归巢的基因(SELL 和 ITGB2)以及向上皮归巢的基因(PXN 和 LINC00861),这些与癌症中 PD-1 呈正相关【25】(图 2f,g,补充图 1m 和图 3,补充表 7)。总之,irColitis 的特征是细胞毒性 ITGAEHi CD8 TRM 和 ITGB2Hi CD8 T 细胞在组织中的扩展,这些细胞可能会再循环,并在双重抗 PD-1/CTLA-4 治疗中比抗 PD-1 单药治疗更强烈扩展,因此可能增加 irColitis 的风险。
组织 CD8 T 细胞受体多样性在 irColitis 中增加¶
ICI 治疗重塑了肿瘤浸润 CD8 T 细胞的 TCR 受体库【26】,但 irColitis 对结肠 TCR 多样性和与血液的 TCR 共享的影响尚不清楚。单细胞 TCR 分析显示,两个组织中的 ITGB2Hi CD8 T 细胞亚型(簇 3 和 11)与其他 CD8 簇共享的 TCR 显著减少(图 2h–i 和扩展数据图 3a)。相比之下,表达 ITGAE 的 CD8 TRM 细胞在多个簇中可变地共享 TCR,表明在 ITGAEHi 细胞的表型光谱中具有共享的起源。
血液 CD8 TCR 多样性与 ICI 疗效和 IRAE 发展呈正相关【12】。我们的分析显示,与对照组相比,病例的组织 CD8(但不是 CD4)T 细胞的 TCR 多样性增加(图 2j 和扩展数据图 3)。我们未观察到患者之间共享的组织 CD8 TCR。病例中组织 CD8 TCR 多样性的增加表明 T 细胞扩展不太可能由单一主导抗原驱动。
扩展的 ITGB2 Hi CD8 T 细胞在 irColitis 中与循环细胞共享克隆¶
为了确定表达循环标志物的 ITGB2Hi CD8 T 细胞群是否与血液 T 细胞具有克隆关系,我们分析了每位患者在组织采集时匹配的外周血样本(图 1a,b 和补充表 2)。PBMCs 经过 scRNA-seq 处理,进行了亚群聚类(方法,图 2k,扩展数据图 4,补充图 1 和图 7,补充表 3 和 4),并与最近发表的数据集中血细胞类型进行比较【27】(扩展数据图 4f,k)。与组织不同,我们在病例与对照之间未发现差异显著的血液免疫群体或 DE 基因(扩展数据图 4,补充图 1 和图 7,补充表 6 和 7)。
TCR 分析显示,个体 CD8 T 细胞克隆中平均有 2% 在结肠和血液中均观察到(扩展数据图 4d 和补充表 8),这种频率在病例和对照之间没有变化。相比之下,与组织中的 ITGAEHi CD8 T 细胞相比,ITGB2Hi 组织 CD8 T 亚群在血液和组织 TCR 共享显著增加(图 2l)。三个血液 CD8 T 亚群(循环簇 12,CX3CR1Hi FGFBP2Hi 簇 5 和激活的 MHC-IIHi GZMKHi 簇 2;扩展数据图 4a–c)与 irColitis 结肠黏膜共享更多 CD8 TCR,而不是对照组(图 2m)。血液簇 5 CX3CR1Hi CD8 T 细胞在转录上与组织簇 11 CX3CR1Hi CD8 T 细胞相似,被描述为再表达 CD45RA 的效应记忆细胞(即 TEMRA)【27】(扩展数据图 4f)。基于 CX3CR1 和 GZMB 的高表达,我们预测血液 CD8 簇 5 和结肠 CD8 簇 11 是血管内 T 效应记忆群体【24,28】。血液 CD8 簇 2 细胞在表型上与表达 GZMK 和 EOMES 的组织 CD8 簇 3 细胞相似,且在表型上与描述的再循环、非细胞毒性效应记忆 CD8 T 细胞相似【24,27】(扩展数据图 4f)。我们预测循环 T 细胞群体在 irColitis 中具有病理作用,因为它们是整合素抑制剂 vedolizumab 的预测靶标,该药物可防止循环免疫细胞进入发炎的胃肠道黏膜,并且在治疗 irColitis 中有效【29】。
ITGAE Hi CD8 TRM 细胞在 irColitis 中上调 IL17A、IL26 和 CXCL13¶
跨 CD8 T 细胞的差异基因表达(DGE)分析显示,几乎所有 irColitis 的 DE 基因都在 ITGAEHi CD8 T 细胞中,而不是在 ITGB2Hi CD8 T 细胞中(图 3a,b,补充图 4a 和补充表 7)。在 CD8 T 细胞亚群中检测到许多 DE 效应基因,突显了共享的转录程序(图 3,补充图 4a 和补充表 7)。多光谱 RNA 显微镜确认在 irColitis 组织的固有层(LP)中强烈富集了共同表达 CD3E 和 IFNG 的 T 细胞(图 3e 和补充图 5)
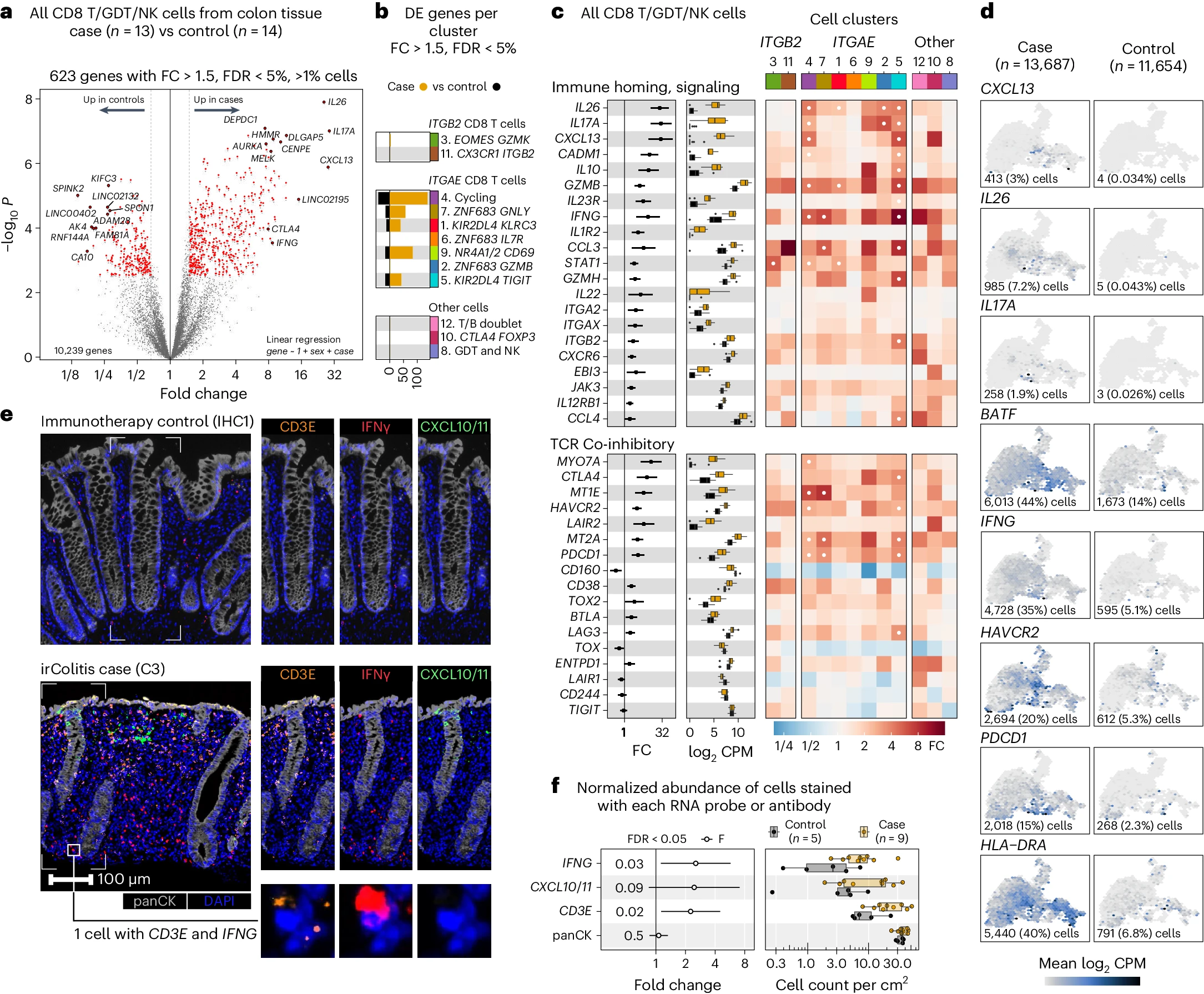
a. 火山图显示所有 CD8 T、GDT 和 NK 细胞的伪批量 DGE,对比 irColitis 病例(n=13)和对照组(n=14)。x 轴表示 FC,y 轴表示 limma 报告的负对数 10 P 值(双侧)。
b. 条形图显示了定义在图 2 中的每个 CD8 T/GDT/NK 细胞簇中的 DE 基因,并跨三个细胞亚群组织:ITGB2Hi CD8 T 细胞、ITGAEHi CD8 T 细胞和具有混合细胞谱系的簇(FC > 1.5 且 FDR < 5%)。
c. 报告了选择基因在结肠组织 CD8 T 和先天性细胞毒淋巴细胞簇中病例(橙色)和对照组(黑色)的 FC 和基因表达(log2CPM)(左列和中列)。热图颜色表示 irColitis 病例与对照组之间的 FC 差异(右列)。白点表示 FDR < 5%。
d. 特征图使用颜色表示 UMAP 嵌入中的基因表达(log2CPM)水平,来自图 2a。每个特征图底部报告了检测到的每个候选基因的细胞数量和百分比。
e. 多光谱荧光面板显示了来自一个对照个体的 CD3E(橙色)、IFNG(红色)、CXCL10/11(绿色)的 RNA 表达水平,全 CK(灰色)和 DAPI(蓝色)的蛋白质免疫荧光(上面板),以及一个 irColitis 病例(中间面板)。右下角的方框突出显示了在一个 irColitis 病例的组织中观察到的在同一 T 细胞中共同表达 CD3E 和 IFNG 的代表性共表达。
f. 从显微图像中定量 RNA(IFNG、CXCL10/11 和 CD3E)和蛋白质(PanCK)信号。报告了 FC(左列,黑色表示 FDR < 5% 且显示未调整的线性模型 P 值)和针对病例(橙色)和对照组(黑色)标准化到组织面积(cm²)的给定标志物阳性细胞的频率(右列)。黑色垂直线(左图)表示 FC = 1。点代表个别患者,箱线图显示中位数和四分位范围。误差条表示 95% 置信区间。
多个 TCR 共抑制/衰竭基因在激活的 T 细胞中上调,在 ITGAEHi GZMBHi CD8 T 效应簇(4、5 和 7)中富集(图 3c,d)。几个 ITGAEHi CD8 T 群体上调趋化因子 CXCL13(促进三级淋巴结构 [TLS] 形成)和多个 Th17 基因(IL17A、IL26、IL23R 和 BATF)(图 3c,d 和补充表 7)。LDLR 在多个 CD8 T 细胞亚群中上调(补充图 4),并显著使 T 细胞向 Th17 样表型极化【30】。IL17A、IL26 和 CXCL13 在 ITGB2Hi CD8 T 细胞中没有表达(图 3c 和补充表 7)。在我们的数据中,1.9% 的 CD8 T 细胞表达 IL17A,这与我们在另一组 irColitis 数据集【15】中检测到的 1.5% 和在炎症性肠病(IBD)数据集【31】中检测到的 1.7-2.8% 一致(补充图 4c)。鉴于 IL-17A 在 IBD 中可能的保护作用【32】,我们推测 IL-17A 表达可能在限制 irColitis 病理中发挥类似作用,这需要在未来的研究中进一步探讨。
总之,我们的结果表明,ITGAEHi GZMBHi CD8 TRM 效应细胞在 irColitis 中经历了显著的转录调控,趋向于 Th17 表型。相比之下,ITGB2Hi CD8 T 细胞具有很少的 DE 基因,表明其组织扩展由重新定位和/或旁观者激活驱动,而不是 TCR 参与。
在 irColitis 中扩展的 IL17A Hi CXCL13 Hi 效应 CD4 T 细胞和 Tregs¶
CD4 T 细胞的亚聚类显示 Th1/Th17 细胞(簇 7)和 FOXP3Hi Tregs,包括 SELLHi 滤泡(簇 5)和激活的 TNFRSF4Hi TNFRSF18Hi(簇 9)Tregs 显著扩展(图 4a–d,扩展数据图 1,补充图 1 和补充表 3、4 和 6)。Th1/17 细胞(簇 7)和 TNFRSF4Hi Tregs(簇 9)上调了涉及干扰素刺激、核酸感应、抗原呈递、细胞代谢、转录、凋亡和自噬的基因(图 4e–g,补充图 4b 和补充表 7)。Th1/Th17 细胞还上调了 Th17 基因(IL17A 和 IL21)、IFNG 和 CXCL13,这些基因在 T 滤泡辅助(Tfh)和循环细胞中也增加(图 4g,h 和补充表 7)。没有 CXCL13Hi CD4 T 细胞表达 CX3CR1(补充表 4),因此这些细胞与类风湿性关节炎中报道的 T 周边辅助 CD4 细胞不同【33】。TNFRSF4Hi Tregs 上调了 Th1 基因(即 TBX21、IL12RB1/2 和 CXCR3)和 TNF 受体基因(TNFRSF4 和 TNFRSF18),这些基因削弱了 Tregs 的抑制功能【34,35】(图 4b,d,g,h 和补充表 7)。因此,富集于 irColitis 的 Tregs 可能通过减弱 Tregs 的免疫抑制活性并表达细胞毒性基因程序来促进炎症。
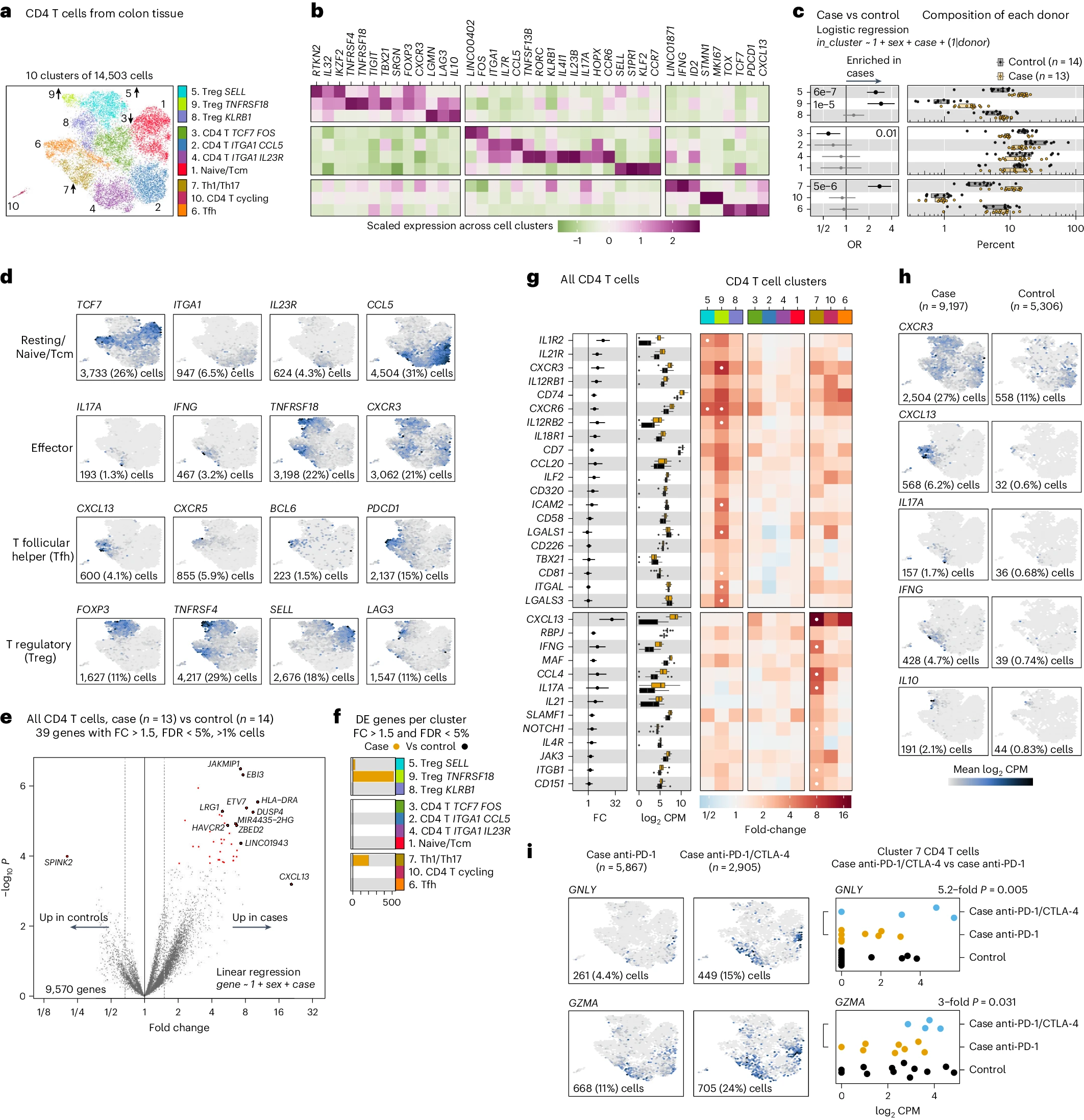
a. CD4 T 细胞的 UMAP 嵌入,按细胞簇进行颜色编码。簇号旁的箭头显示观察到的丰度差异方向。
b. 选择基因的标准化表达(均值为零,单位方差),显示在各细胞簇中的相对表达。热图的行与每个簇对齐。
c. 在各亚群中,病例(橙色,n=13)和对照(灰色,n=14)之间的细胞亚群丰度差异。箱线图显示患者细胞类型组成,每个点代表一个患者。每个患者的组成报告为每个细胞簇中的细胞百分比。误差条表示逻辑回归 OR 的 95% 置信区间,以显示每个细胞簇中 irColitis 病例细胞的差异丰度。显示未调整的似然比检验双侧 P 值。
d,h. 候选基因的特征图使用颜色表示 UMAP 嵌入中的基因表达(log2CPM)水平。每个候选基因的检测表达细胞的数量和百分比报告在每个特征图的底部。
e. 火山图显示所有 CD4 T 细胞的伪批量 DGE,对比病例与对照。x 轴表示 FC,y 轴表示来自 limma 的负对数 10 P 值。
f. 条形图显示每个 CD4 T 细胞簇中的 DE 基因(a 中定义的簇,FC > 1.5 且 FDR < 5%)。
g. 报告选择基因在 CD4 T 细胞簇中病例(橙色)和对照(黑色)的 FC 和基因表达(log2CPM)水平(左列和中列)。热图颜色表示病例与对照之间的 FC 差异(右列)。白点表示 FDR < 5%。
h. 特征图分别展示来自 irColitis 病例和对照的细胞。
i. 在 a 中的 UMAP 嵌入中,病例接受抗 PD-1 或双重抗 PD-1/CTLA-4 治疗的细胞中 GNLY 和 GZMA 的表达水平。点图显示伪批量表达,每个点代表一个患者的所有 CD4 T 细胞。双重抗 PD-1/CTLA-4 治疗与抗 PD-1 单药治疗病例的 FC 和 P 值来自 limma。误差条表示 95% 置信区间;箱线图显示中位数和四分位范围。
最后,与接受抗 PD-1 治疗的患者相比,接受双重抗 PD-1/CTLA-4 抑制的患者具有独特的效应 Th1/17 T 细胞(簇 7),这些细胞上调了细胞毒基因 GNLY 和 GZMA(图 4i)。这些细胞类似于人类癌症和病毒感染中的细胞毒 CD4 T 细胞【36】,并预测在 irColitis 中具有病理作用。
在独立数据集中验证 irColitis 特异性特征¶
最近的两项研究检测到 irColitis 中的 CD8 TRM 扩展;然而,他们未报告 CD8 CXCL13、IL17A 或 IL26 的表达,也未报告扩展的 ITGB2Hi S1PR1Hi CD8 T 细胞。为了评估与我们研究的差异,我们使用我们的计算方法重新分析了一个数据集【15】。重新分析验证了两个扩展的 ITGB2Hi CD8 T 细胞群,分别表达 EOMES 和 CX3CR1,并在 irColitis 患者中增加了 CD8 T IL17A、IL26 和 CXCL13 的表达(补充图 6)。虽然我们在两个数据集中都发现了增加的 CD4 T CXCL13 表达,但只有我们的数据集显示了 CD4 T IL17A 的表达(图 4,补充图 6 和补充表 7)。我们在独立的 irColitis 数据集中检测到扩展的 CXCL13Hi T 细胞、IL17AHi IL26Hi CD8 T 细胞和 ITGB2Hi CD8 T 细胞,支持了这些发现的可重复性。
ISGHI MPs 在 irColitis 中扩展,而 B 细胞亚群保持不变¶
MPs 的亚聚类显示 irColitis 中富集的三个群体——VCANHi 炎性单核细胞(簇 5)、CD68HiC1QAHiCXCL10Hi MPs(簇 6)和表达组织蛋白酶基因的 CD68HiC1QAHi MPs(簇 2),这些基因在 IBD 中的巨噬细胞中上调【37】(扩展数据图 1 和 5,补充图 1 和补充表 3、4 和 6)。如同在 IBD 中【31】,传统的 CD1CHi 树突状细胞(cDC2)在 irColitis 病例中表现不足。irColitis 中的大多数 DE MP 基因是 ISGs(扩展数据图 5 和补充表 7)。RNA-ISH 确认 ISGs CXCL10/11 主要由 irColitis 固有层中的 panCK−细胞表达(图 3e,f),可能对应于 CXCL10Hi CD68Hi 细胞(簇 6)。MPs 还上调了 CD274(即 PD-L1)、MHC-I 基因和涉及 TCR 共刺激的基因(CD80)(扩展数据图 5 和补充表 7),反映出高度活化的 MPs 对 TCR 信号传导具有复杂的预测效应。
与 MPs 相反,组织和血液 B 细胞在病例与对照之间只有少量差异显著的群体和 DE 基因(扩展数据图 1 和 6,补充图 1 和图 7,补充表 3、4 和 7)。与 IBD 中浆细胞优先级转换到 IgG 不同【38】,我们在病例与对照之间未观察到 IgG 到 IgA 浆细胞比例的差异(扩展数据图 6F)。最后,来自 irColitis 组织和血液的 B 细胞受体(BCRs)在多样性上与对照无差异(扩展数据图 3c,d)。值得注意的是,一名患者在 B 细胞消耗后接受抗 PD-1 治疗时发展为 irColitis(扩展数据图 6 和补充表 1、2;患者 C14*)。在此患者中,扩展的 GZMBHi IFNGHi CD8 T 细胞的存在和结肠黏膜 B 细胞的缺失表明,B 细胞在所有情况下都不是 irColitis 发展的必要因素,B 细胞可能是轻微的疾病贡献者。
ICI 治疗对照组观察到的组织免疫变化¶
将 ICI 治疗对照组与健康个体进行比较,以了解 ICIs 在没有 irColitis 的情况下如何影响结肠免疫(扩展数据图 2 和补充图 1m)。与健康对照组相比,ICI 治疗对照组的 KIRHi CD8 T 细胞(簇 1 和 5)减少,这些细胞由 Treg 转录调控因子(ID3 和 IKZF2)、T 细胞衰竭基因(TIGIT)定义,类似于自身免疫中描述的调节性 CD8 T 细胞【39】(图 2a-d 和扩展数据图 2)。相比之下,ICI 治疗未改变 CD4 Treg 丰度,尽管在 TNFRSF9Hi Tregs 中抑制了 ADAR(扩展数据图 2)。ADAR 促进 T 细胞自我耐受并保护免受结肠炎【40】。在 MPs 亚群中,ICI 对照组的组织 VCANHi 炎性单核细胞扩展,而血液 CD14Hi 单核细胞相对减少,这表明 ICIs 在没有 irColitis 的情况下促进了单核细胞从血液向结肠黏膜的募集(扩展数据图 2 和补充图 2)。此外,暴露于 ICIs 导致组织 cDC1 丰度和 HLA-DOB 表达下降(扩展数据图 2)。DCs 中的 HLA-DO 增加 MHC 抗原多样性并防止自身免疫【41】,因此其抑制可能促进自身免疫。我们怀疑这些对 ICI 治疗的集体反应可能代表亚临床炎症的早期阶段。为什么这些患者未发展为 irColitis 需要进一步研究。
炎症血清蛋白与 irColitis 状态不一致¶
分析了病例与对照的血清中的 39 种分泌因子。这些因子包括结肠黏膜 T 细胞上调的对应基因(CXCL13、IFN-γ、IL-7、IL-10、IL-17α、IL-22 和 CCL3/4)和 MPs(GM-CSF、CXCL1/2/8/9/10、IL-1α/β、IL-1RA、IL-15 和 IL-27)(补充表 7 和 9)。这些因子中没有一个在检测上有差异(补充表 9),这表明外周血并不能很好地反映 irColitis 患者观察到的组织炎症环境。
irColitis 与 ISG 介导的上皮周转和屏障功能缺陷相关¶
由于上皮损伤是 irColitis 的组织学特征【4】,我们接下来寻求定义与疾病相关的结肠上皮和间质改变。为了最大限度地恢复上皮,我们从冷冻的黏膜标本中进行了单核 RNA 测序(snRNA-seq)(图 1 和 5a-c,扩展数据图 7,补充图 1 和补充表 1-4)。在 irColitis 中,我们观察到 LGR5Hi 干细胞(簇 8)的比例下降,这些细胞强烈上调了 ISGs(即 STAT1);吸收性上皮细胞(簇 5 和 6);成熟的 CEACAM7Hi 上皮细胞(簇 20);刷细胞(簇 19);和扩展的过渡增殖细胞(簇 3 和 14),表明上皮周转增加(图 5b,d-g 和补充表 6)。

a. 五个细胞家族中的上皮/间质亚群的标记基因标准化相对表达(左)。
b. 误差条表示每个亚群病例(橙色;n=12)和对照(灰色;n=14)之间差异丰度的逻辑回归 OR 的 95% 置信区间,未经调整的似然比检验 P 值。b,j, 箱线图显示患者细胞类型组成,报告为每个簇中来自患者的细胞核的百分比;每个点代表一个患者。
c. 上皮/间质亚群的 UMAP,以 a 中的颜色显示,箭头显示关联方向。插图显示六边形中的细胞核。颜色代表来自病例的细胞的富集倍数。UMAP 中显示的隐窝轴得分(方法)。
d. 伪批量 DGE 火山图,显示病例与对照之间所有上皮/间质细胞核的对比。x 轴表示 FC,y 轴表示负对数 10 P 值(limma)。
e. 每个簇的隐窝轴得分箱线图(左)(方法)。条形图显示每个亚群的 DE 基因数量(右)(FC > 1.5 和 FDR < 5%),按 a 和 b 中的顺序排列。
f,i. 特征图显示病例(左)和对照(右)中的基因表达(log2CPM)水平,投射在 c 的 UMAP 上。报告了每个基因的细胞核数量和表达百分比。
g. 报告按主题组织的基因在病例(橙色)和对照(黑色)中的 FC 和基因表达(log2CPM)(两左列)。热图颜色表示病例与对照之间的 FC 差异。白点表示 FDR < 5%。
h. 对 ICI 对照(左)和病例(右)的结肠黏膜染色,DAPI(蓝色)、panCK(灰色)、CD8A(水蓝色)、FOXP3(黄色)、PD-1(橙色)、PD-L1(绿色)和 CD68(粉色)。右侧面板突出显示了单个细胞中 PD-L1±CD68 和 PD-L1±panCK 的共检测(1 和 2)以及单个细胞中 PD-1±CD8A 的共检测(3 和 4)。
i. 左,抗 PD-1(左)和抗 PD-1/CTLA-4(右)治疗的病例之间跨亚群的 DE 基因。右,CXCL1、DUOX2、HIF1A 和 NOS2 在所有上皮/间质亚群中的伪批量表达水平,按对照(黑色)、抗 PD-1 治疗的病例(橙色)或抗 PD-1/CTLA-4 治疗的病例(蓝色)分层。
j. 表达 VWF 的内皮细胞核(簇 13)的丰度分析。对照(黑色;n=13)、抗 PD-1 治疗的病例(橙色;n=8)或抗 PD-1/CTLA-4 治疗的病例(蓝色;n=4)。b,g,j, 箱线图显示中位数和四分位范围。误差条表示 95% 置信区间。
上皮和间质群体中 irColitis 患者最上调的基因是 ISGs(即 STAT1, CD274/PD-L1 和 NOS2)(图 5d–h),这些基因在双重 PD-1/CTLA-4 阻断治疗中比单一疗法更强烈诱导(图 5i 和补充表 7)。在成熟上皮细胞(簇 2 和 11)和杯状细胞(簇 10)中特异性上调的基因涉及 TCR 信号传导、细胞迁移和代谢(图 5f,g,扩展数据图 7 和补充表 7)。使用隐窝轴评分【42】,我们预测 ISG 特征最强的细胞类型(簇 2、10 和 11)位于上皮隐窝的顶部(图 5a,c,e,f,g)。簇 20 表达隐窝顶部标志物(即 SELENOP),但在表达较低的 ISGs 方面与簇 2 和 11 不同。由于簇 20 在 irColitis 中显著减少,我们怀疑干扰素可能诱导簇 20 细胞转变为簇 2 和 11 的亚群。
irColitis 特异性转录程序影响上皮屏障功能,包括上调 CEACAM 粘附基因和下调与 IBD 相关的水通道蛋白基因和溶质载体基因(SCL22A5 和 SLC22A23)【43,44】,这可能会损害结肠水分吸收(图 5d,f,g,扩展数据图 7 和补充表 7)。干扰素可以下调 AQP 基因【45】,这与在隐窝顶部上皮簇中观察到的高 ISG 和低 AQP 表达一致。与临床组织学一致【3,4】,irColitis 上皮细胞也显示出凋亡基因特征(TNFRSF10A、CASP1 和 CASP8)。
为了进一步评估上皮 ISG 隐窝轴梯度,我们使用蛋白质免疫荧光检测 PD-L1,后者由干扰素强烈诱导【46】。在 irColitis 患者中,PD-L1 在上皮隐窝的顶部容易检测到,但在底部不易检测到(图 5h 和补充图 8 和 9),并且在固有层中也是如此。我们的 scRNA-seq 显示 CD274 在 ISGHi MPs 中强烈上调(补充表 7),表明在固有层中检测到的 PD-L1 可能来自 CD68+ MPs,而在 irColitis 中,这些 MPs 定位于靠近腔隙的位置(图 5h 和补充图 8 和 9)。隐窝顶部上皮细胞中的干扰素信号传导可以诱导免疫趋化因子和减弱免疫反应的因子,如 CD274/PD-L1 和 MHC 基因。总体来看,这些数据支持 PD-1/CTLA-4 抑制可能导致干扰素诱导的免疫耐受程序受损和腔隙上皮屏障功能障碍。
irColitis 中的内皮和成纤维细胞重塑¶
大多数间质群体,包括内皮细胞(簇 13)和肌成纤维细胞(簇 21 和 22),在病例中更为丰富(图 5a,b)。鉴于上皮细胞上调了缺氧和血管生成基因(HIF1A、SERPINE1 和 VEGFA)(扩展数据图 7 和补充表 7),低氧张力可能驱动这一背景下的新生血管形成。在接受双重抗 PD-1/CTLA-4 治疗的患者中,上皮细胞中的 HIF1A 表达和内皮细胞的丰度显著增加,而单一疗法中则不明显(图 5i,j),虽然尚不清楚血管生成是否有助于或改善了 irColitis。
与 IBD 不同【31】,CCL19Hi 炎性成纤维细胞(簇 23)在 irColitis 中并不丰富(图 5b)。然而,隐窝顶部成纤维细胞簇 7(PDGFRA 和 BMP4/5/7)上调了涉及成纤维细胞(FGF7)和上皮(BMP7)分化的基因。此外,这一簇独特地表达了 OSMR,后者在 IBD 中的成纤维细胞中上调【31】,以及 ROBO1,后者激活肠干细胞自噬【47】(扩展数据图 7 和补充表 4 和 7)。
ICAM 和 CXCR3 配体在 irColitis 中的潜在作用¶
鉴于 PD-1/PD-L1 在耐受中的中心作用【48】以及大多数患者接受 PD-1 抑制剂治疗(图 1b),我们绘制了分别表达 PD-1 配体 CD274(PD-L1)或 PDCD1LG2(PD-L2)的细胞与 PDCD1 表达细胞之间的相互作用。结果预测 PDCD1 表达的 T 细胞与 CD274 表达的上皮细胞和 CD274/PDCD1LG2 表达的 MPs(巨噬细胞和单核细胞但不是 DCs)相互作用(扩展数据图 7b 和 8a,b)。鉴于在 irColitis 中上皮隐窝顶部的 PD-L1 升高(图 5h 和补充图 8 和 9),PD-1 抑制可能优先破坏上皮隐窝顶部的 PD-1/PD-L1 相互作用,导致局部未受控制的细胞毒性 T 细胞反应和 2 型干扰素释放。
我们接下来分析了 1,826 对精心整理的基因对【49】,基于配体 - 受体基因对表达预测潜在的细胞 - 细胞相互作用,并识别出 irColitis 中富集的 431 对 DE 配体 - 受体对(扩展数据图 8 和 9 及补充表 10 和 11)。我们首先检查了 ITGAEHi GZMBHi 和 ITGB2Hi CD8 T 细胞(图 2b),重点关注 MP、上皮和/或间质群体中高度上调的基因和 T 细胞分泌因子(CXCL13、IL26 和 IL17A)(图 3a 和 4e 及扩展数据图 8c,d)。预测 ITGB2–ICAM1/2/3 对促进 ITGB2Hi CD8 T 细胞(簇 3 和 11)与各种黏膜细胞类型之间的相互作用。相比之下,CXCR3 趋化因子配体 - 受体对预测 MPs 或上皮隐窝顶部细胞与 ITGAEHi GZMBHi TRM 或 ITGB2Hi CD8 T 细胞之间的相互作用有限(扩展数据图 8)。类似地,Th17 细胞因子 IL17A 和 IL26 预测仅促进 ITGAEHi GZMBHi(而非 ITGB2Hi)CD8 TRM 细胞与结肠黏膜之间的对话(扩展数据图 8c,d)。有趣的是,每个患者中衰竭的 IL17A 表达 HAVCR2Hi CD8 T 细胞(簇 5)的丰度与 SPINK5HiCCL20Hi 上皮细胞(簇 18)的丰度在病例中强烈相关(扩展数据图 8f 及补充表 12)。SPINK5 由 IL-17 诱导,可能加强屏障功能【50】。由于 IL-17A 阻断可加重 IBD【32】,我们推测 IL-17A 可能在 irColitis 中遏制细胞毒性。
我们还在 irColitis 中识别了 166(1,826 对中)的显著差异配体 - 受体对,这些对在所有上皮/间质细胞和每个组织免疫谱系之间(扩展数据图 9a,b)。一些基因对被预测促进 T 细胞向内皮细胞的归巢(即 CX3CR1/CX3CL1)。此外,ITGB2HiCX3CR1Hi CD8 T 细胞的丰度与 CX3CL1Hi 内皮细胞的丰度强烈相关(扩展数据图 8g 及补充表 12),表明这些细胞类型之间可能存在相互作用。最后,irColitis 病例中的 T 细胞和 MPs 上调了诱导 TLSs 的细胞因子/趋化因子受体基因(LTA/B 和 CCL19/20)。
总体来看,这些结果预测了 ICAM 和 CXCR3 配体在 irColitis 中结肠黏膜 CD8 T 细胞募集和滞留中的重要作用。个别 T 细胞在 irColitis 中是被激活还是被抑制,可能反映了 TCR 共抑制/刺激配体和归巢配体在局部结肠黏膜环境中的空间分布。
识别治疗 irColitis 的潜在药物靶点¶
我们接下来定义了由 FDA 批准的 IBD 药物和临床试验中的药物破坏的配体 - 受体对的细胞类型表达模式(图 6a,b 及补充表 13)。TNF、ITGA4 和 ITGB7 在淋巴细胞中特别丰富,而 ITGA4/ITGB7 配体在成纤维细胞(例如,FN1)和内皮细胞(例如,MADCAM1)中也有表达,这与 vedolizumab 破坏免疫/间质相互作用一致(图 6a,b 及补充表 4)。JAK 抑制剂 tofacitinib 的主要靶点 JAK1/3 在 T 细胞中丰富,尽管 JAK 广泛表达(图 6b)。S1P 受体在 ITGB2Hi CD8 T 细胞(簇 3 和 11)、SELLHi KLF2Hi CD4 T 细胞(簇 1)、滤泡 B 细胞(簇 2)和内皮细胞(簇 13)中显示出显著的富集(图 6b)。此外,内皮细胞表达 CX3CL1(图 6b,c),在临床试验中,单克隆抗体(E6011)存在时,其与 CX3CR1 表达的免疫细胞结合受损(参考文献 51)。一些正在 IBD 试验中测试的药物靶点(即 HIF1A 和 NLRX1)在上皮和间质细胞中特别丰富(图 6b,c)。