Statistical method scDEED for detecting dubious 2D single cell embeddings and optimizing t SNE and UMAP hyperparameters
摘要¶
二维(2D)嵌入方法对单细胞数据可视化至关重要。流行的方法如 t- 分布邻域嵌入(t-SNE)和统一流形近似与投影(UMAP)通常用于可视化细胞簇;然而,众所周知,t-SNE 和 UMAP 的二维嵌入可能无法可靠地反映细胞簇之间的相似性。针对这一挑战,我们提出了一种统计方法,scDEED,用于检测由二维嵌入方法生成的可疑细胞嵌入。通过基于细胞二维嵌入邻域与预嵌入邻域之间的相似性计算每个细胞嵌入的可靠性评分,scDEED 将低可靠性评分的细胞嵌入标记为可疑嵌入,而高可靠性评分的细胞嵌入则被视为可信嵌入。此外,通过最小化可疑细胞嵌入的数量,scDEED 为优化嵌入方法的超参数提供了直观的指导。我们在多个数据集上展示了 scDEED 在检测可疑细胞嵌入和优化 t-SNE 和 UMAP 超参数方面的有效性。
引言¶
在蓬勃发展的单细胞生物学领域,二维(2D)数据可视化是不可或缺的探索性步骤,使研究人员能够检查单个细胞之间的相似性和差异,从而识别离散细胞类型和连续细胞轨迹的潜在存在。二维数据可视化是通过嵌入方法(也称为降维方法)实现的。在为单细胞数据开发的众多嵌入方法中,t-SNE 和 UMAP 是最受欢迎的,因为它们可以增强同类细胞的相似性并增加不同细胞类型的对比度。
然而,在二维空间中增强相似细胞的接近度是以扭曲所有细胞之间的整体距离为代价的。在强调局部细胞簇和保持全局细胞拓扑结构之间的权衡中,t-SNE 和 UMAP 侧重于局部,而经典的主成分分析(PCA)通过优化保持细胞的全局方差,位于全局端。尽管 t-SNE 和 UMAP 很受欢迎,但已经有警告信息指出使用 t-SNE 和 UMAP 嵌入来推断细胞簇距离存在风险,这与全局细胞拓扑结构有关。
与这些警告信息一致,已经提出了从保持全局细胞拓扑结构的角度优化 t-SNE 和 UMAP 超参数的策略,并且开发了新的嵌入方法来改进 t-SNE 和 UMAP。然而,这些优化策略或新方法都不能保证结果细胞嵌入在多大程度上能准确保持细胞簇之间的距离。因此,研究人员仍然对特定细胞簇的邻近簇在二维嵌入空间中的可信度感到困惑。一个相关的问题是,二维嵌入空间中距离较远的簇是否真的不同。此外,二维嵌入空间中的可视化簇是否可信,还是应该划分为多个簇,这仍然值得怀疑。
为了解决这些问题,我们提出了 scDEED(单细胞可疑嵌入检测器),一种决定每个细胞二维嵌入是否可疑或可信的统计方法。scDEED 的核心思想是评估一个细胞在二维嵌入前后是否有类似顺序的中程邻居。基于这个思想,scDEED 为每个细胞分配一个“可靠性评分”,其较大值表明该细胞的近期到中程邻居在嵌入后得到了很好的保留。然后,scDEED 将每个细胞的可靠性评分与可靠性评分的零分布(所有细胞都有随机邻居时的评分)进行比较,并在细胞的可靠性评分比 95% 的零分布评分差(或好)时,识别该细胞的嵌入为可疑(或可信)。scDEED 的结果将帮助研究人员避免使用可疑细胞嵌入来解释细胞相似性,并对使用可信细胞嵌入充满信心。
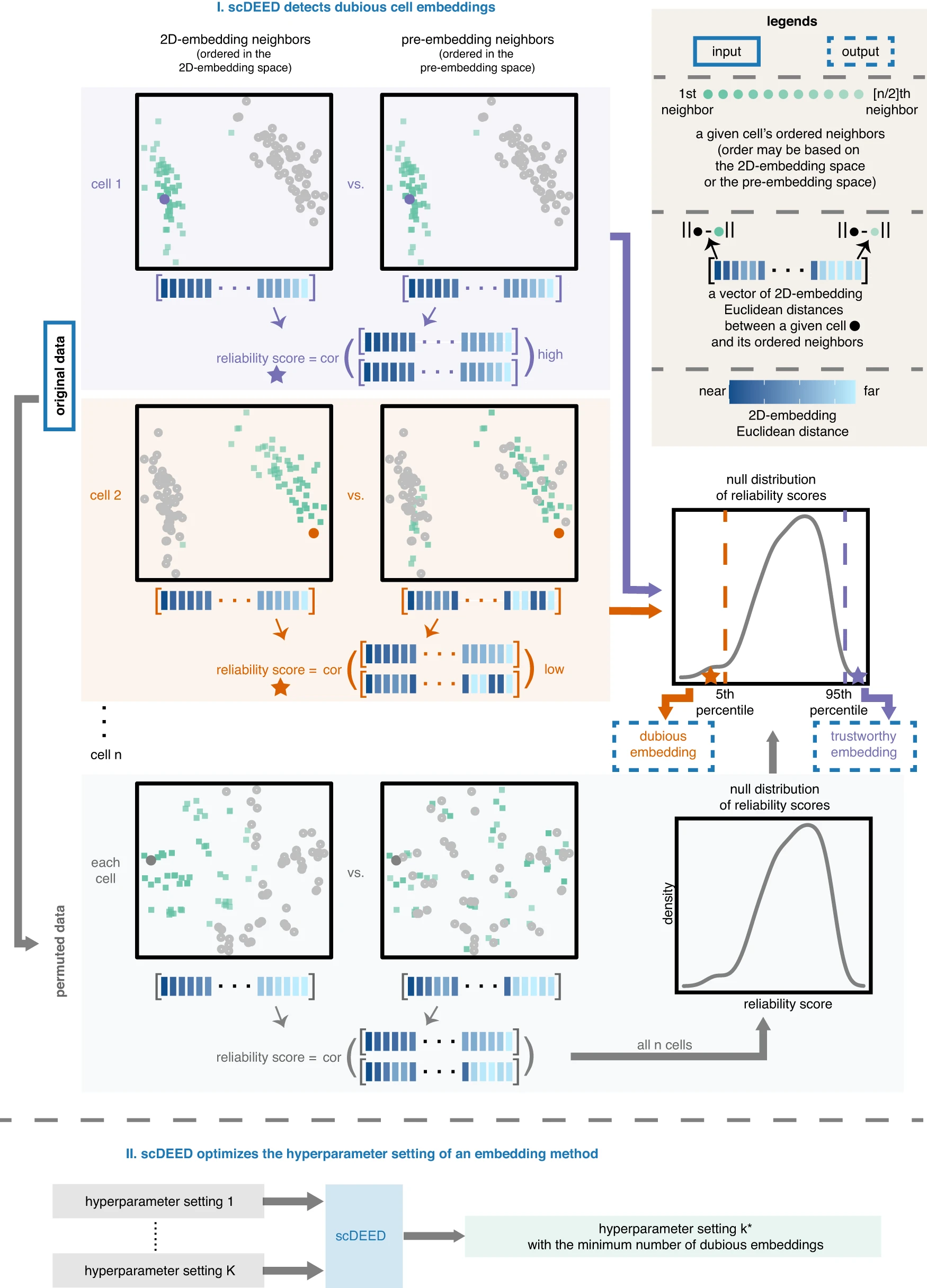
功能 I 通过计算可靠性评分来决定每个细胞的嵌入是否可信或可疑。该可靠性评分定义为细胞在二维嵌入空间中与其最近 50% 邻近细胞的距离与同一细胞在预嵌入空间中与其最近 50% 邻近细胞的距离之间的皮尔逊相关系数(每个空间中的距离按从第 1 个邻居到第 [n/2] 个邻居的顺序排列,其中 n 是细胞总数)。与通过排列得到的可靠性评分的零分布相比,细胞 1 的可靠性评分(用紫色星星标记)落在最高的 5% 内,因此它有一个可信的嵌入;相反,细胞 2 的可靠性评分(用橙色星星标记)落在最低的 5% 内,因此它有一个可疑的嵌入。功能 I 的实现使得功能 II 可以通过最小化可疑嵌入的数量来优化嵌入方法(如 t-SNE 或 UMAP)的超参数设置。
这种可疑嵌入检测功能还赋予 scDEED 另一种用途:通过最小化可疑嵌入的数量来优化二维嵌入方法的超参数设置(图 1II)。与现有的 t-SNE 和 UMAP 优化策略不同,scDEED 提供了用户以直观和图形化方式优化超参数的灵活性(用户可以看到每个超参数设置下哪些细胞嵌入是可疑的),而无需修改嵌入方法的算法。此外,scDEED 对可疑细胞嵌入的定义将其与 DynamicViz 区分开来,后者通过最小化多个引导样本中细胞嵌入的欧几里得距离的方差来优化超参数。与 DynamicViz 检查二维嵌入的稳定性不同,scDEED 评估二维嵌入空间中细胞近期到中程邻居的保留情况。
值得注意的是,scDEED 兼容所有二维嵌入方法,这里我们展示了其在 t-SNE 和 UMAP 中的应用,以示范其使用。这种通用性使 scDEED 与 EMBEDR 不同,后者基于 t-SNE 损失函数为每个细胞分配质量评分,因此不适合比较不同方法的嵌入。相反,scDEED 可以在各种分辨率下比较不同方法的嵌入,包括单个细胞、细胞类型和所有细胞。
在本研究中,我们将 scDEED 应用于多个 scRNA-seq 数据集。结果显示,scDEED 成功地在原始研究中识别出了可疑的细胞嵌入。此外,scDEED 优化的超参数比原始研究中更好地保留了细胞类型的生物学关系。我们还展示了 scDEED 在优化 t-SNE 的困惑度超参数方面相对于 EMBEDR 的优势,尽管 EMBEDR 是基于 t-SNE 损失函数设计的。
结果¶
scDEED 方法的简要描述¶
图 1I 展示了 scDEED 方法,该方法通过比较每个细胞在预嵌入空间和二维嵌入空间中的邻居来评估一种二维嵌入方法(如 t-SNE 或 UMAP),两个邻居集均由欧几里得距离和预定的邻居大小(即细胞数量乘以“相似度百分比”,scDEED 唯一的超参数,默认设置为 50%;详细设置见方法部分)定义。在 t-SNE 和 UMAP 的实际应用中(如在 Seurat 管道中),预嵌入空间由对数变换归一化基因表达水平的前几个主成分(PC)定义,通常为 20 到 50 个主成分。由于每个细胞与二维嵌入邻居的欧几里得距离对于数据的视觉检查至关重要,scDEED 为每个细胞定义了一个“可靠性评分”,用于衡量细胞在嵌入前后与有序邻居的二维欧几里得距离的一致性。具体而言,为每个细胞构建两个有序距离向量:预嵌入距离向量和二维嵌入距离向量,它们的长度均等于邻居大小。预嵌入距离向量包含细胞与预嵌入邻居之间的二维欧几里得距离,按这些邻居在预嵌入空间中从最近到最远的顺序排列。二维嵌入距离向量包含细胞与二维嵌入邻居之间的二维欧几里得距离,按这些邻居在二维嵌入空间中从最近到最远的顺序排列。然后,细胞的可靠性评分定义为两个向量之间的皮尔逊相关系数。
为了构建可靠性评分的零分布,scDEED 采用了一种排列策略:独立地排列每个基因在细胞间的表达水平。排列后,所有细胞的关系被破坏;所有细胞变得可交换,每个细胞的邻居成为所有细胞的随机集合。因此,在排列数据上计算的所有细胞的可靠性评分形成了零分布,即如果二维嵌入破坏了细胞在预嵌入空间中的邻居,并在二维嵌入空间中随机分配邻居给细胞时的可靠性评分分布。基于该零分布,定义了两个可靠性评分的临界值:(1)对应于零分布 95 百分位的可信临界值;(2)对应于零分布 5 百分位的可疑临界值。因此,对于原始数据中的每个细胞,如果其可靠性评分大于或等于可信临界值,则 scDEED 将其二维嵌入标记为可信。相反,如果细胞的可靠性评分小于或等于可疑临界值,则其二维嵌入被标记为可疑。不满足这些标准的细胞则保持未标记。值得注意的是,标记为可信(或可疑)的细胞嵌入的百分比不一定是 5%,因为百分位临界值是基于零分布定义的,而不是基于原始数据计算的可靠性评分。
scDEED 对细胞嵌入的可信或可疑识别使用户能够识别二维嵌入空间中可能是嵌入过程伪影而不是代表生物学意义的细胞类型的潜在伪造细胞簇。它还可以突出显示全局定位可能具有误导性的细胞簇。t-SNE 和 UMAP 中缺乏全局保留和随机定位簇是一个众所周知的问题。通过识别可疑细胞嵌入,scDEED 旨在解决这个问题。在以下部分中,我们将展示如何通过识别可疑细胞嵌入来揭示二维可视化中的可疑细胞类型关系的示例。
为了帮助用户获得更可信的数据二维可视化,scDEED 提供了一种方法,通过网格搜索来优化二维嵌入方法的超参数设置,以最小化可疑细胞嵌入的数量(例如,t-SNE 的困惑度;UMAP 的 min.dist 和 n.neighbors)。
我们注意到,scDEED 是一种灵活的方法,适用于任何二维嵌入方法,不限于 t-SNE 和 UMAP。在我们的结果中,我们重点展示了 scDEED 在 t-SNE 和 UMAP 中的应用,以展示其在增强二维可视化的可靠性方面的效果,从而得出具有生物学意义的结论。
scDEED 增强了 Hydra 数据集的 t-SNE 可视化¶
Hydra scRNA-seq 数据集是首个成年 Hydra 珊瑚虫的细胞图谱。通过 Drop-seq 对 24,985 个 Hydra 细胞的转录组进行了测序。在原始研究中,Hydra 细胞通过 t-SNE 进行可视化,困惑度超参数设置为 40。
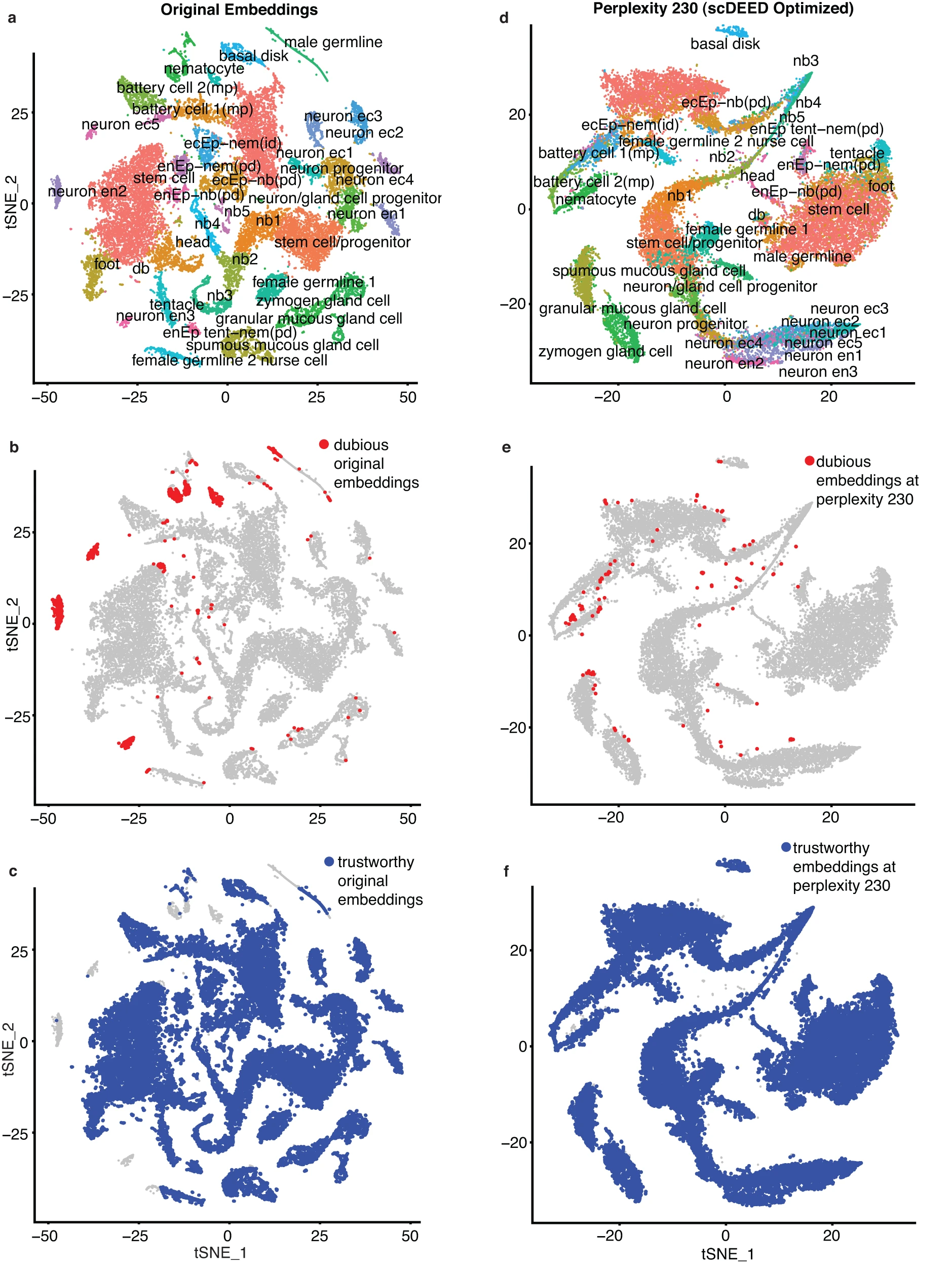
a. 原始研究中困惑度为 40 的 Hydra 数据集的 t-SNE 图。 b. scDEED 在困惑度为 40 时定义的可疑细胞嵌入。 c. scDEED 在困惑度为 40 时定义的可信细胞嵌入。 d. scDEED 优化的困惑度为 230 的 Hydra 数据集的 t-SNE 图。 e. scDEED 在困惑度为 230 时定义的可疑细胞嵌入。 f. scDEED 在困惑度为 230 时定义的可信细胞嵌入。
我们首先将 scDEED 应用于原始 t-SNE 可视化以检测可疑细胞嵌入。结果显示,有 4.77% 的细胞嵌入是可疑的(图 2b),而 92.84% 的细胞嵌入是可信的(图 2c)。其余的细胞嵌入的可靠性评分介于零可靠性评分的 5% 和 95% 之间,既不可信也不可疑。有趣的是,大多数可疑细胞嵌入表现为小簇,这表明这些簇可能不代表不同的细胞类型。此外,有三个值得注意的例子。首先,注释为刺细胞的细胞中包含许多可疑嵌入细胞(补充图 S1a)。我们通过显示这些细胞的基因表达谱与其他具有可信嵌入的刺细胞显著不同来验证这一发现(补充图 S1b)。其次,注释为神经元 ec1(外胚层神经元 1)的细胞被分为两个不相邻的簇(图 3a),但其中一个簇由可疑细胞嵌入组成(图 2b)。我们检查了这两个簇的基因表达谱,发现它们几乎无法区分(图 3c)。第三,注释为雄性生殖细胞的细胞有许多可疑嵌入(补充图 S2a)。检查具有可疑嵌入的雄性生殖细胞的基因表达谱,我们发现这些细胞应该属于两个簇(补充图 S2b),而不是原始可视化所示的连续体。这些结果证实了 scDEED 有效地识别了值得进一步研究的可疑细胞嵌入。
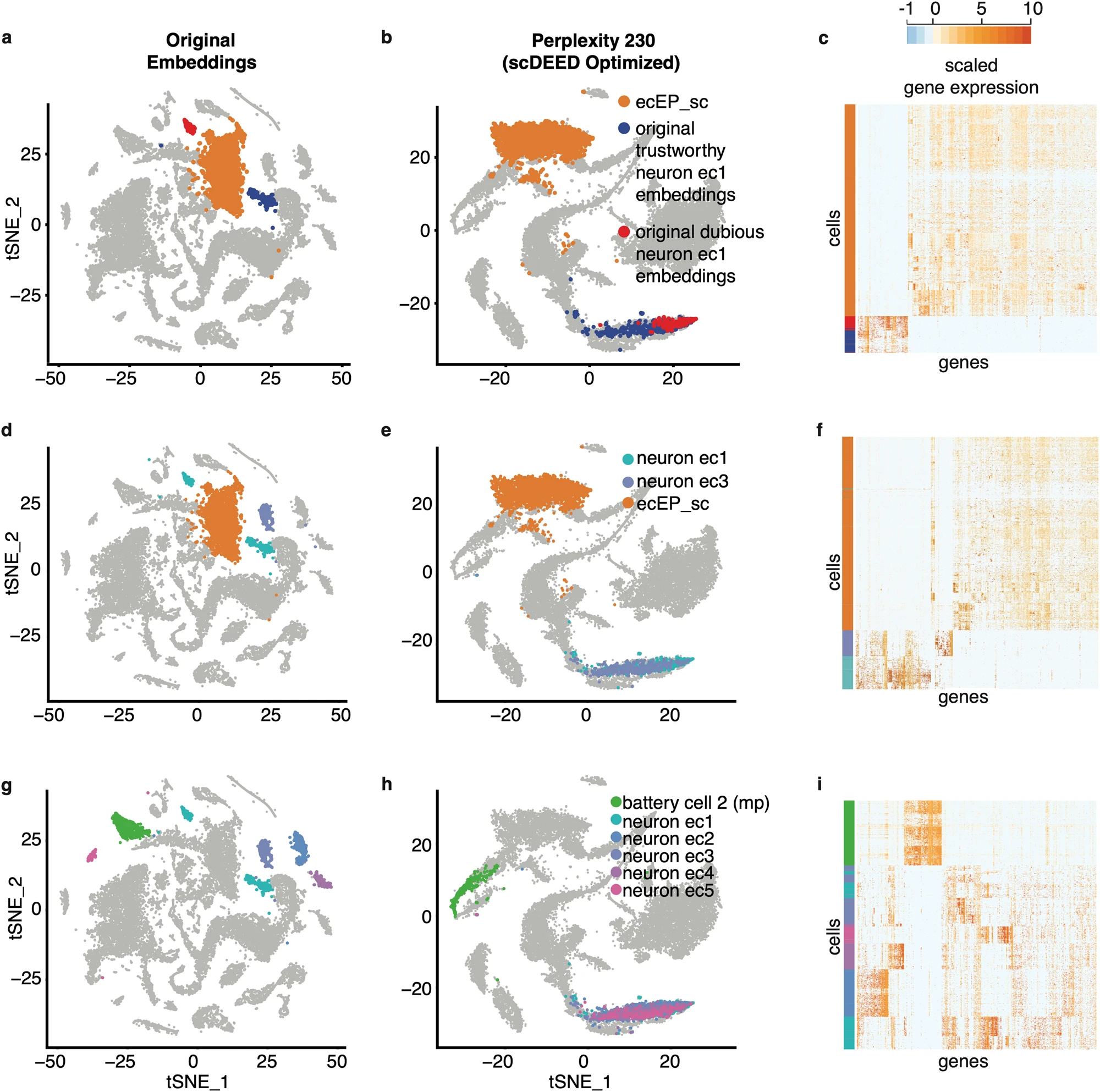
a–b 比较 t-SNE 图,其中突出显示了 ecEP_sc(外胚层上皮单细胞)、神经元 ec1 中的可信细胞嵌入和神经元 ec1 中的可疑细胞嵌入,分别在原始困惑度 40(a)和 scDEED 优化的困惑度 230(b)。c 基因表达热图,其中 (a) 和 (b) 中的突出显示细胞按 R 函数 heatmap.2() 找到的默认层次聚类排序。d–e 比较 t-SNE 图,其中突出显示了神经元 ec1、神经元 ec3 和 ecEP_sc 细胞。在原始困惑度 40 时(d),神经元 ec1 细胞分为两个独立的簇,并且与 ecEP_sc 细胞的距离与神经元 ec3 细胞的距离相似;在优化的困惑度 230 时(e),神经元 ec1 和神经元 ec3 细胞统一为一个远离 ecEP_sc 细胞的簇。f 基因表达热图,其中 (d) 和 (e) 中的突出显示细胞按 R 函数 heatmap.2() 找到的默认层次聚类排序。g–h 比较 t-SNE 图,其中突出显示了神经元 ec1、神经元 ec2、神经元 ec3、神经元 ec4、神经元 ec5 和电池细胞 2(mp)细胞。在原始困惑度 40 时(g),神经元 ec1、神经元 ec2、神经元 ec3、神经元 ec4 和神经元 ec5 细胞在围绕电池细胞 2(mp)细胞的不同簇中;在优化的困惑度 210 时(h),五个神经元 ec 簇统一为一个远离电池细胞 2(mp)细胞的簇。i 基因表达热图,其中 (g) 和 (h) 中的突出显示细胞按 R 函数 heatmap.2() 找到的默认层次聚类排序。
接下来,我们使用 scDEED 通过最小化可疑细胞嵌入的数量来优化 t-SNE 的困惑度超参数(图 2d);优化后的困惑度为 230(补充图 S3a,左)。在这个优化的困惑度下,可疑细胞嵌入从 4.77% 减少到 0.51%(图 2e),可信细胞嵌入从 92.84% 增加到 99.1%(图 2f)。值得注意的是,这个优化的困惑度 230 接近于 Kobak 和 Berens 建议的 251 困惑度,即细胞数量除以 100 的值。与这个建议的困惑度相比,scDEED 的优化困惑度是数据驱动的,因此考虑了除细胞数量以外的其他数据特征。除了找到最小化可疑细胞嵌入数量的困惑度外,我们还考虑了“kneedle”方法,通过在散点图中可疑细胞嵌入数量(y 轴)与困惑度(x 轴)的肘点处找到困惑度。结果的“kneedle”困惑度为 170(补充图 S3a,右),其细胞嵌入与 scDEED 优化的 230 困惑度输出的细胞嵌入类似(补充图 S3b)。
通过 scDEED 的优化,t-SNE 可视化(图 2d)与原始 t-SNE 可视化(图 2a)相比展示了几个关键差异。首先,在原始困惑度为 40 的情况下(图 3a),神经元 ec1 细胞的两个簇在 scDEED 优化的困惑度 230 下变为一个连续体(图 3b)。以 ecEP_sc(外胚层上皮单细胞)作为参考细胞类型,我们观察到一个更清晰的画面:在原始困惑度下,ecEP_sc 是两个分离的神经元 ec1 簇的近邻(在二维嵌入空间中具有相似的欧几里得距离)(图 3a);然而,在 scDEED 优化的困惑度下,ecEP_sc 远离统一的神经元 ec1 连续体(图 3b)。基因表达谱支持 scDEED 优化的可视化:统一的神经元 ec1 细胞确实具有相似的基因表达,并与 ecEP_sc 有很大区别(图 3c)。
其次,在原始困惑度下,注释为神经元 ec1 和神经元 ec3 的细胞是与 ecEP_sc 细胞相似地接近的独立簇(图 3d);然而,在 scDEED 优化的困惑度下,神经元 ec1 和神经元 ec3 细胞统一为一个远离 ecEP_sc 的簇(图 3e)。再次,scDEED 优化的可视化得到了这些细胞基因表达谱的支持(图 3f),这确认了神经元 ec1 和神经元 ec3 细胞确实相似并且与 ecEP_sc 细胞有区别。
第三,在原始困惑度下,外胚层神经元显示为五个亚型标记的六个簇:神经元 ec1、神经元 ec2、神经元 ec3、神经元 ec4 和神经元 ec5(图 3g)。相反,在 scDEED 优化的困惑度下,这五个亚型被统一为一个簇(图 3h)。以电池细胞 2(mp)作为参考细胞类型,我们发现 mp 相对于五个亚型的位置在 scDEED 优化困惑度后发生了巨大变化:在原始困惑度下,mp 比其他四个亚型更接近神经元 ec5(图 3g),但在优化的困惑度下,mp 远离所有五个亚型(图 3h)。同样,基因表达谱支持了 scDEED 优化的可视化(图 3i)。为了进一步证明这五个亚型应该被统一,我们计算了每个亚型和统一簇的 ROGUE 值,这是一种细胞簇纯度指标,范围为 0 到 1,值越高表示簇越纯。五个亚型的 ROGUE 值平均为 0.768,标准差为 0.055,而统一簇的 ROGUE 值为 0.744。因此,统一五个亚型并没有显著降低亚型的纯度,因此被认为是合理的。以上结果证实了 scDEED 在这个 Hydra 数据集上有效地优化了 t-SNE 的困惑度超参数。
scDEED 增强了 CAR-T 数据集的 t-SNE 可视化¶
CAR-T scRNA-seq 数据集包含来自 10 名接受 CD19 CAR-T 免疫治疗患者的 62,167 个 CD8+ 嵌合抗原受体修饰 -T(CAR-T)细胞。在原始研究中,CAR-T 细胞通过 t-SNE 进行可视化,困惑度为 30(图 4a,重现原始研究的图 5A,并突出显示了细胞簇)。
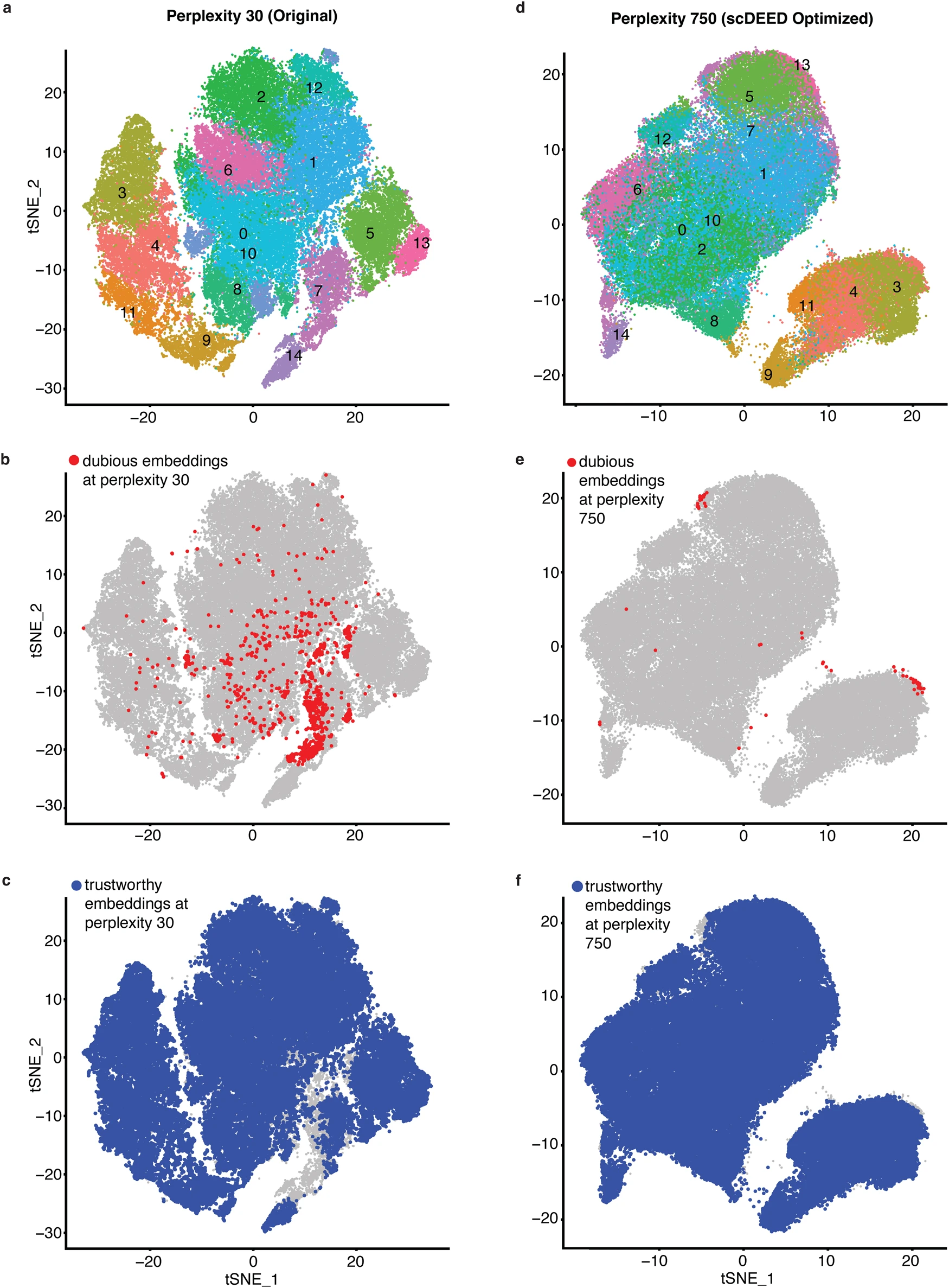
a 原始研究中困惑度为 30 的 CAR-T 数据集的 t-SNE 图。b scDEED 在原始困惑度 30 下定义的可疑嵌入。c scDEED 在原始困惑度 30 下定义的可信嵌入。d scDEED 优化后的困惑度 750 的 CAR-T 数据集的 t-SNE 图。e scDEED 在优化困惑度 750 下定义的可疑嵌入。f scDEED 在优化困惑度 750 下定义的可信嵌入。
我们首先将 scDEED 应用于原始可视化以检测可疑细胞嵌入。结果显示,有 1.81% 的细胞嵌入是可疑的(图 4b),而 83.99% 的细胞嵌入是可信的(图 4c)。大多数可疑细胞嵌入位于簇 7;在原始困惑度为 30 的情况下,这些可疑细胞嵌入在簇 7 中与可信细胞嵌入在视觉上不可区分(图 5a),这表明需要进一步研究。为了理解簇 7 中可疑和可信细胞嵌入之间的差异,我们进行了差异基因表达分析,发现这些细胞组确实具有不同的基因表达谱(图 5c;补充表 1)。特别是,许多核糖体基因在可信嵌入的细胞中表达更高,这与较高的转录活性有关。这一观察结果与可信嵌入细胞中早期刺激阶段的 CAR-T 细胞更丰富这一事实一致(可信嵌入细胞中有 48.05%,而可疑嵌入细胞中有 28.51% 的细胞来自早期刺激阶段;p 值=3.304e-12)。由于模拟后早期阶段的 CAR-T 细胞预期会比其他 CAR-T 细胞具有更高的转录活性,因此可信嵌入细胞具有比可疑嵌入细胞更高的核糖体基因表达是合理的。
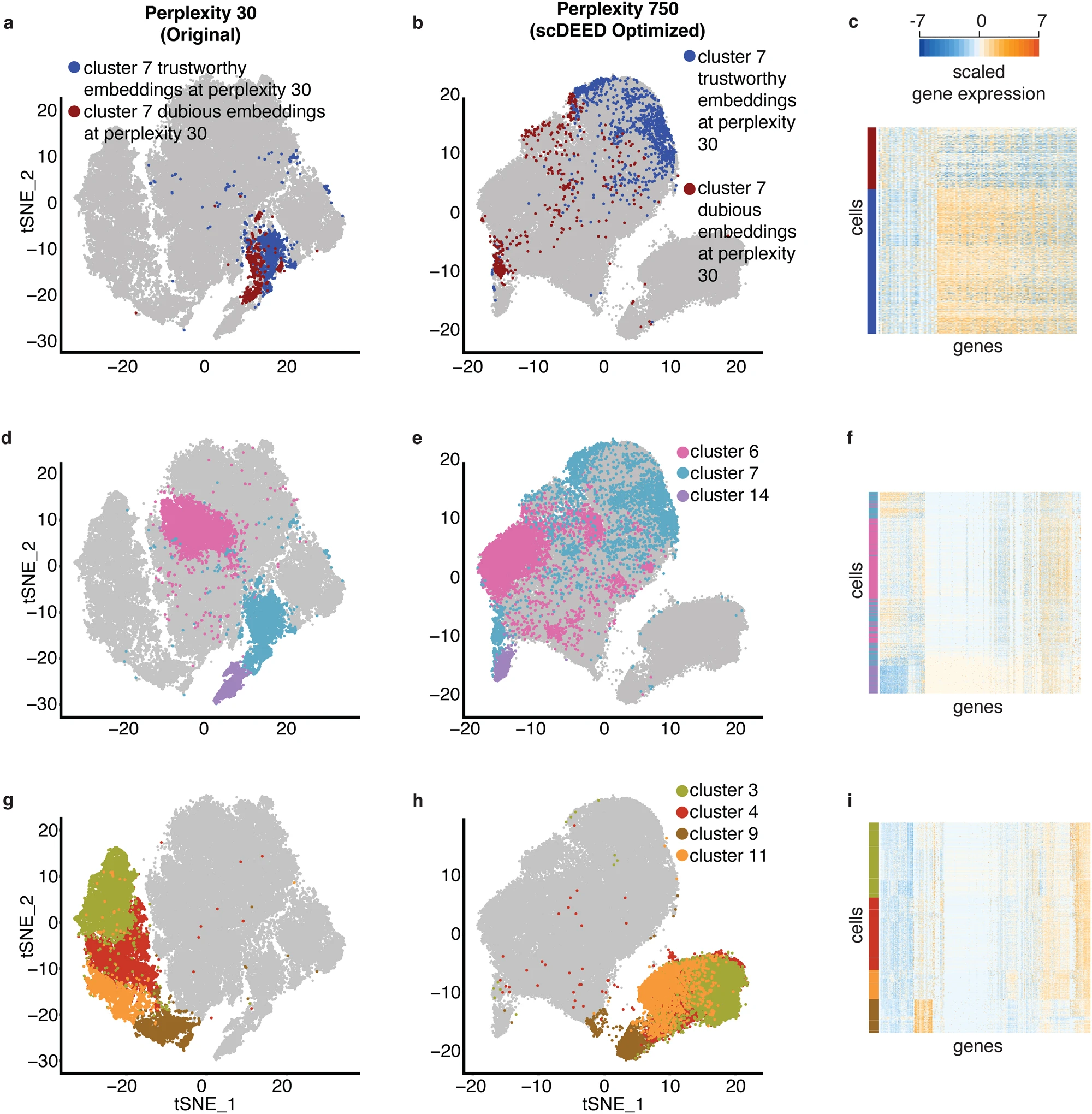
原始困惑度为 30(a)和 scDEED 优化的困惑度为 750(b)下簇 7 中可疑和可信细胞嵌入的比较 t-SNE 图。c (a) 和 (b) 中突出显示细胞的基因表达热图。原始研究中困惑度为 30(d)和 scDEED 优化的困惑度为 750(e)下簇 6、7 和 14 的比较 t-SNE 图。我们重新着色了簇 7 以便于在(d–f)中的可视化。f (d) 和 (e) 中突出显示细胞的基因表达热图,细胞按 R 函数 heatmap.2() 找到的默认层次聚类排序。原始研究中困惑度为 30(g)和 scDEED 优化的困惑度为 750(h)下簇 3、4、9 和 11 的比较 t-SNE 图。我们重新着色了这些簇以便于在(g–i)中的可视化。i (g) 和 (h) 中突出显示细胞的基因表达热图。
接下来,我们使用 scDEED 通过最小化可疑细胞嵌入的数量来优化 t-SNE 的困惑度超参数;结果的困惑度为 750(补充图 S4a,左)。在这个优化的困惑度下,可疑细胞嵌入从 1.81% 减少到 0.09%(图 4e),可信细胞嵌入从 83.99% 增加到 95.87%(图 4f)。“kneedle”方法得出的困惑度为 170(补充图 S4a,右),其细胞嵌入与优化的 750 困惑度的细胞嵌入类似(补充图 S4b)。
在 scDEED 优化的 750 困惑度下,t-SNE 可视化(图 4d)与原始 t-SNE 可视化(图 4a)相比表现出几个关键差异。首先,在原始困惑度 30 的情况下,簇 7 中可疑嵌入和可信嵌入的细胞仍在一个簇中(图 5a),而在 scDEED 优化的困惑度下,这些细胞分离得很远(图 5b)。以簇 5 和簇 14 作为参考,我们观察到簇 7 中可疑嵌入的细胞靠近簇 14,而簇 7 中可信嵌入的细胞靠近簇 5(补充图 S5)。我们使用细胞轨迹重建方法 STREAM 验证了这一可视化(补充图 S5b):重建的细胞轨迹显示簇 7 中可疑嵌入的细胞与簇 14 细胞在一个分支(分支 S6),而簇 7 中可信嵌入的细胞与簇 5 细胞在另一个分支(分支 S4)。
其次,在原始困惑度下,簇 7 位于簇 14 和簇 6 之间;特别是簇 7 紧挨着簇 14,并与簇 6 有一定距离(图 5d)。然而,在我们优化困惑度后,这一模式发生了变化。特别是在 scDEED 优化的困惑度下,簇 7 中大多数可疑嵌入细胞在簇 14 旁边的一个小簇中,但其余簇 7 细胞(包括可信嵌入的簇 7 细胞)不再位于簇 14 和簇 6 之间(图 5e)。这一优化的可视化与基因表达谱一致(图 5f)。
第三,在阶段 IP(无刺激)下的四个簇,即簇 3、簇 4、簇 9 和簇 11(原始研究的图 5A),在原始困惑度下几乎没有重叠(图 5g)。然而,在 scDEED 优化的困惑度下,簇 9 保持独立,而其他三个簇(簇 3、簇 4 和簇 11)有较大的重叠(图 5h)。基因表达谱提供了支持性证据,显示簇 9 与其他三个簇不同(图 5i)。为了定量确认这一结果,我们计算了簇 3、簇 3 和簇 4 组合以及簇 3、簇 4 和簇 11 组合的 ROGUE 值,分别为 0.887、0.885 和 0.886。这些 ROGUE 值表明三个簇之间没有明显的分离。此外,当我们将簇 3、簇 4、簇 9 和簇 11 组合在一起时,ROGUE 值降至 0.832,这表明簇 9 的基因表达谱与簇 3、簇 4 和簇 11 不同。总之,ROGUE 值与 scDEED 优化困惑度下的 t-SNE 可视化一致。
第四,在原始困惑度下,簇 8 和簇 9 与簇 14 的距离相似(补充图 S6a 左)。然而,在优化困惑度后,簇 8 和簇 9 的相对位置发生了变化,簇 8 位于簇 14 和簇 9 之间(补充图 S6a 右)。同样,它们的基因表达谱及相应的细胞层次聚类支持了优化困惑度下的 t-SNE 可视化(补充图 S6b)。
scDEED 增强了 Alveolar 数据集的 UMAP 可视化¶
Alveolar 数据集是为了研究博来霉素诱导的肺损伤后再生过程中细胞动力学而收集的。通过 Dropseq 测量,Alveolar 数据集包含 28 只小鼠的 29,297 个细胞,每只小鼠约有 1000 个细胞。在原始研究中,细胞通过 UMAP 进行可视化,超参数设置为 min.dist=0.3 和 n.neighbors=10(图 6a,重现原始研究的图 1a,并突出显示注释的细胞簇)。
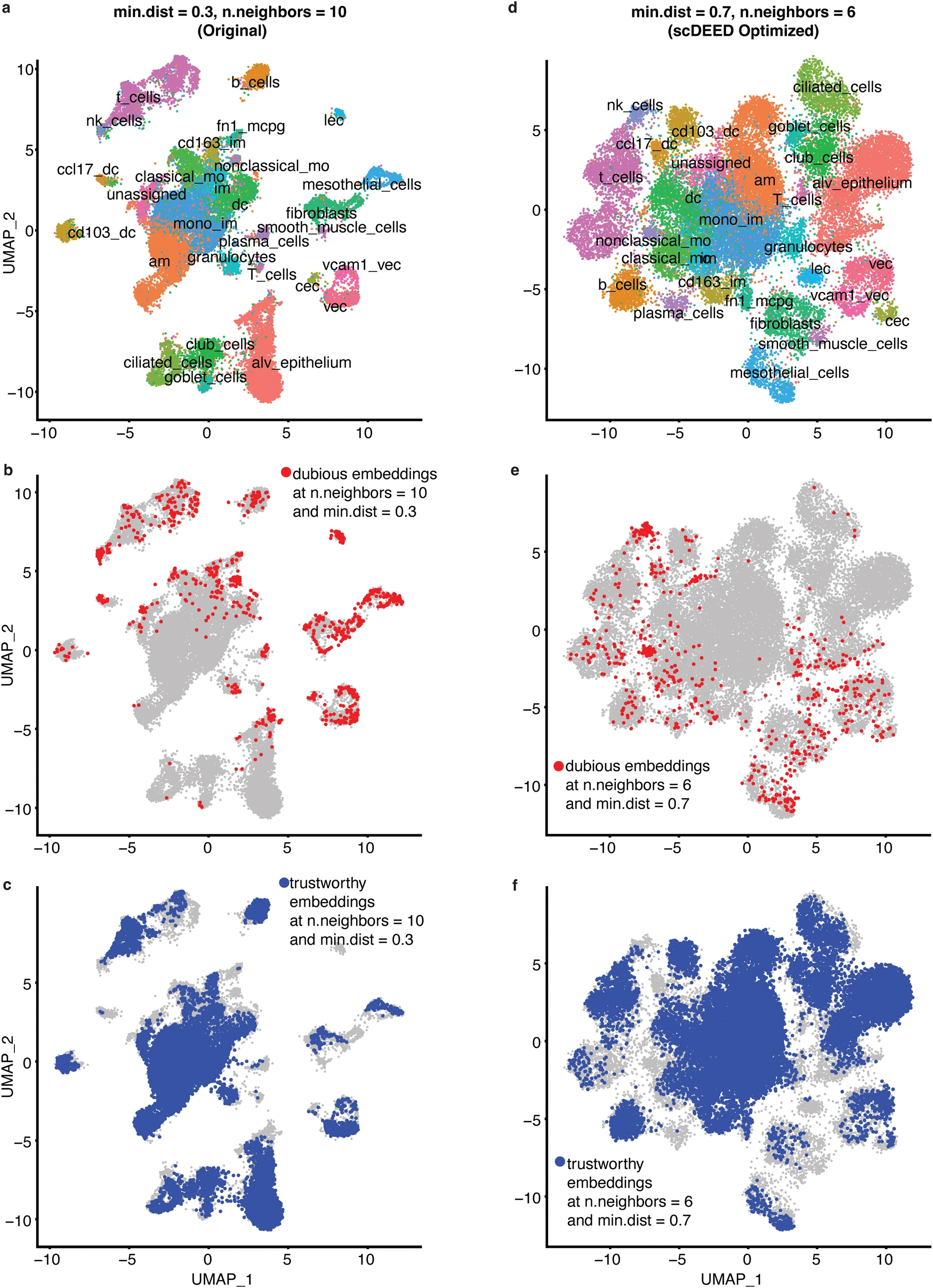
a 原始研究中超参数设置为 min.dist=0.3 和 n.neighbors=10 的 Alveolar 数据集的 UMAP 图。b scDEED 在原始超参数下定义的可疑嵌入。c scDEED 在原始超参数下定义的可信嵌入。d scDEED 联合优化超参数后(min.dist=0.7 和 n.neighbors=6)的 Alveolar 数据集的 UMAP 图。e scDEED 在优化超参数下定义的可疑嵌入。f scDEED 在优化超参数下定义的可信嵌入。
我们将 scDEED 应用于原始可视化以检测可疑细胞嵌入。结果显示,3.14% 的细胞嵌入是可疑的(图 6b),而 50.71% 的细胞嵌入是可信的(图 6c)。与 t-SNE 不同,UMAP 有两个超参数:min.dist 和 n.neighbors,它们共同决定二维细胞嵌入。scDEED R 包允许用户边际或联合优化 min.dist 和 n.neighbors,其目标是最小化可疑嵌入的数量。在边际优化中,我们通过将另一个超参数固定在原始值来优化 min.dist 或 n.neighbors。因此,边际优化考虑的超参数组合较少,因此比联合优化更高效。
通过边际优化,我们获得了 n.neighbors=5(min.dist=0.3)和 min.dist=0.6(n.neighbors=10)(补充图 S7)。特别是,第一个超参数集(min.dist=0.3 和 n.neighbors=5)将可疑嵌入减少到 2.28%,将可信嵌入增加到 56.73%;第二个超参数集(min.dist=0.6 和 n.neighbors=10)将可疑嵌入减少到 2.45%,将可信嵌入增加到 54.75%。当我们联合优化两个超参数时,获得了 min.dist=0.7 和 n.neighbors=6(图 6d),进一步将可疑嵌入减少到 2.04%(图 6e),将可信嵌入增加到 58.84%(图 6f)。
在包含许多可疑细胞嵌入的细胞簇中,我们关注以下簇的相对位置:lec、b_cells、vcam1_vec、vec 和 t_cells。我们注意到原始 UMAP 可视化(图 7a, d)与三个优化可视化(图 7b, e 中的联合超参数优化;补充图 S8a–b 中的边际超参数优化)之间的两个关键差异;请注意,在这三个优化可视化中,细胞类型的相对位置比在原始可视化中的位置更相似。
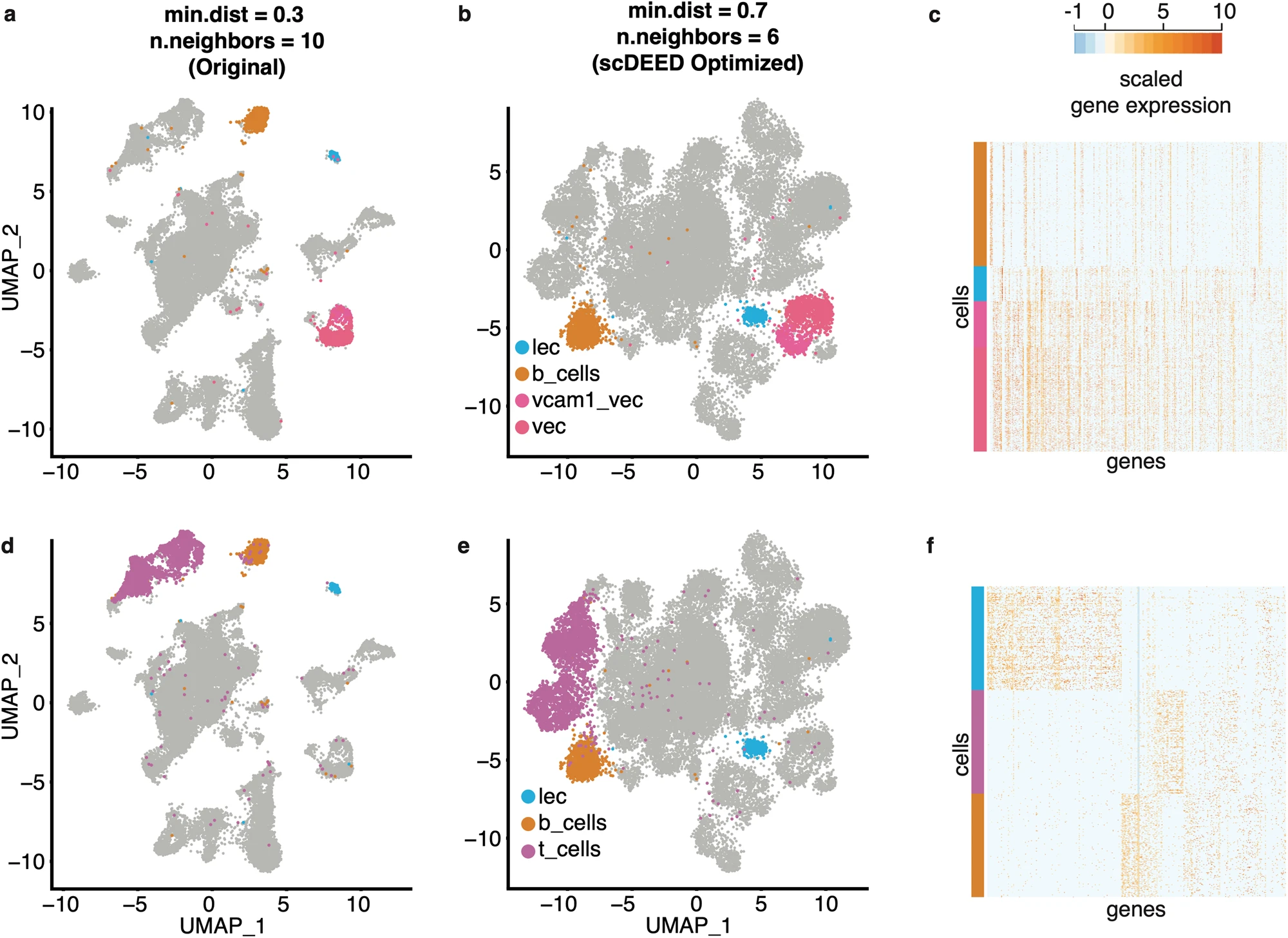
原始超参数 min.dist=0.3 和 n.neighbors=10(a)与 scDEED 联合优化超参数 min.dist=0.7 和 n.neighbors=6(b)下 Alveolar 数据集中突出显示 lec、b_cells、vcam1_vec 和 vec 细胞的比较 UMAP 图。c (a) 和 (b) 中突出显示细胞的基因表达热图。原始超参数(d)与 scDEED 联合优化超参数(e)下 Alveolar 数据集中突出显示 lec、b_cells 和 t_cells 细胞的比较 UMAP 图。f (d) 和 (e) 中突出显示细胞的基因表达热图。注意我们随机抽样了 b_cells 和 t_cells(分别从 911 和 2709 个细胞中抽样)以使每个簇有 256 个细胞(与 lec 中的细胞数相同),以便生成有视觉信息的热图。
首先,在原始超参数设置(min.dist=0.3 和 n.neighbors=10)下,lec 簇靠近 b_cells 簇,远离 vcam1_vec 和 vec 簇(图 7a)。相比之下,在三个优化的超参数设置下,lec 不再与 b_cells 相邻,而是靠近 vcam1_vec 和 vec,b_cells 则远离(图 7b 和补充图 S8a)。我们在基因表达谱中找到了支持证据,显示 b_cells 簇与 lec、vcam1_vec 和 vec 簇有显著区别(图 7c)。
其次,在原始超参数设置下,b_cells 簇位于 t_cells 和 lec 簇之间,距离大致相等(图 7d)。相比之下,在三个优化的超参数设置下,b_cells 和 t_cells 簇彼此靠近,但远离 lec 簇(联合超参数优化见图 7e;边际超参数优化见补充图 S8b)。再次,为了评估 b_cells、t_cells 和 lec 簇的相对位置,我们检查了基因表达谱。图 7f 显示,lec 在三个簇中最为独特,这支持了 scDEED 发现的优化可视化结果。
scDEED 增强了 Samusik 数据集的 UMAP 可视化¶
Samusik 数据集来自对骨髓造血的质谱流式细胞术研究。它包含超过 86,000 个细胞、38 个细胞表型特征和 24 个注释的细胞类型。在原始研究中,细胞通过 UMAP 可视化,超参数设置为 min.dist=0.2 和 n.neighbors=15(图 8a,重现原始研究的图 2a)。
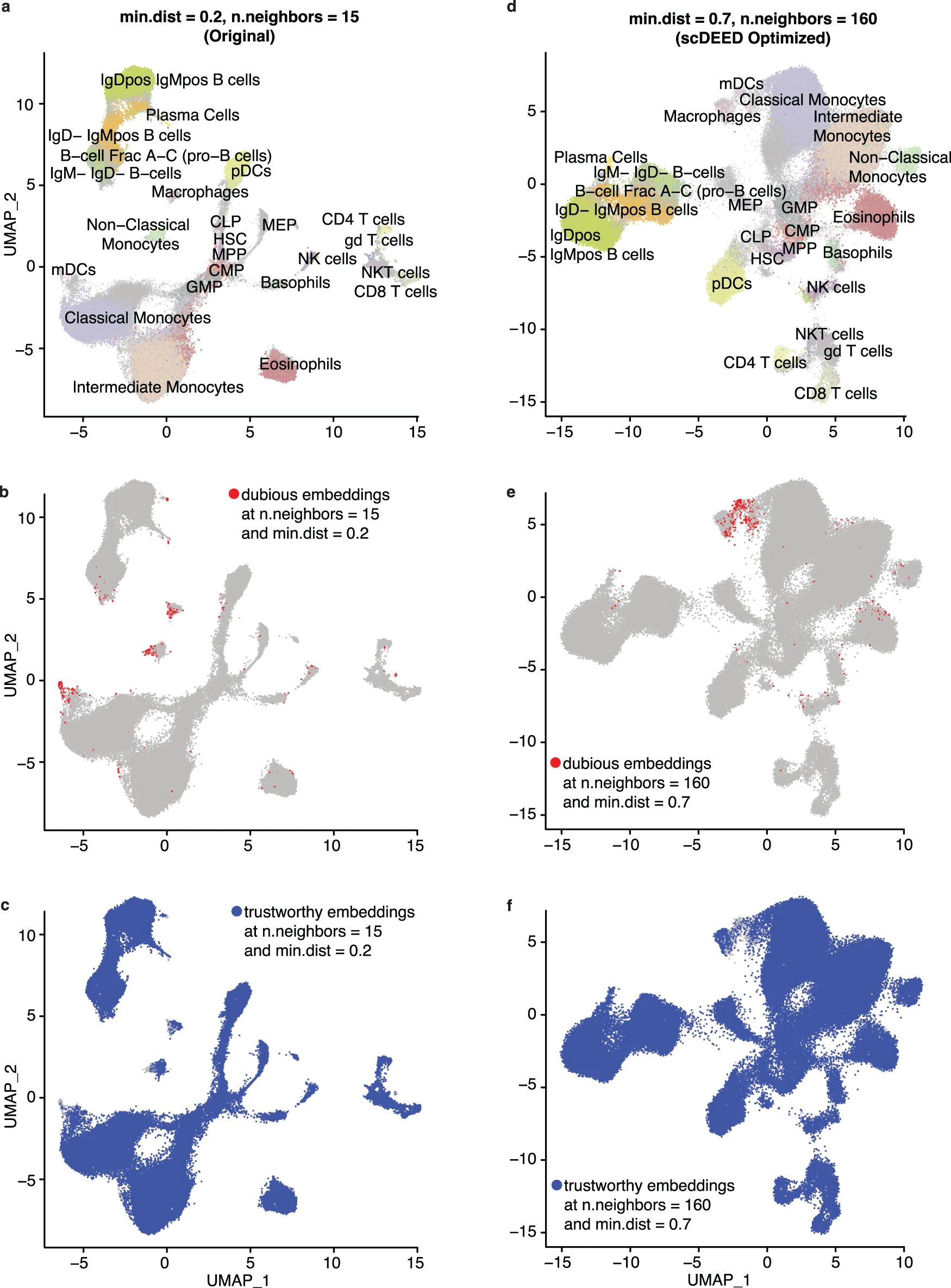
a 原始研究中超参数设置为 min.dist=0.2 和 n.neighbors=15 的 Samusik 数据集的 UMAP 图。b scDEED 在原始超参数下定义的可疑嵌入。c scDEED 在原始超参数下定义的可信嵌入。d scDEED 联合优化超参数后(min.dist=0.7 和 n.neighbors=160)的 Samusik 数据集的 UMAP 图。e scDEED 在优化超参数下定义的可疑嵌入。f scDEED 在优化超参数下定义的可信嵌入。
我们首先将 scDEED 应用于原始可视化,发现 1.13% 的细胞嵌入是可疑的(图 8b),98.74% 的细胞嵌入是可信的(图 8c)。接下来,我们通过最小化可疑细胞嵌入的数量,应用 scDEED 边际优化 min.dist 和 n.neighbors。从边际优化中,我们得到了 n.neighbors=160(min.dist=0.2)和 min.dist=0.05(n.neighbors=15)(补充图 S9)。特别是,第一个超参数集(min.dist=0.2 和 n.neighbors=160)将可疑嵌入减少到 0.68%,将可信嵌入增加到 99.19%;第二个超参数集(min.dist=0.05 和 n.neighbors=15)将可疑嵌入减少到 0.837%,将可信嵌入增加到 99.028%。当我们联合优化两个超参数时,得到了 min.dist=0.7 和 n.neighbors=160(图 8d)。这个超参数集进一步将可疑嵌入减少到 0.641%(图 8e),将可信嵌入增加到 99.269%(图 8f)。正如预期的那样,联合优化实现了最低比例的可疑细胞嵌入和最高比例的可信细胞嵌入。同时,联合优化和 n.neighbors 的边际优化共享相同的 n.neighbors=160,并且在可视化上相似(联合超参数优化见图 8d;边际超参数优化见补充图 S9b)。
在包含可疑细胞嵌入的细胞类型中,我们关注以下细胞类型的相对位置:非经典单核细胞(ncm)、中间单核细胞、嗜碱性粒细胞、浆细胞样树突细胞(pDCs)、髓系树突细胞(mDCs)和巨噬细胞。值得注意的是,联合优化和 n.neighbors 的边际优化(图 8d 和补充图 S9b 左)与原始 UMAP 可视化(图 8a)相比表现出两个关键差异。
首先,在原始超参数设置下(min.dist=0.2 和 n.neighbors=15),巨噬细胞位于 pDCs 和 ncm 之间,距离大致相等,并且远离 mDCs(图 9a)。相比之下,在边际优化的 n.neighbors 设置(min.dist=0.2 和 n.neighbors=160)和联合优化的超参数设置(min.dist=0.7 和 n.neighbors=160)下,巨噬细胞变得邻近 mDCs,但远离 ncm 和 pDCs(图 9b)。基因表达谱证实,巨噬细胞与 mDCs 比与 ncm 和 pDCs 更相似(图 9c)。
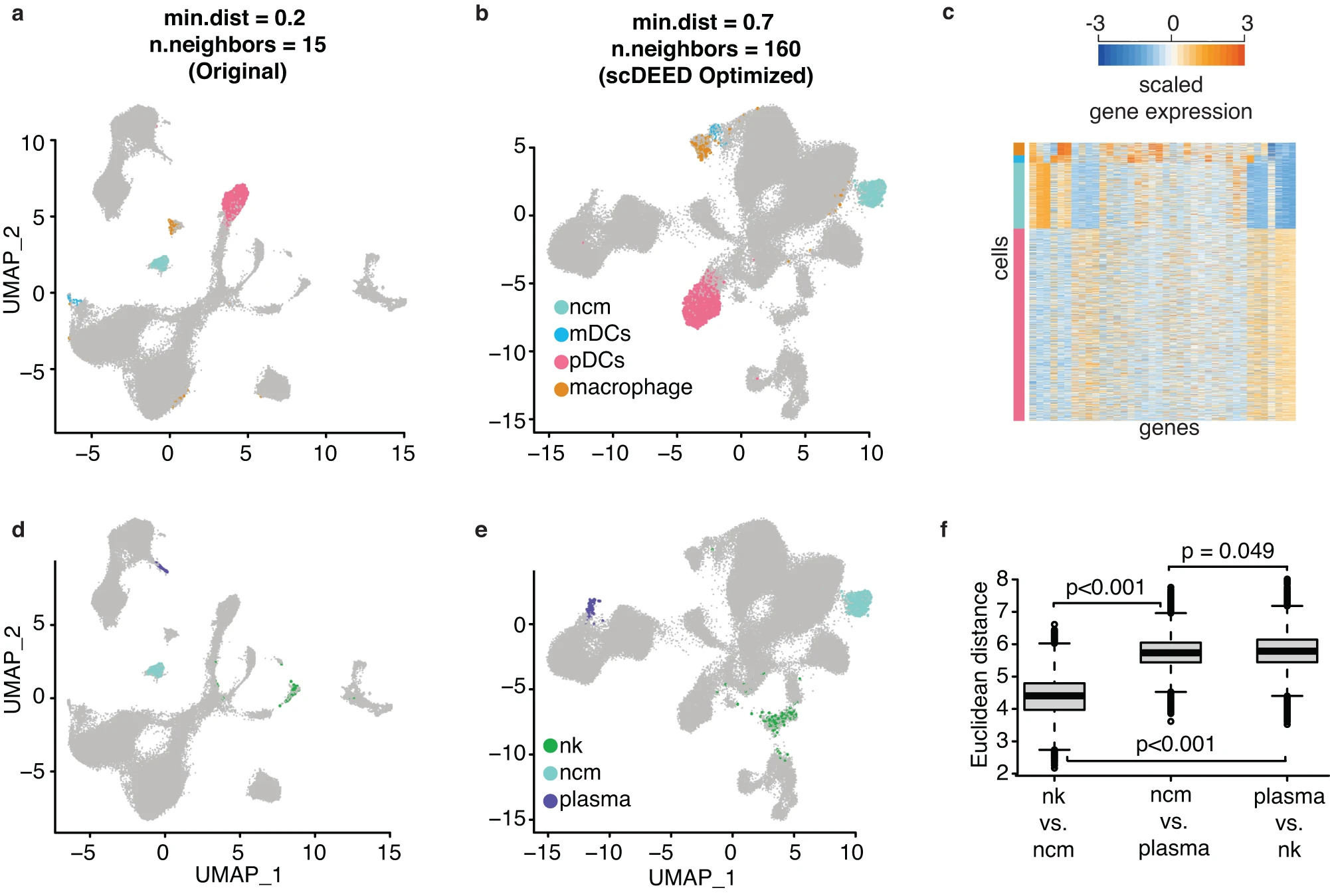
原始超参数 min.dist=0.2 和 n.neighbors=15(a)与 scDEED 联合优化超参数 min.dist=0.7 和 n.neighbors=160(b)下突出显示非经典单核细胞(ncm)、髓系树突细胞(mDCs)、浆细胞样树突细胞(pDCs)和巨噬细胞的 Samusik 数据集的比较 UMAP 图。c (a) 和 (b) 中突出显示细胞的基因表达热图。原始超参数(d)与 scDEED 优化超参数(e)下突出显示 NK 细胞(nk)、ncm 和浆细胞的 Samusik 数据集的比较 UMAP 图。f 比较 ncm、nk 和浆细胞之间的距离。箱线图的中心线、边界和须分别表示中位数、第一和第三四分位数以及在箱线范围 1.5 倍的最小值和最大值。显示了箱线图之间比较的两样本 t 统计量 p 值,零分布基于对三种细胞类型的 1000 次随机划分计算,保持三种细胞类型的大小(理论 t 分布不应使用,因为距离不是独立的)。两样本 t 统计量如下:(nk vs. ncm, n=192,024 对) vs (ncm vs. 浆细胞, n=119,888 对)=-728.580 (p<0.001),(nk vs. ncm, n=192,024 对) vs (浆细胞 vs. nk, n=22,302 对)=-370.042 (p<0.001),(ncm vs. 浆细胞, n=119,888 对) vs (浆细胞 vs. nk, n=22,302 对)=-13.094 (p=0.049)。f 证实(e)比(d)更好地保留了三个簇的相对距离。源数据作为上传到 Zenodo 的源数据文件提供。
其次,在原始超参数设置下,查看浆细胞、非经典单核细胞(ncm)和 NK 细胞三种细胞类型之间的成对距离,我们发现浆细胞与 ncm 之间的距离与 ncm 和 NK 细胞之间的距离相似,而浆细胞与 NK 细胞之间的距离最大(图 9d)。相比之下,在联合优化的超参数设置下(min.dist=0.7 和 n.neighbors=160),ncm 和 NK 细胞之间的距离成为三对成对距离中最小的,而其他两对距离变得相似(图 9e)。为了评估这三种细胞类型的相对位置,我们计算了 38 维特征空间中(UMAP 嵌入前)每两种细胞类型之间的细胞间欧几里得距离,确认 NK 细胞和 ncm 比浆细胞更接近彼此(图 9f)。
注意,上述两种差异在边际优化超参数 min.dist(min.dist=0.05 和 n.neighbors=15)得到的嵌入中不那么明显。可能的原因是这个超参数集在三个优化超参数集中的可疑嵌入比例最高,可信嵌入比例最低。
scDEED 改进了 t-SNE 和 UMAP 之间的一致性¶
人类 PBMC 数据集是为了比较多种 scRNA-seq 技术而收集的。它包含 31,021 个带有细胞类型标签的细胞,基因表达水平以每 10,000 个转录本的对数转化 UMI 计数为单位。我们通过 R 包“SeuratData”访问数据集“pbmcsca.SeuratData”。
我们考虑了在数据集中测量超过 500 个细胞的三种 scRNA-seq 技术:Dropseq、inDrops 和 SeqWell。对于每种技术,我们使用默认超参数或 scDEED 优化的超参数,通过 t-SNE 和 UMAP 对其测量的细胞进行可视化(图 10)。我们观察到,优化后的超参数在细胞类型之间的相对距离方面,在不同的 scRNA-seq 技术之间以及 t-SNE 和 UMAP 之间更加一致。具体来说,我们有以下三点观察结果。
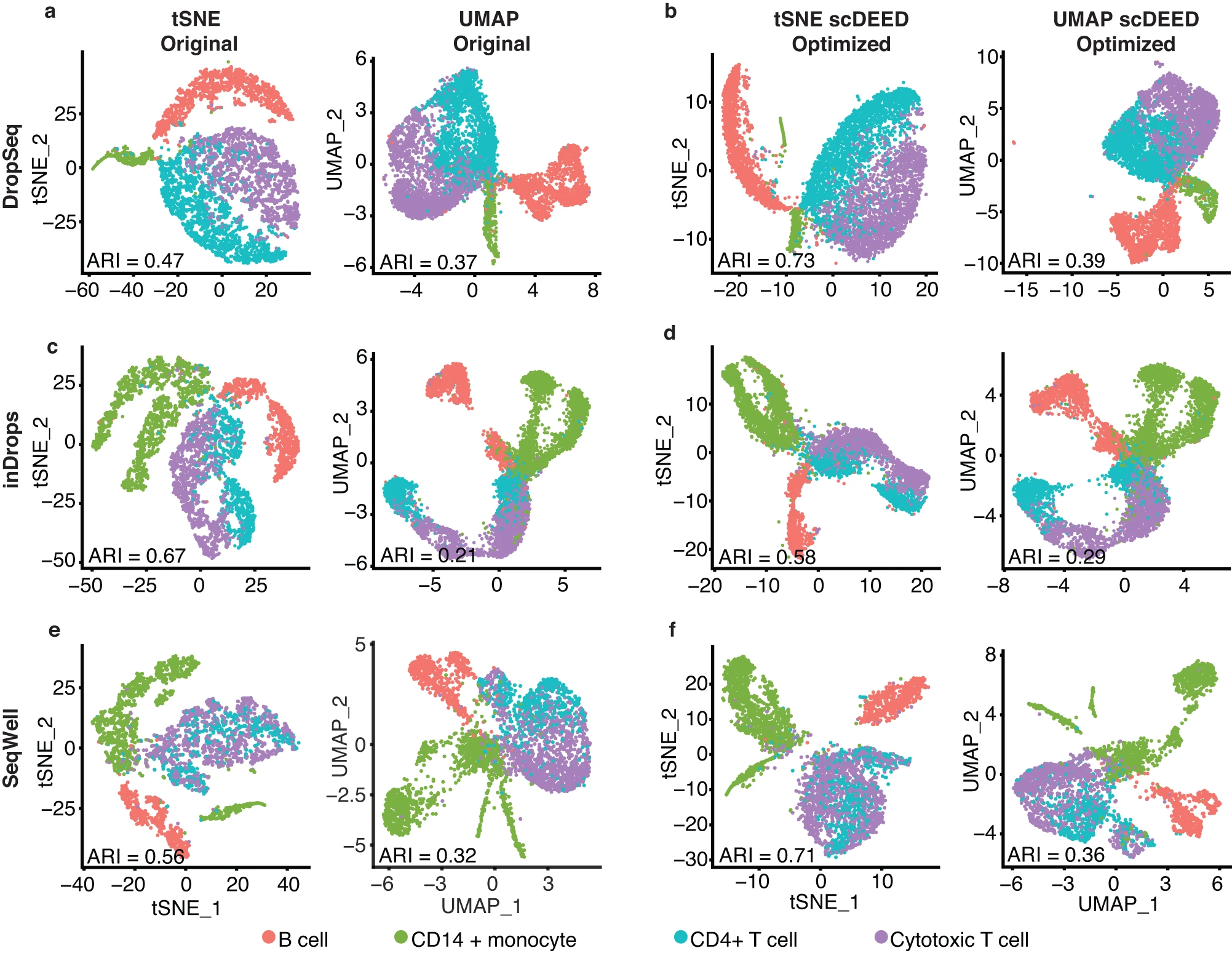
a DropSeq 数据集在原始超参数下的 t-SNE 和 UMAP 图,困惑度=30(左)和 min.dist=0.3 以及 n.neighbors=30(右)。b DropSeq 数据集在 scDEED 优化超参数下的 t-SNE 和 UMAP 图,困惑度=290(左)和 min.dist=0.5 以及 n.neighbors=5(右)。c inDrops 数据集在原始超参数下的 t-SNE 和 UMAP 图,困惑度=30(左)和 min.dist=0.3 以及 n.neighbors=30(右)。d inDrops 数据集在 scDEED 优化超参数下的 t-SNE 和 UMAP 图,困惑度=320(左)和 min.dist=0.5 以及 n.neighbors=80(右)。e SeqWell 数据集在原始超参数下的 t-SNE 和 UMAP 图,困惑度=30(左)和 min.dist=0.3 以及 n.neighbors=30(右)。f SeqWell 数据集在 scDEED 优化超参数下的 t-SNE 和 UMAP 图,困惑度=140(左)和 min.dist=0.2 以及 n.neighbors=7(右)。通过应用谱聚类识别与每组嵌入中的细胞类型数量相同的细胞簇,我们发现六种情况中的五种(t-SNE 和 UMAP 嵌入的三种 scRNA-seq 技术)中,优化后的嵌入导致了更高的调整兰德指数(ARI),这表明优化后的嵌入更好地代表了细胞类型。
首先,在默认超参数下可视化 Dropseq 数据时,t-SNE 和 UMAP 显示了四种细胞类型之间不同的相对距离:t-SNE 按顺时针顺序排列了细胞类型细胞毒性 T 细胞、CD4+ T 细胞、CD14+ 单核细胞和 B 细胞(图 10a,左),但 UMAP 交换了 CD14+ 单核细胞和 B 细胞的顺序(图 10a,右)。相比之下,在 scDEED 优化的超参数下,t-SNE 和 UMAP 具有相同的四种细胞类型的逆时针顺序:细胞毒性 T 细胞、CD4+ T 细胞、B 细胞和 CD14+ 单核细胞(图 10b)。也就是说,scDEED 优化的超参数提高了 t-SNE 和 UMAP 之间的一致性。
其次,在默认超参数下可视化 inDrops 数据时,t-SNE 按逆时针顺序排列了细胞毒性 T 细胞、CD4+ T 细胞、B 细胞和 CD14+ 单核细胞,而 UMAP 按顺时针顺序排列了这四种细胞类型(图 10c)。相比之下,在 scDEED 优化的超参数下,t-SNE 和 UMAP 都按顺时针顺序排列了四种细胞类型(图 10d)。值得注意的是,这个顺时针顺序与 Dropseq 数据的 t-SNE 和 UMAP 可视化中的逆时针顺序一致。因此,在 scDEED 优化后,四种细胞类型的相对距离在 Dropseq 和 inDrops 之间变得一致。
第三,在默认的困惑度超参数下可视化 Seqwell 数据时,t-SNE 将 CD14+ 单核细胞类型分成了两个簇,细胞毒性 T 细胞和 CD4+ T 细胞位于它们之间(图 10e,左);然而,在 scDEED 优化的困惑度下,这两个 CD14+ 单核细胞簇连接在一起(图 10f,左)。优化后的 t-SNE 和 UMAP 可视化具有一致的逆时针顺序的四种细胞类型:细胞毒性 T 细胞、CD4+ T 细胞、B 细胞和 CD14+ 单核细胞(图 10f)。因此,在 scDEED 优化后,四种细胞类型的相对距离在三种 scRNA-seq 技术之间变得一致。
最后,我们验证了 scDEED 优化后的二维细胞嵌入更好地与细胞类型对齐。为此,我们对原始嵌入和 scDEED 优化的嵌入应用谱聚类,以识别细胞簇并通过计算调整兰德指数(ARI)检查与细胞类型的一致性。我们选择谱聚类是因为它能够识别非球形形状的簇。作为细胞簇标签和细胞类型标签之间一致性的度量,ARI 调整了由于随机机会引起的一致性。我们发现,在六种情况中的五种(t-SNE 和 UMAP 嵌入的三种 scRNA-seq 技术),优化后的嵌入导致更高的 ARI,这表明优化后的嵌入更好地代表了细胞类型。
scDEED 改进了 RNA 速度的 t-SNE 可视化¶
我们对齿状回数据集进行了 RNA 速度分析。RNA 速度向量在高维空间中计算,并用于可视化二维向量场。可视化的二维向量是基于二维嵌入空间中定义的小邻域细胞计算的。因此,二维嵌入影响速度可视化。
补充图 S10a 显示了原始研究中的 t-SNE 可视化的一部分细胞,补充图 S10c 显示了 scDEED 优化的困惑度下相同细胞的 t-SNE 可视化。特别值得注意的是从 nIPC 到神经元亚型 1 和 2(Neuro1 和 Neuro2),然后到未成熟颗粒细胞,最后到成熟颗粒细胞的轨迹。在 scDEED 优化的困惑度下,这条速度轨迹在 t-SNE 可视化中变得更加明显(补充图 S10d)而不是在原始困惑度下(补充图 S10b)。除了轨迹中的细胞外,成熟颗粒细胞继续保持接近零的速度,这与成熟颗粒细胞处于分化末端的事实一致。因此,scDEED 的优化困惑度改进了 RNA 速度的 t-SNE 可视化。
scDEED 在优化 t-SNE 和 UMAP 方面优于 EMBEDR¶
我们将 scDEED 与基于 t-SNE 损失函数设计的 EMBEDR 方法进行了比较,比较了三个数据集:(1)Hydra 数据集(用于“结果”部分“scDEED 增强 Hydra 数据集的 t-SNE 可视化”),(2)Tabula Muris 联盟骨髓数据集(用于 EMBEDR 论文),以及(3)由 scDesign3 生成的合成数据。我们从三个方面比较了 scDEED 和 EMBEDR:在默认 t-SNE 困惑度下检测可疑嵌入,优化 t-SNE 困惑度,以及计算时间。
在 Hydra 数据集上,在原始 t-SNE 困惑度 40 下,与 scDEED(图 2b;补充图 S11a)不同,EMBEDR 发现大多数细胞嵌入是可疑的(补充图 S11b)。我们认为 EMBEDR 的结果不太可能合理,因为如果大多数细胞嵌入是可疑的,那么 Hydra 研究中使用的 t-SNE 可视化将毫无意义;然而,事实并非如此。
除了可疑嵌入的数量外,scDEED 和 EMBEDR 在 Hydra 数据集上的检测结果还有两个显著差异。首先,在刺细胞中,scDEED 结果中的可疑嵌入细胞与可信嵌入细胞相比更为明显(补充图 S11c 和 e;补充图 S1)比在 EMBEDR 结果中(补充图 S11d 和 f)。为了量化 scDEED 和 EMBEDR 结果之间的差异,我们分别计算了 scDEED 和 EMBEDR 结果中可疑嵌入细胞和可信嵌入细胞之间的成对距离;然后,我们使用单边 Wilcoxon 秩和检验比较了这两组成对距离,并获得了小于 10^-16 的 p 值,表明 scDEED 发现的可疑嵌入和可信嵌入更为不同,因此更合理。其次,对于分为两个不相邻簇的神经元 ec1 细胞(图 3a),scDEED 将一个簇识别为可疑,另一个簇识别为可信(补充图 S11a)。相比之下,EMBEDR 在两个簇中都识别出可疑细胞嵌入(补充图 S11g),但 EMBEDR 识别的可疑嵌入和可信嵌入在基因表达水平上没有明显差异(补充图 S11i)。由于 scDEED 和 EMBEDR 都在各自优化的嵌入中统一了两个簇(scDEED 见图 3b;EMBEDR 见补充图 S11h),因此在原始困惑度 40 下,scDEED 的可疑检测结果比 EMBEDR 的更合理,因为每个簇中的神经元 ec1 细胞应该是共同可疑(远离相似细胞)或共同可信(接近相似细胞)。
关于在 Hydra 数据集上优化困惑度超参数,由于 EMBEDR 没有默认的候选困惑度值列表,我们为 EMBEDR 提供了 scDEED 的默认候选困惑度值,EMBEDR 选择了最高的候选困惑度 410(补充图 S12)。这一结果是预期的,因为 EMBEDR 的损失函数是 t-SNE 损失函数(即 KL 散度),据报道,t-SNE 损失函数随着困惑度的增加而趋于减少。这就是说,EMBEDR 预计会选择最大的候选困惑度,这是一个概念上不理想的特性。
虽然 scDEED 和 EMBEDR 在 Hydra 数据集上找到了不同的优化困惑度值,但结果的 t-SNE 可视化非常相似(补充图 S12)。因此,我们通过评估关于保持邻近信息的两个指标进一步比较了这两个 t-SNE 可视化。第一个指标是 K 最近邻(KNN),反映了局部信息的保持,即预嵌入空间中 K=10 最近邻居保持在二维嵌入空间中 K=10 最近邻居集合中的平均比例(为每个细胞计算一个比例,并对所有细胞的比例取平均)。第二个指标是 K 最近簇(KNC),反映了全局信息的保持,即预嵌入空间中 K=4 最近簇保持在二维嵌入空间中 K=4
最近簇集合中的平均比例(为每个细胞计算一个比例,并对所有细胞的比例取平均)。对于 Hydra 数据集,由 EMBEDR 优化的嵌入和由 scDEED 优化的嵌入具有相同的 KNC 为 0.44,而 KNN 对 scDEED(0.77)比对 EMBEDR(0.75)稍好。因此,这两组优化的嵌入共享相似的信息保留水平。
尽管 scDEED 和 EMBEDR 在 Hydra 数据集上生成的优化嵌入相似,scDEED 在计算效率方面远优于 EMBEDR。在不使用并行化的情况下,scDEED 在 4 小时内完成了分析(可疑嵌入检测和困惑度优化),而 EMBEDR 使用所有可用处理器(EMBEDR 的默认设置)完成了 18.5 小时。
在 EMBEDR 论文中使用的 Tabula Muris 联盟骨髓数据集上,EMBEDR 报告的最佳 t-SNE 困惑度为 1000,我们能够使用 4821 个细胞的处理数据重现 EMBEDR 的优化可视化(补充图 S13b)。与 EMBEDR 不同,scDEED 找到了一个最佳 t-SNE 困惑度为 100(补充图 S13a)。这个例子再次突显了 EMBEDR 对高困惑度值的偏好。需要注意的是,scDEED 的最佳困惑度 100 落在建议的困惑度范围 1-10% 的细胞数量之间,而 EMBEDR 的最佳困惑度 1000 远远超出了这个范围。
此外,在 Tabula Muris 联盟骨髓数据集上,比较 EMBEDR 和 scDEED 的优化可视化显示了 KLS 细胞簇和粒细胞簇位置的显著差异(补充图 S13a,b)。虽然这两个簇在 EMBEDR 的优化可视化中有很大的分离(补充图 S13b),但在 scDEED 的优化可视化中它们共享一些邻近细胞(补充图 S13a)。我们随机挑选了一个靠近粒细胞的 KLS 细胞,并检查了它在 50 维预嵌入 PC 空间中的 50 个最近邻细胞。我们发现,在 scDEED 的优化可视化中,这个 KLS 细胞靠近它的 50 个最近邻细胞(补充图 S13c),但在 EMBEDR 的优化可视化中则不然(补充图 S13d),这表明 scDEED 的优化可视化更好地保留了邻近信息。
最后,我们使用模拟器 scDesign3 从基于真实 scRNA-seq 数据集(小鼠小肠上皮细胞)拟合的模型中生成了 20 个模拟的 scRNA-seq 数据集。对于每个模拟数据集,我们使用 scDEED 或 EMBEDR 来优化 t-SNE 困惑度或边际优化 UMAP 的 n.neighbors 超参数。我们将 UMAP 的 min.dist 超参数固定为 0.1 以匹配 EMBEDR 算法,该算法仅允许优化 t-SNE 困惑度或 UMAP 的 n.neighbors。比较 scDEED 和 EMBEDR 优化的超参数(补充图 S14a-d),我们发现 EMBEDR 倾向于选择最高的困惑度值(与之前的真实数据结果一致)和最高的 n.neighbors 值,而 scDEED 没有表现出这种概念上不理想的现象。此外,scDEED 将可疑嵌入的数量减少到零,而 EMBEDR 由于使用 t-SNE 损失函数作为标准,始终偏向 t-SNE。因此,虽然 scDEED 可以公平地比较二维嵌入方法,EMBEDR 在设计上偏向于 t-SNE。对 KNC 和 KNN 指标的比较(补充图 S14f)显示,scDEED 在保留局部信息方面优于 EMBEDR(即,scDEED 导致的 KNN 值显著高于 EMBEDR;单边配对 t 检验 p 值=7.88×10^-15,n=20 对,分别对应 20 个模拟数据集),而 scDEED 在保留全局信息方面与 EMBEDR 相似(即,scDEED 和 EMBEDR 具有相似的 KNC 值;双边配对 t 检验 p 值=0.208,n=20 对,分别对应 20 个模拟数据集)。这些结果与之前呈现的 Hydra 数据集的结果一致;对于 Hydra 数据集,EMBEDR 和 scDEED 优化的嵌入的 KNC 相同,但 KNN 对 scDEED 比 EMBEDR 更高。因此,scDEED 在保留邻近信息方面表现更好。
讨论¶
scDEED 是一种灵活的统计方法,用于检测由 t-SNE 和 UMAP 等二维嵌入方法生成的可疑细胞嵌入。scDEED 基于二维嵌入前后有序邻居之间的统计显著低相似性来检测可疑嵌入。基于检测到的可疑嵌入,scDEED 通过最小化可疑细胞嵌入的数量,优化嵌入方法的超参数设置(例如,t-SNE 的困惑度和 UMAP 的 min.dist 和 n.neighbors)。
使用多个 scRNA-seq 数据集及其发表研究中的嵌入,我们展示了 scDEED 检测到的可疑细胞嵌入相对于其他细胞嵌入确实具有可疑位置。我们还展示了 scDEED 优化的超参数设置生成的细胞嵌入比发表研究中使用的原始超参数设置更好地与生物学知识和预嵌入细胞距离对齐。
默认情况下,scDEED 设置了一个保守的阈值来检测可疑细胞嵌入:如果细胞的可靠性评分不大于零可靠性评分的 5 百分位,则该细胞被标记为可疑。然而,scDEED R 包的用户可以根据他们的知识和偏好增加或减少百分位阈值,以便标记更多或更少的细胞嵌入为可疑。
最小化可疑细胞嵌入的数量还可以帮助优化其他超参数,例如随机种子和学习率,这些参数已知对 t-SNE 和 UMAP 可视化有影响。此外,检测可疑细胞嵌入可能有助于辨别数据集的拓扑结构。例如,如果随着超参数值的增加,可疑细胞嵌入的数量表现出复杂的趋势,则细胞可能具有复杂的拓扑结构,用户在解释二维可视化时应谨慎。
我们的结果表明,scDEED 在可疑嵌入检测、超参数优化和计算时间方面优于竞争方法 EMBEDR。EMBEDR 使用 t-SNE 损失函数,因此偏向 t-SNE 而不是其他嵌入方法。即使对于 t-SNE,EMBEDR 的优化也是不理想的,因为它本质上偏向高困惑度值。另一种方法 DynamicViz 仅从稳定性角度评估细胞嵌入,无法根据预嵌入细胞距离判断细胞嵌入是否可疑。此外,EMBEDR 和 DynamicViz 都在计算上非常昂贵,因为它们需要多次引导然后嵌入。相比之下,scDEED 没有这些限制:scDEED 对所有嵌入方法公平,在检测可疑嵌入方面有效且计算高效。
我们对 scDEED 的评估中一个有趣的观察是,scDEED 优化后,t-SNE 和 UMAP 可能有更相似的可视化结果。这一发现质疑了 t-SNE 在全局信息保留方面不如 UMAP 的普遍看法,并表明缺乏超参数优化可能是一个影响因素。需要注意的是,虽然我们在 scRNA-seq 数据上使用 t-SNE 和 UMAP 作为概念验证展示了 scDEED 的实用性,但 scDEED 也适用于其他数据类型(例如,多模态检测)和其他嵌入方法。
我们期望 scDEED 成为单细胞研究人员生成和解释可视化图的重要计算工具,这些图在基于观察的科学发现中起着至关重要的作用。然而,仍有一些未解决的问题。首先,我们缺乏对基因表达谱中可疑嵌入和可信嵌入差异的理解。我们的实验证据表明,这种差异取决于检测到可疑嵌入时的超参数值。例如,在 Hydra 数据集中,在低 t-SNE 困惑度下,可疑嵌入的细胞比可信嵌入的细胞表现出更高的稀疏性。然而,这一趋势在困惑度超过 scDEED 选择的最佳困惑度后会逆转。在最佳困惑度下,可信和可疑嵌入的细胞表现出统计上相似的稀疏水平(如补充图 S15 所示)。这一结果可能表明在优化超参数下,可疑嵌入和可信嵌入之间的差异更多地归因于随机性而不是生物信号。作为未来的研究方向,探索这种差异随超参数值的变化可能提供另一种选择超参数值的方法。其次,理解优化的超参数设置如何与 t-SNE 和 UMAP 的算法相关仍是一个未解决的问题。例如,t-SNE 使用困惑度在预嵌入空间中定义细胞的邻居选择概率,并且 t-SNE 的优化尝试找到细胞的二维嵌入,以便细胞的邻居选择概率在二维嵌入空间中得到良好保留。因此,一个未解决的问题是优化的困惑度是否会在某种意义上导致更合理的邻居选择概率,从而更好地与细胞的拓扑结构对齐。
方法¶
scDEED 算法¶
给定一个基因 - 细胞矩阵(经过适当的归一化和对数转换,即 PCA 的输入,由用户自行决定),该矩阵有 n 列(对应 n 个细胞),以及一个二维嵌入方法,例如 t-SNE 和 UMAP,具有给定的超参数设置,scDEED 通过以下六个步骤找到可疑的细胞嵌入。
步骤 1:scDEED 通过对原始数据矩阵中的每个基因(即独立打乱每行中的 n 个值)独立地置换 n 个细胞来构建一个置换数据矩阵。
步骤 2(可选):在大多数单细胞数据分析管道中,例如 Python 包 Scanpy 和 R 包 Seurat,用户在应用二维嵌入方法之前,对原始数据矩阵执行选定的 K(PC 的数量)PCA。类似地,scDEED 要求用户输入 K,并在原始数据矩阵和置换数据矩阵上执行 K 的 PCA。用户可以根据其首选方法选择 K。在本研究的结果中,我们根据原始研究或基于肘图选择 K(如果原始研究未提供 K)。
步骤 3:scDEED 将二维嵌入方法(具有给定的超参数设置和 rand.seed=100)应用于原始和置换的预嵌入矩阵。这些矩阵要么是原始数据(如果未执行步骤 2),要么是 K 个 PC(因此每个矩阵的维度为 K×n)。因此,每个细胞获得两个二维嵌入,一个是原始的,一个是置换的。
步骤 4:基于二维嵌入前后的原始数据,scDEED 基于细胞 i=1,…,n 的 x%(默认 x=50,这是 scDEED 的唯一超参数)在二维嵌入空间和预嵌入空间(如果执行了步骤 2 则为 PC 空间,否则为原始空间)中最接近的邻居,定义每个细胞的可靠性评分。在每个空间中,基于欧几里得距离定义邻居。给定两组邻居,scDEED 计算细胞 i 在二维嵌入空间中到有序邻居(从最近到最远)的欧几里得距离,得到两个长度为 x%×n(四舍五入到最接近的整数)的距离向量。最后,scDEED 将可靠性评分定义为两个距离向量的皮尔逊相关系数。即每个细胞的可靠性评分范围为 -1 到 1;较高的可靠性评分表示细胞的有序邻居在二维嵌入前后的一致性更好。我们使用皮尔逊相关系数,因为二维嵌入空间中欧几里得距离的实际值很重要(对于我们的可视化和解释),而不仅仅是使用斯皮尔曼相关系数的欧几里得距离的排名。
步骤 5:基于二维嵌入前后的置换数据,scDEED 应用步骤 4 中的相同程序来获得 n 个细胞的零可靠性评分。由于置换,细胞之间的相似性被破坏,二维嵌入不保留任何生物邻居关系。因此,每个细胞的邻居纯粹由随机机会决定,其可靠性评分反映了二维嵌入前后有序邻居之间的随机一致性。利用 n 个零可靠性评分,scDEED 找出调用细胞可靠性评分低或高的阈值。
步骤 6:scDEED 将可靠性评分小于或等于 n 个零可靠性评分的 5 百分位数的细胞的嵌入定义为可疑嵌入。另一方面,scDEED 将可靠性评分大于或等于 n 个零可靠性评分的 95 百分位数的细胞的嵌入定义为可信嵌入。
在上述步骤之后,scDEED 报告给定参数设置下的可疑细胞嵌入的数量。通过候选超参数设置的网格搜索,scDEED 找到最小化可疑细胞嵌入数量的设置。
在 scDEED R 包中,默认设置了以下候选超参数值,但用户可以指定自己的候选超参数值。对于 t-SNE,默认候选困惑度值为 20、50、…、380、410、450、500、…、750 和 800。对于 UMAP,默认的 n.neighbors 值为 5、6、…、29、30、35、40、45 和 50;默认的 min.dist 值为 0.0125、0.05、0.1、0.2、…、0.7 和 0.8。
置换策略有效性的分析¶
我们通过两次置换 inDrops 数据集(使用不同的随机种子)来说明置换在消除细胞 - 细胞关系方面的效果。首先,我们检查了原始 inDrops 数据集和两个置换数据集的 PCA 图(补充图 S16a)。我们观察到,在原始 PCA 图中,注释的细胞类型是可区分的,而在两个置换的 PCA 图中,所有细胞类型都混合在一起。接下来,我们检查了三个数据集的 t-SNE 图(困惑度为 40),并观察到在置换数据集中细胞类型模式的相似丧失(补充图 S16b)。最后,我们检查了三个数据集中的基因表达水平(补充图 S16c)。我们观察到,在原始数据集中,注释的细胞类型表现出基因表达谱的聚类模式;然而,这些模式在置换数据集中消失了。因此,我们得出结论,置换在消除真实数据中的细胞 - 细胞关系方面是有效的。
scDEED 唯一超参数“相似百分比”的敏感性分析¶
scDEED 算法的步骤 4 和 5 需要 scDEED 的唯一超参数 x,即“相似百分比”(即最接近的邻居的百分比,或邻域大小)。默认值为 x=50,这意味着将一半的细胞视为邻居。直观上,较小的 x 值定义较小的邻域大小,因此更强调保留局部结构。为了研究 x 对 scDEED 性能的影响,我们在使用 t-SNE 作为嵌入方法的 Hydra 数据集上检查了 x=5、20、35、50、65、80 和 95。
首先,我们检查了在范围内的 t-SNE 困惑度值下应用 scDEED 时每个 x 发现的可疑和可信细胞嵌入的数量(补充图 S17a-c)。在较小的相似百分比 x=5 下,可疑细胞嵌入的数量在困惑度值范围内相对稳定,这是预期的结果,因为 t-SNE 旨在保留细胞的局部邻域。在较大的相似百分比 x=95 下,可信嵌入的数量在大多数困惑度值下少于其他 x 值;这一结果也是预期的,因为 t-SNE 不是为了保留细胞的全局拓扑结构。因此,使用过小或过大的 x 不能反映 t-SNE 保留中等范围邻居或相邻细胞簇相对位置的能力。这个结果证明了 scDEED 中 x=50 的默认值是合理的。
其次,我们观察到,在任何 x 值下,当困惑度值超过一个阈值(对于 Hydra 数据集大约是 170)时,可疑细胞嵌入的数量趋于稳定,不再进一步减少(补充图 S17a)。这一结果提供了证据,表明 scDEED 没有偏向于大困惑度值。值得注意的是,在原始困惑度下,过小或过大的 x 值发现的可疑细胞嵌入数量少于接近默认 x=50 的 x 值,再次表明过小或过大的 x 值不适合检测可疑细胞嵌入(补充图 S17d)。最重要的是,接近 50 的 x 值导致的优化困惑度值是相似的,确认了 scDEED 在默认 x=50 下的稳定性(补充图 S17e)。
为了进一步解释上述观察结果,我们检查了置换数据和原始数据中每个 x 的可靠性评分分布。我们发现,尽管随着 x 的增加,零可靠性评分(即置换细胞的可靠性评分)的分布略微单调向右偏移,但在各 x 值之间保持稳定(补充图 S17f)。在原始数据上,未置换细胞的可靠性评分分布在 x=95 时最集中在较低的评分上(补充图 S17g)。这解释了为什么 x=95 找到的可信细胞嵌入最少。
第三,我们检查了从原始 Hydra 嵌入中检测到的在 x=5、50 或 95 时的可疑或可信细胞嵌入。原始嵌入中的几个簇在 x=50 和 95 时被检测为可疑,但在 x=5 时没有(补充图 S18a)。此外,仅在 x=5 时,几乎所有细胞嵌入都被发现是可信的(补充图 S18b)。我们认为,在 x=5 时不同的检测结果是由于检查过小的邻域无法揭示相对于其他簇位置可疑的簇。
第四,我们进一步检查了给定困惑度值在不同 x 值下检测到的可疑细胞嵌入的相似性。我们专注于可疑细胞嵌入,因为它们的数量是 scDEED 用于优化的标准。我们考虑了覆盖广泛范围的三个困惑度值:困惑度 40(用于原始嵌入)、困惑度 230(在 x=50 时由 scDEED 优化)和困惑度 410(scDEED 包中的最大候选困惑度值)(补充图 S19)。给定每个困惑度值,我们使用 scDEED 在每个 x 值下检测一组可疑细胞嵌入;然后我们计算每对 x 值之间的 Jaccard 指数(补充图 S19)。我们观察到,在过小的 x 值下检测到的可疑嵌入与在其他 x 值下检测到的可疑嵌入之间几乎没有一致性,这是一个不理想的结果,因为我们期望可疑嵌入对 x 值具有合理的鲁棒性。相比之下,中高 x 值(x=50、65 和 80)之间的相似性较高,特别是 x=50。我们还检查了接近 50 的三个 x 值(x=40、50、60),并确认它们各自优化的可视化高度相似(补充图 S20)。
综上所述,上述敏感性分析结果支持我们默认选择 x=50 的决定。类似的理论依据在参考文献 27 中描述,该文献发现有效的降维需要强调中等范围的邻居。
通过“kneedle”进行替代超参数优化¶
我们实现了“kneedle”方法,而不是在候选超参数值的默认网格上寻找最小化可疑嵌入数量的超参数值,该方法在可疑嵌入数量(即 y 轴)与超参数值(即 x 轴)的关系图中搜索肘点作为超参数值。我们在两个数据集上研究了这种替代优化方法,发现结果的 t-SNE 可视化与 scDEED 使用的网格搜索全局最小化方法得到的可视化高度相似(补充图 S3-4, 20)。
我们还比较了三种 x 值(40、50 和 60)在 Hydra 数据集上使用的两种超参数优化方法(scDEED 使用的全局最小化方法和“kneedle”方法)(补充图 S20)。我们发现,对于每个 x 值,这两种方法的优化困惑度值导致的 t-SNE 可视化高度相似。结果确认了基于可疑嵌入数量的 scDEED 优化可视化对优化方法不敏感。
t-SNE 和 UMAP 的实现¶
我们使用 R 包 Seurat(版本 3.2.3)中的函数 RunTSNE() 和 RunUMAP() 分别执行 t-SNE 和 UMAP。scDEED 优化的超参数是 RunTSNE() 函数中的 perplexity 以及 RunUMAP() 函数中的 n.neighbors 和 min.dist。在运行 RunTSNE() 和 RunUMAP() 时,我们使用“seed.use = 100”,其余参数保持默认。
评估 Hydra 数据集中细胞簇的纯度¶
我们使用 R 包 Rogue(版本 2.0.0)中的函数 CalculateRogue() 来计算 ROGUE 统计量,该统计量衡量细胞簇的纯度。ROGUE 值越大,细胞簇越纯(或越同质)。在 Hydra 数据集中,五个神经外胚层细胞簇的 ROGUE 值分别为 0.710(神经元 ec1)、0.784(神经元 ec2)、0.714(神经元 ec3)、0.793(神经元 ec4)和 0.839(神经元 ec5)。
ARI 的计算¶
我们使用 R 包 aricode(版本 1.0.2)中的函数 ARI() 来计算调整后的 Rand 指数(ARI),该指数表示两组标签(例如,一组细胞的簇标签和一组细胞的注释类型标签)之间的一致性,并调整标签的一致性随机机会。ARI 的范围是 0 到 1,1 表示完全一致,0 表示没有超出随机机会的一致性。
热图中的基因选择¶
在每个热图中,除非另有说明,我们绘制了在热图中显示的细胞中表达方差最大的前 300 个基因(基于 PCA 步骤前的基因表达值)。
DE 基因识别¶
使用 R 包 Seurat(版本 4.3.0.1)中的 FindMarkers() 函数和 Wilcoxon 秩和检验(默认设置)来查找差异表达(DE)基因。
本地和全局保留的评估指标¶
我们使用 ref. 20 中的以下两个指标评估信息保留。
KNN 反映了本地信息的保留,即预嵌入 PC 空间中 K=10 最近邻在二维嵌入空间中仍保留在 K=10 最近邻集合中的平均比例。KNN 也在 ref. 44 中使用。
K 最近簇(KNC)反映了全局信息的保留,即预嵌入 PC 空间中 K=4 最近簇在二维嵌入空间中仍保留在 K=4 最近簇集合中的平均比例。请注意,对于 KNC,我们与 ref. 20 不同,定义每个簇中心为中位数而不是均值,因为中位数对异常值更稳健。
数据集¶
只要有预处理的数据集可用,就直接在本研究中使用它们。否则,数据集按照生成数据的原始研究中的方式进行预处理。以下是每个数据集的预处理细节。预处理后的数据和代码可以在 Zenodo 上找到(https://zenodo.org/record/7216361#.ZDNgd-zMLJ8)。
Hydra¶
数据集 Hydra/Hydra_Seurat_Whole_Transcriptome.rds(来自原始研究)包含由 Drop-seq 测序的 n=25,052 个单个 Hydra polyp 细胞的转录组,这些细胞带有簇标签,并且经过 Seurat28 处理的 33,391 个基因的缩放表达水平。原始研究中的数据在 NCBI GEO 归档,编号为 GSE121617。按照原始研究,我们在 scDEED 的步骤 2 中使用了 K=31 个 PC,并且我们使用了 Seurat R 包中的默认 RunTSNE(dims=1:5) 作为二维嵌入方法。预处理代码在 Hydra/data_processing_hydra.Rmd 中,预处理数据集在 Hydra/Hydra.rds 中。
我们运行 scDEED,使用候选困惑度值 10, 30, …, 390 和 410,以及原始研究中使用的值 40。运行时间为 2.80 小时(见“计算环境”)。
CAR-T¶
在患有 B 细胞恶性肿瘤的患者中,淋巴细胞耗竭化疗后输注 CD19 特异性嵌合抗原受体修饰的 T(CAR-T)细胞已知会产生抗肿瘤反应。生成该数据集以了解输注产品(IP)中 CAR-T 细胞的克隆组成,特别是包含 10 名接受 CD19 特异性 CAR-T 细胞的患者样本,代表了年龄、性别、不良事件、临床结果、淋巴细胞耗竭治疗和细胞剂量方面的人群。使用 10x Genomics 平台,单细胞 RNA 测序数据是从表达截短的人类表皮生长因子受体(EGFRt)的 IP 和输注后早期(第 7-14 天)、晚期(第 26-30 天)和非常晚期(第 83-112 天)时间点的 CD8+ CAR-T 细胞中生成的,共 n=62,167 个细胞。该数据集位于文件 CART/raw.expMatrix.csv 中,从基因表达汇编(GEO)下载,编号为 GSE125881。按照原始研究,我们在 scDEED 的步骤 2 中使用了 K=15 个 PC,并且我们使用了 Seurat R 包中的默认 RunTSNE(dims=1:5) 作为二维嵌入方法。预处理代码在 CART/data_processing_CART.Rmd 中,预处理数据集在 CART/seuratObj_v3.RData 中。
我们运行 scDEED,使用候选困惑度值 20, 50, …, 380, 410, 450, 500, …, 750 和 800,以及原始研究中使用的值 30。运行时间为 14.37 小时(见“计算环境”)。
Alveolar¶
该数据集旨在了解在博来霉素诱导的肺损伤后再生过程中细胞间的通信。它包含来自小鼠的全器官单细胞悬液,来自损伤后六个时间点和未损伤对照肺,每个时间点平均有四只复制小鼠。使用 Dropseq 工作流程对每只小鼠大约 1000 个细胞进行了单细胞转录组测序,最终数据集中包含 n=29,297 个细胞。原始数据集可在 NCBI GEO 中找到,编号为 GSE141259。按照原始研究,我们在 scDEED 的步骤 2 中使用了 K=50 个独立成分,以获得应用 UMAP 前的预嵌入空间,使用 Seurat R 包中的 RunUMAP(dims=1:50)。与 RunTSNE 不同,Seurat 要求用户为 UMAP 指定输入维度。预处理代码在 Alveolar/data_processing_Alveolar.Rmd 中,预处理数据集在 Alveolar/Seurat_v3.RData 中。
我们运行 scDEED,使用候选 n.neighbors 值 5, 6, …, 9, 10, 15, 20, …, 45, 50, 80, 160, …, 240 和 320;以及候选 min.dist 值 0.0125, 0.05, 0.1, 0.2, …, 0.7 和 0.8。运行时间为 n.neighbors 的边际优化 1.37 小时,min.dist 的边际优化 38.59 分钟,联合优化 12.42 小时(见“计算环境”)。
Samusik¶
从骨髓样本中收集细胞,并使用细胞表面标记物进行 CyTOF 分析。数据进行了标准化,并注释了簇和手动门控群体。去除了双细胞和中性粒细胞。原始数据集可在 [https://figshare.com/s/9c3a0136f12b97f1dadd]14 中找到。最终数据集包含 n=841,644 个细胞。按照原始研究,我们使用 p=38 个遗传标记作为预嵌入空间(省略了可选步骤 2),然后使用 R 命令 RunUMAP(features=feature_list) 应用 UMAP,其中 feature_list 指的是 38 个标记。预处理代码在 Samusik/data_processing_samusik01.Rmd 中,预处理数据集在 Samusik/samusik01_seurat.Rdata 中。
我们运行 scDEED,使用与 Alveolar 数据集相同的候选 n.neighbors 和 min.dist 值。运行时间为 n.neighbors 的边际优化 12.43 小时,min.dist 的边际优化 4.89 小时,联合优化 3.77 天(见“计算环境”)。
Human PBMC¶
该数据集由 Broad Institute 收集,以比较七种单细胞/单核测序方法。原始研究基于经典细胞标记手动注释细胞。在这里,我们专注于三种测序方法(inDrops、DropSeq 和 SeqWell)和四种常见的细胞类型细胞毒性 T 细胞、CD4+ T 细胞、CD14+ 单核细胞和 B 细胞。这导致 inDrops 的 n=5858 个细胞,DropSeq 的 n=5801 个细胞,SeqWell 的 n=3626 个细胞。整个数据集可在 R 包 SeuratData 中作为 pbmcsca.SeuratData 获得(也可在 NCBI GEO 中找到,编号为 GSE132044)。我们分析的子集数据在 Across_Techniques/Seurat.Rdata 中。我们在 scDEED 的步骤 2 中使用了 K=50 个 PC。二维嵌入空间使用 RunUMAP(dims=1:50) 和 RunTSNE(dims=1:5) 获得。
我们运行 scDEED,使用候选困惑度值 5, 10, …, 135 和 140;n.neighbors 值 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 80, 160 和 240;以及 min.dist 值 0.0125, 0.05, 0.1, 0.3, 0.5, 0.7 和 0.9。
Marrow¶
该数据集在 EMBEDR 论文中使用,是 Mus. musculus 单细胞转录组的一部分。细胞从小鼠中收获,并使用荧光激活细胞分选(FACS)进行分选。使用 Smart-seq2 协议和 Illumina 测序进行测序。原始数据集可在 [https://figshare.com/projects/Tabula_Muris_Transcriptomic_characterization_of_20_organs_and_tissues_from_Mus_musculus_at_single_cell_resolution/27733] 找到。原始数据集包含 n=5037 个细胞。按照 EMBEDR 的 GitHub 上可用的预处理笔记本,我们获得了 n=4821 个细胞(Marrow/Marrow_processed.csv)。这与 EMBEDR 报告的预处理后的 n=4771 个细胞不同(在 EMBEDR 出版物中)。然而,EMBEDR 作者的代码表明所有 n=5037 个细胞都用于分析。
尽管存在这种差异,我们的预处理 n=4821 个细胞和 17,303 个基因很好地复制了 EMBEDR 结果。为了公平比较,处理后的数据用于所有分析。按照 EMBEDR 教程,我们在应用 EMBEDR 优化之前使用了 K=50 个 PC 作为预嵌入空间。对于 scDEED 优化,我们在 scDEED 的步骤 2 中使用了 K=16 个 PC(从肘图中选择)。低维空间和可视化使用默认 Seurat R 命令 RunTSNE(dims=1:5) 获得。
我们运行 scDEED,使用候选值 10, 15, 20, 30, 40, 50, 60, 80, 100, 150, 200, 300, 350, 500, 600, 800, 1000, 1300, 1700, 2200, 2900 和 3700,以模拟 EMBEDR 论文原始图 4 中的 Keff 值。运行时间为 10 分钟(见“计算环境”)。
实施 t-SNE 和 UMAP¶
我们使用 R 包 Seurat(版本 3.2.3)中的函数 RunTSNE() 和 RunUMAP() 分别执行 t-SNE 和 UMAP。scDEED 优化的超参数是 RunTSNE() 函数中的 perplexity 以及 RunUMAP() 函数中的 n.neighbors 和 min.dist。在运行 RunTSNE() 和 RunUMAP() 时,我们使用“seed.use = 100”,其余参数保持默认。
评估 Hydra 数据集中细胞簇的纯度¶
我们使用 R 包 Rogue(版本 2.0.0)中的函数 CalculateRogue() 来计算 ROGUE 统计量,该统计量衡量细胞簇的纯度。ROGUE 值越大,细胞簇越纯(或越同质)。在 Hydra 数据集中,五个神经外胚层细胞簇的 ROGUE 值分别为 0.710(神经元 ec1)、0.784(神经元 ec2)、0.714(神经元 ec3)、0.793(神经元 ec4)和 0.839(神经元 ec5)。
ARI 的计算¶
我们使用 R 包 aricode(版本 1.0.2)中的函数 ARI() 来计算调整后的 Rand 指数(ARI),该指数表示两组标签(例如,一组细胞的簇标签和一组细胞的注释类型标签)之间的一致性,并调整标签的一致性随机机会。ARI 的范围是 0 到 1,1 表示完全一致,0 表示没有超出随机机会的一致性。
热图中的基因选择¶
在每个热图中,除非另有说明,我们绘制了在热图中显示的细胞中表达方差最大的前 300 个基因(基于 PCA 步骤前的基因表达值)。
DE 基因识别¶
使用 R 包 Seurat(版本 4.3.0.1)中的 FindMarkers() 函数和 Wilcoxon 秩和检验(默认设置)来查找差异表达(DE)基因。
本地和全局保留的评估指标¶
我们使用 ref. 20 中的以下两个指标评估信息保留。
KNN 反映了本地信息的保留,即预嵌入 PC 空间中 K=10 最近邻在二维嵌入空间中仍保留在 K=10 最近邻集合中的平均比例。KNN 也在 ref. 44 中使用。
K 最近簇(KNC)反映了全局信息的保留,即预嵌入 PC 空间中 K=4 最近簇在二维嵌入空间中仍保留在 K=4 最近簇集合中的平均比例。请注意,对于 KNC,我们与 ref. 20 不同,定义每个簇中心为中位数而不是均值,因为中位数对异常值更稳健。
数据集¶
只要有预处理的数据集可用,就直接在本研究中使用它们。否则,数据集按照生成数据的原始研究中的方式进行预处理。以下是每个数据集的预处理细节。预处理后的数据和代码可以在 Zenodo 上找到(https://zenodo.org/record/7216361#.ZDNgd-zMLJ8)。
Hydra¶
数据集 Hydra/Hydra_Seurat_Whole_Transcriptome.rds(来自原始研究)包含由 Drop-seq 测序的 n=25,052 个单个 Hydra polyp 细胞的转录组,这些细胞带有簇标签,并且经过 Seurat28 处理的 33,391 个基因的缩放表达水平。原始研究中的数据在 NCBI GEO 归档,编号为 GSE121617。按照原始研究,我们在 scDEED 的步骤 2 中使用了 K=31 个 PC,并且我们使用了 Seurat R 包中的默认 RunTSNE(dims=1:5) 作为二维嵌入方法。预处理代码在 Hydra/data_processing_hydra.Rmd 中,预处理数据集在 Hydra/Hydra.rds 中。
我们运行 scDEED,使用候选困惑度值 10, 30, …, 390 和 410,以及原始研究中使用的值 40。运行时间为 2.80 小时(见“计算环境”)。
CAR-T¶
在患有 B 细胞恶性肿瘤的患者中,淋巴细胞耗竭化疗后输注 CD19 特异性嵌合抗原受体修饰的 T(CAR-T)细胞已知会产生抗肿瘤反应。生成该数据集以了解输注产品(IP)中 CAR-T 细胞的克隆组成,特别是包含 10 名接受 CD19 特异性 CAR-T 细胞的患者样本,代表了年龄、性别、不良事件、临床结果、淋巴细胞耗竭治疗和细胞剂量方面的人群。使用 10x Genomics 平台,单细胞 RNA 测序数据是从表达截短的人类表皮生长因子受体(EGFRt)的 IP 和输注后早期(第 7-14 天)、晚期(第 26-30 天)和非常晚期(第 83-112 天)时间点的 CD8+ CAR-T 细胞中生成的,共 n=62,167 个细胞。该数据集位于文件 CART/raw.expMatrix.csv 中,从基因表达汇编(GEO)下载,编号为 GSE125881。按照原始研究,我们在 scDEED 的步骤 2 中使用了 K=15 个 PC,并且我们使用了 Seurat R 包中的默认 RunTSNE(dims=1:5) 作为二维嵌入方法。预处理代码在 CART/data_processing_CART.Rmd 中,预处理数据集在 CART/seuratObj_v3.RData 中。
我们运行 scDEED,使用候选困惑度值 20, 50, …, 380, 410, 450, 500, …, 750 和 800,以及原始研究中使用的值 30。运行时间为 14.37 小时(见“计算环境”)。
Alveolar¶
该数据集旨在了解在博来霉素诱导的肺损伤后再生过程中细胞间的通信。它包含来自小鼠的全器官单细胞悬液,来自损伤后六个时间点和未损伤对照肺,每个时间点平均有四只复制小鼠。使用 Dropseq 工作流程对每只小鼠大约 1000 个细胞进行了单细胞转录组测序,最终数据集中包含 n=29,297 个细胞。原始数据集可在 NCBI GEO 中找到,编号为 GSE141259。按照原始研究,我们在 scDEED 的步骤 2 中使用了 K=50 个独立成分,以获得应用 UMAP 前的预嵌入空间,使用 Seurat R 包中的 RunUMAP(dims=1:50)。与 RunTSNE 不同,Seurat 要求用户为 UMAP 指定输入维度。预处理代码在 Alveolar/data_processing_Alveolar.Rmd 中,预处理数据集在 Alveolar/Seurat_v3.RData 中。
我们运行 scDEED,使用候选 n.neighbors 值 5, 6, …, 9, 10, 15, 20, …, 45, 50, 80, 160, …, 240 和 320;以及候选 min.dist 值 0.0125, 0.05, 0.1, 0.2, …, 0.7 和 0.8。运行时间为 n.neighbors 的边际优化 1.37 小时,min.dist 的边际优化 38.59 分钟,联合优化 12.42 小时(见“计算环境”)。
Samusik¶
从骨髓样本中收集细胞,并使用细胞表面标记物进行 CyTOF 分析。数据进行了标准化,并注释了簇和手动门控群体。去除了双细胞和中性粒细胞。原始数据集可在 [https://figshare.com/s/9c3a0136f12b97f1dadd]14 中找到。最终数据集包含 n=841,644 个细胞。按照原始研究,我们使用 p=38 个遗传标记作为预嵌入空间(省略了可选步骤 2),然后使用 R 命令 RunUMAP(features=feature_list) 应用 UMAP,其中 feature_list 指的是 38 个标记。预处理代码在 Samusik/data_processing_samusik01.Rmd 中,预处理数据集在 Samusik/samusik01_seurat.Rdata 中。
我们运行 scDEED,使用与 Alveolar 数据集相同的候选 n.neighbors 和 min.dist 值。运行时间为 n.neighbors 的边际优化 12.43 小时,min.dist 的边际优化 4.89 小时,联合优化 3.77 天(见“计算环境”)。
Human PBMC¶
该数据集由 Broad Institute 收集,以比较七种单细胞/单核测序方法。原始研究基于经典细胞标记手动注释细胞。在这里,我们专注于三种测序方法(inDrops、DropSeq 和 SeqWell)和四种常见的细胞类型细胞毒性 T 细胞、CD4+ T 细胞、CD14+ 单核细胞和 B 细胞。这导致 inDrops 的 n=5858 个细胞,DropSeq 的 n=5801 个细胞,SeqWell 的 n=3626 个细胞。整个数据集可在 R 包 SeuratData 中作为 pbmcsca.SeuratData 获得(也可在 NCBI GEO 中找到,编号为 GSE132044)。我们分析的子集数据在 Across_Techniques/Seurat.Rdata 中。我们在 scDEED 的步骤 2 中使用了 K=50 个 PC。二维嵌入空间使用 RunUMAP(dims=1:50) 和 RunTSNE(dims=1:5) 获得。
我们运行 scDEED,使用候选困惑度值 5, 10, …, 135 和 140;n.neighbors 值 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 80, 160 和 240;以及 min.dist 值 0.0125, 0.05, 0.1, 0.3, 0.5, 0.7 和 0.9。
Marrow¶
该数据集在 EMBEDR 论文中使用,是 Mus. musculus 单细胞转录组的一部分。细胞从小鼠中收获,并使用荧光激活细胞分选(FACS)进行分选。使用 Smart-seq2 协议和 Illumina 测序进行测序。原始数据集可在 [https://figshare.com/projects/Tabula_Muris_Transcriptomic_characterization_of_20_organs_and_tissues_from_Mus_musculus_at_single_cell_resolution/27733] 找到。原始数据集包含 n=5037 个细胞。按照 EMBEDR 的 GitHub 上可用的预处理笔记本,我们获得了 n=4821 个细胞(Marrow/Marrow_processed.csv)。这与 EMBEDR 报告的预处理后的 n=4771 个细胞不同(在 EMBEDR 出版物中)。然而,EMBEDR 作者的代码表明所有 n=5037 个细胞都用于分析。
尽管存在这种差异,我们的预处理 n=4821 个细胞和 17,303 个基因很好地复制了 EMBEDR 结果。为了公平比较,处理后的数据用于所有分析。按照 EMBEDR 教程,我们在应用 EMBEDR 优化之前使用了 K=50 个 PC 作为预嵌入空间。对于 scDEED 优化,我们在 scDEED 的步骤 2 中使用了 K=16 个 PC(从肘图中选择)。低维空间和可视化使用默认 Seurat R 命令 RunTSNE(dims=1:5) 获得。
我们运行 scDEED,使用候选值 10, 15, 20, 30, 40, 50, 60, 80, 100, 150, 200, 300, 350, 500, 600, 800, 1000, 1300, 1700, 2200, 2900 和 3700,以模拟 EMBEDR 论文原始图 4 中的 Keff 值。运行时间为 10 分钟(见“计算环境”)。
DG¶
该齿状回数据集用于阐明回旋细胞系谱。分析了 10x Genomics 处理的数据,该数据在 R 包 Velocyto(版本 0.6)的教程中使用(Velocyto/10×43.1_loom)。教程可在以下网址找到:
[https://htmlpreview.github.io/?https://github.com/satijalab/seurat.wrappers/blob/master/docs/velocity.html]。
细胞使用基于原始论文中的标记基因的 Louvain 聚类在默认分辨率(0.8)下进行注释(Velocyto/data_annotated.Rds)。该数据集由 n=3396 个细胞和 92,135 个特征组成,跨越剪接和未剪接的检测,可在以下网址访问:
[http://pklab.med.harvard.edu/velocyto/DG1/10X43_1.loom]。对于 scDEED 优化,我们在 scDEED 的步骤 2 中使用了 K=12 个 PC,并使用默认的 Seurat R 命令 RunTSNE(dims=1:5) 获得低维空间。最终可视化使用命令 RunTSNE(dims=1:12)。
我们运行 scDEED,使用候选困惑度值 20, 50, …, 380, 410, 450, 500, 600, 700 和 800。
模拟数据¶
20 个模拟数据集(Simulated_Data/Simulated_data_1.Rds,…,Simulated_Data/Simulated_data_20.Rds)由 scDesign343 生成,训练于 R 包 scDesign248 的内置小鼠小肠上皮细胞数据集(GEO 访问编号 GSE9233249)。为了增加三种细胞类型(肠细胞、祖细胞、TA.Early 和干细胞)之间的距离,我们独立地在所有基因中置换每种细胞类型的基因表达平均值(每个基因在每种细胞类型中都有一个平均参数值)。为了确保基因表达不会大幅偏离指定的簇平均值,将 100 个最大的离散参数除以 150。总共,我们有 10,000 个基因和 n=7217 个细胞。对于 scDEED 优化,我们在 scDEED 的步骤 2 中使用了 K=12 个 PC。为了获得二维嵌入空间,我们使用了 RunUMAP(dims=1:12) 和默认的 RunTSNE(dims=1:5)。对于 EMBEDR,我们也使用了 K=12 个 PC 进行优化。
为了公平比较 t-SNE 和 UMAP,我们计算了最终的二维嵌入,这些嵌入用于 KNN 和 KNC 比较,全部使用 K=12 个 PC 作为预嵌入空间。
我们运行 scDEED,使用候选困惑度值 20, 50, …, 380, 410, 450, 500, …, 750 和 800(scDEED 中的默认设置)。
RNA 速度¶
使用 Velocyto(版本 0.6)按照默认设置进行 RNA 速度分析,教程可在以下网址找到:
[https://htmlpreview.github.io/?https://github.com/satijalab/seurat.wrappers/blob/master/docs/velocity.html]。
与 EMBEDR 和 DynamicViz 的比较¶
EMBEDR 作为 Python 包可用。超参数扫描按照可用的教程进行,使用默认设置,包括使用所有可用处理器的选项。将 EMBEDR 应用于 Hydra 数据集时,我们使用建议的 25 个数据嵌入和 15 个零嵌入。将 EMBEDR 应用于 20 个模拟数据集时,为了节省计算时间,我们使用了 5 个数据嵌入和 10 个零嵌入。由于 EMBEDR 要求用户提供候选超参数列表,我们使用了 scDEED 中的默认困惑度和 n.neighbors 值列表。EMBEDR 不扫描 min.dist,因此为了公平比较,我们在使用 scDEED 时将 min.dist 固定为 0.1(EMBEDR 的默认设置)。
EMBEDR 将细胞分类为嵌入良好或噪声。为了在术语上与 EMBEDR 和 scDEED 保持一致,我们认为嵌入良好的细胞具有可信嵌入,而噪声细胞具有可疑嵌入。具体来说,我们将 EMBEDR p 值高于 0.1 的细胞定义为可疑嵌入,根据 EMBEDR 论文,这些细胞的噪声水平相似。
DynamicViz 也作为 Python 包可用。参数扫描未内置,但可以迭代进行。我们能够成功地将此包用于 t-SNE,但在 UMAP 中出现了一些错误。由于这一点以及 DynamicViz 和 scDEED 在可疑细胞嵌入定义上的概念差异,我们决定在分析中省略 DynamicViz。
统计与可重复性¶
没有使用统计方法预先确定样本大小。有关可重复性,请参阅 Zenodo 存储(https://zenodo.org/record/7216361#.ZDNgd-zMLJ8),获取生成图形的所有代码以及处理过的数据集。
R 包的版本¶
- Seurat 版本 4.3.0.1:除了 EMBEDR 分析外,所有 t-SNE 和 UMAP 分析。
- SeuratData 版本 0.2.2:用于数据集“pmbcsca.SeuratData”。
- doParallel 版本 1.0.15;foreach 版本 1.5.0:用于并行计算和循环。
- ggsci 版本 2.9:用于绘图。
- Rogue 版本 2.0.0:用于评估细胞簇的纯度。
- distances 版本 0.1.8:用于快速计算向量之间的成对距离。
- velocyto.R 版本 0.6:用于 RNA 速度分析。
- scDesign3 版本 0.99.6:用于数据模拟。
其他包: - Rfast 版本 1.9.9;VGAM 版本 1.1.3;pracma 版本 2.2.9;ggplot2 版本 3.3.2;SeuratWrappers 版本 0.3.0;aricode 版本 1.0.2
计算环境¶
所有算法和代码在一台配备 3.6 GHz Intel Core i9 处理器、64GB 内存和 Mojave 10.14 系统的 iMac 上执行。进行本文中的数据分析时,使用了六个核心。
报告摘要¶
有关研究设计的更多信息,请参阅链接到本文的 Nature Portfolio Reporting Summary。
数据可用性¶
支持本研究主要发现的所有相关数据都在本文及其补充信息文件中提供。所有处理过的数据集可在 [https://zenodo.org/record/7216361#.ZDNgd-zMLJ8] 找到。原始数据集如下。Hydra 数据集通过 NCBI GEO 访问编号 GSE121617 获得。CAR-T 数据集通过 NCBI GEO 访问编号 GSE125881 获得。Alveolar 数据集通过 NCBI GEO 访问编号 GSE141259 获得。Samusik 数据集来自 [https://figshare.com/s/9c3a0136f12b97f1dadd]。Human PBMC 数据集通过 R 包 SeuratData 版本 0.2.2 获得,也可以通过 NCBI GEO 访问编号 GSE132044 获得。Marrow 数据集来自 [https://figshare.com/projects/Tabula_Muris_Transcriptomic_characterization_of_20_organs_and_tissues_from_Mus_musculus_at_single_cell_resolution/27733]。齿状回数据集来自 [http://pklab.med.harvard.edu/velocyto/DG1/10X43_1.loom]。用 NCBI GEO 访问编号 GSE92332 训练 scDesign2 模拟数据。通过 Zenodo(https://zenodo.org/record/7216361#.ZDNgd-zMLJ8)提供的本文的可重复性材料(包括数据集、R 和 Python 脚本以及中间结果)。通过 Zenodo(https://zenodo.org/records/10511446)提供本文的数据源文件(Excel 和 CSV 文件足以生成点图、线图和箱线图)。
代码可用性¶
scDEED R 包可在 GitHub 存储库 [https://github.com/JSB-UCLA/scDEED](Zenodo doi:[https://zenodo.org/badge/latestdoi/402656304])上获得。计算机代码可在 Zenodo(https://zenodo.org/record/7216361#.ZDNgd-zMLJ8)上获得。