Systematic dissection of tumor normal single cell ecosystems across a thousand tumors of 30 cancer types
Abstract¶
肿瘤微环境的复杂性在癌症治疗中带来了重大挑战。本文为了全面调查肿瘤 - 正常生态系统,整合分析了来自 1070 例肿瘤和 493 例正常样本的 490 万单细胞转录组,结合了全癌种 137 个空间转录组学、8887 个 TCGA 样本和 1261 个使用检查点抑制剂治疗的肿瘤样本。我们定义了组成肿瘤 - 正常生态系统的多种细胞状态,并鉴定了不同细胞类型和器官的标志性基因特征。我们的图谱刻画了由 AKR1C1 或 WNT5A 标记的炎症成纤维细胞在细胞间相互作用和空间共定位模式上的差异。共现分析揭示了包括三级淋巴结构 (TLS) 成分在内的干扰素富集社区状态,这些状态在肿瘤、邻近正常组织和健康正常组织之间表现出不同的重连线。通过我们的肺癌队列 (n=497) 验证了干扰素富集社区状态对免疫治疗的良好反应。空间转录组的解卷积区分了 TLS 富集和非富集细胞类型在免疫治疗有利成分中的作用。我们对肿瘤 - 正常生态系统的系统性解剖为理解肿瘤内外异质性提供了更深的见解。
Introduction¶
肿瘤是由恶性细胞及多种组织浸润的基质和免疫细胞组成的高度异质性实体,这些细胞共同形成了肿瘤微环境 (TME)1。单细胞 RNA 测序 (scRNA-seq) 技术的出现为高分辨率表征 TME 中的广泛异质性提供了无偏和系统的分子分析 2–4。
TME 内的分子和细胞异质性共同影响肿瘤的各个方面,包括进展、转移和治疗反应 1。随着肿瘤内和肿瘤间异质性证据的积累,各种试图在全癌种水平上编制共识基因特征的努力正在进行。例如,最近发布了一份 T 细胞、髓系细胞和恶性细胞的全癌种图谱 5–8。尽管这些全癌种分析很好地表征了感兴趣的细胞类型,但 TME 组件之间的复杂相互作用以及与配对正常组织的区别尚未得到充分理解,导致对肿瘤异质性的视角有限,忽略或过度简化了潜在的重要分子和细胞相互作用。事实上,TME 表型不仅仅被二元化为抗肿瘤或促肿瘤,而是代表了互动的细胞组织或生态系统 9。针对肿瘤生态系统中潜在的肿瘤特异性相互作用是一种有吸引力的策略,可以产生协同的治疗效果 9,10。因此,解剖不同癌症和组织类型中复杂和多层次的生态系统对于开发更有效的治疗策略至关重要。
基于检查点阻断的癌症免疫治疗作为一种有前途的治疗策略已经出现,并对癌症治疗产生了深远的影响。然而,肿瘤生态系统的异质性仍然是主要挑战之一,即检查点抑制剂在不同癌症类型和患者中的疗效差异 11。最近的研究强调了干扰素特征和三级淋巴结构 (TLS)——一种具有淋巴样结构的免疫细胞异位聚集体,是免疫治疗反应的关键决定因素 12–14。然而,与 TLS 相关的有利于免疫治疗的成分的详细理解仍然难以捉摸。因此,全面的全癌种肿瘤生态系统解剖对于区分富含 TLS 和非富含 TLS 的细胞类型,以及理解这些成分如何塑造肿瘤免疫和揭示它们之间的相互作用至关重要。这些见解将使我们更好地理解不同癌症类型患者在免疫治疗中观察到的差异反应。
在此,我们构建了涵盖 30 种癌症类型和 490 万细胞的未分类肿瘤 - 正常单细胞转录组图谱,这些细胞来自 1070 个肿瘤和 493 个正常样本。我们的分析结合了多种分析方法,包括 AND- 门算法和单细胞分辨率的非负矩阵分解 (NMF) 可视化,以揭示肿瘤/正常生态系统之间的区别。我们概述了不同细胞类型和器官的标志性基因特征。我们的分析揭示了炎症成纤维细胞的异质性,包括表达 CXCL1/3/8 的 AKR1C1+ 和 WNT5A+ 炎症成纤维细胞,这些细胞在器官分配、组织偏好、细胞间相互作用和空间共定位模式上表现出显著差异。通过分析细胞状态的共现模式,我们发现了包括 CCL19+ 成纤维细胞和 LAMP3+ 树突状细胞在内的干扰素富集社区的肿瘤特异性重连线,这些成分在免疫治疗队列 (n=1261) 中具有不同的临床意义,包括我们的肺癌 (LC) 队列 (n=497)。此外,我们通过空间转录组分析将富含 TLS 的细胞类型和非富含 TLS 的细胞类型区分开来,并得出了一种预测免疫治疗有利反应的 TLS 特征。总之,我们的全癌种元图谱为理解肿瘤 - 正常生态系统提供了更深入的见解,并为诊断和治疗策略的开发提供了宝贵资源。我们已将全面分析的数据集存储在 Zenodo 存储库 (DOI:10.5281/zenodo.10651059),我们的图谱可以在 https://cellatlas.kaist.ac.kr/ecosystem/进行交互式可视化。
Results¶
构建全癌种肿瘤 - 正常单细胞元图谱¶
为了生成肿瘤和正常生态系统的全面普查,我们选择了已发表的关于癌症、邻近正常和健康正常样本的 scRNA-seq 数据集,这些数据集未针对特定细胞类型进行富集。结果,构建了一个涵盖 30 种不同癌症类型的肿瘤 - 正常单细胞转录组图谱,共包含 104 个数据集(补充数据 1)。经过数据整理,这个元图谱涵盖了来自 999 名捐赠者的 1070 个肿瘤和 493 个正常样本的 490 万个细胞(图 1A)。乳腺癌(BRCA)是最丰富的癌症类型,其次是肺癌(LC)、头颈癌(HNSC)和肝细胞癌(HCC)。
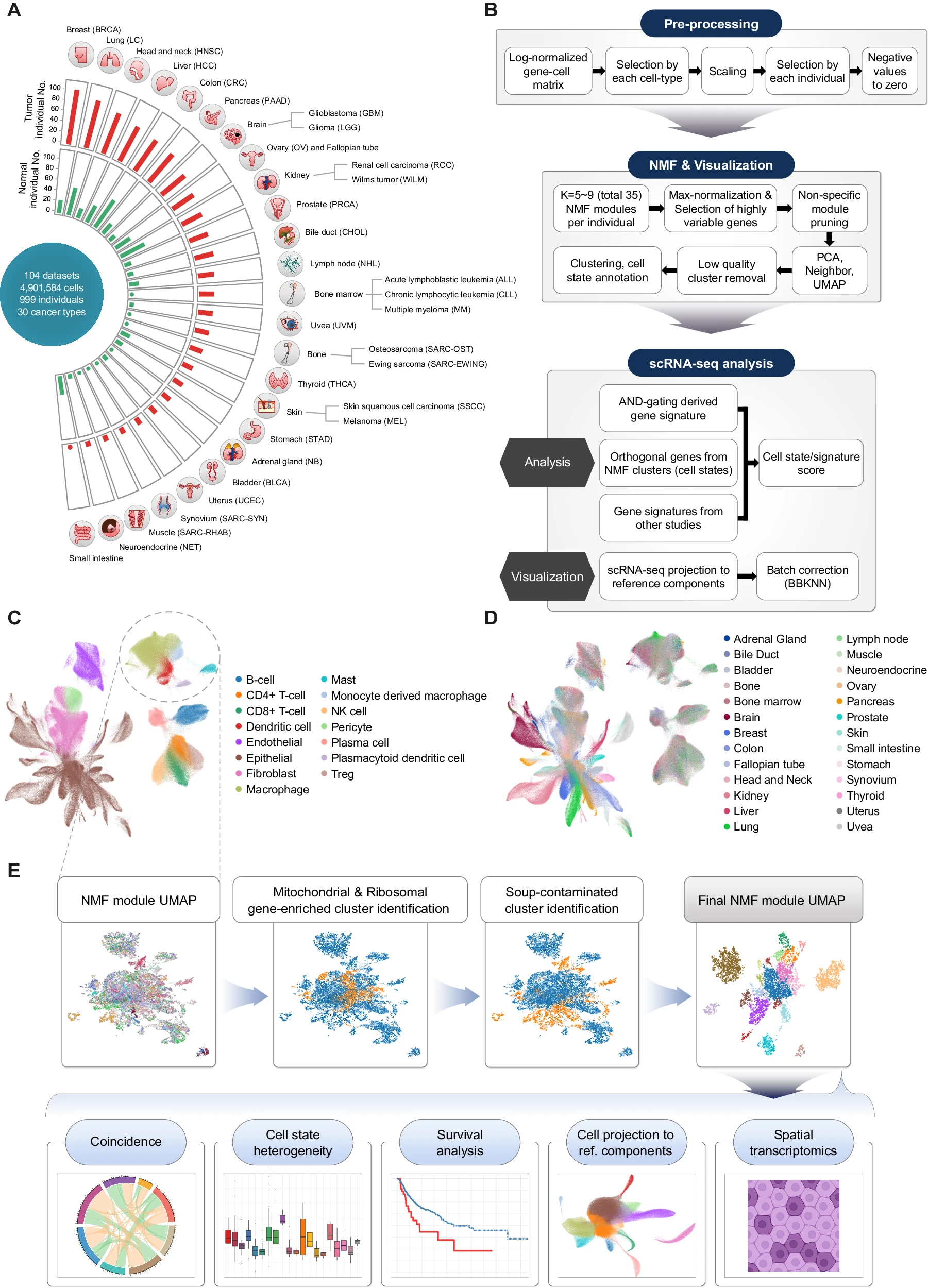
A. 本研究收集的 30 种癌症类型的 scRNA-seq 队列概述。 B. 肿瘤 - 正常单细胞转录组图谱和 NMF 处理的工作流程。 C. 用 UMAP 可视化部分肿瘤 - 正常单细胞图谱,以细胞类型着色。 D. 用 UMAP 可视化部分肿瘤 - 正常单细胞图谱,以器官来源着色。 E. NMF 模块的图形聚类示意图,带有自动化的汤效应检测算法及后续分析。
构建全癌种肿瘤 - 正常单细胞元图谱¶
为了描述不同癌症和组织类型中的多种生态系统,我们将收集到的所有单细胞数据集进行整合,并在全球范围内进行注释。然后,应用 AND- 门算法(参见方法)表征各种器官的肿瘤和正常组织中普遍存在的细胞类型特异性差异表达基因。随后,将注释后的基因表达矩阵按主要细胞类型拆分,并使用非负矩阵分解(NMF)分析分解为各种细胞状态 (图 1B–D 和补充图 S1A, B)。为了最大化稀有细胞状态的恢复,通过扫描多个参数收集每个样本的 NMF 模块,生成的模块进行聚类并投影到统一流形近似投影(UMAP)中,以搜索反复出现的一致性模块。
在 NMF 模块(细胞状态)的聚类中,我们识别出代表环境 RNA 或双重污染的模块,并通过自动化管道从进一步分析中移除这些聚类 (图 1E 和补充图 S1C)。富含核糖体/线粒体基因的 NMF 模块聚类也被过滤掉。NMF 模块的最终 UMAP 表示展示了多个样本中反复出现的细胞状态的总体结构 (图 1E)。利用 NMF 模块的 UMAP 表示,可以通过其来源(例如,组织类型、器官等)直观地检查每个细胞状态的特征。基于具有最高平均 NMF 权重的基因定义并注释细胞状态(补充数据 2)。这使我们能够使用单细胞分辨率下派生的细胞状态过滤器解剖肿瘤单细胞和批量转录组数据(图 1E,细胞状态异质性),并监测它们在正常和肿瘤样本中的共现,以识别细胞类型之间的潜在相互作用及其对肿瘤微环境(TME)的贡献(图 1E,共现)。我们还应用细胞状态特征对批量 RNA-seq 队列(TCGA,免疫检查点治疗)进行解卷积,以检查其临床意义(图 1E,生存分析)。最后,我们使用我们的细胞状态作为参考组件投影细胞,以评估细胞状态与细胞类型之间的一致性 15(图 1E,细胞投影到参考组件),并使用我们的细胞类型解卷 11 种癌症类型(n=137)的空间转录组学(图 1E 和补充数据 3,空间转录组学)。
识别肿瘤 - 正常生态系统的通用标志基因特征¶
为了系统地表征在肿瘤和正常组织中反复上调的标志基因,以及在不同器官组成的 TME 的主要细胞类型中反复上调的标志基因,我们实施了 AND- 门算法(参见方法)(补充图 S2A 和补充数据 4)。对于 CD8+ T 细胞,协同刺激分子(CD27)和免疫检查点或衰竭标记如 CXCL13、PDCD1、TIGIT、CTLA4、LAG3 和 TNFRSF9 在肿瘤中常见上调,而 IL7R、PTGER2 和 PTGER4 在正常组织中上调(图 2A,B)。值得注意的是,胰腺肿瘤组织的 CD8+ T 细胞未显示 PDCD1 和 LAG3 的上调,这可能解释了目前免疫检查点抑制剂在胰腺癌(PAAD)中不可适用性,而在其他癌症类型中适用 16。同样,肿瘤相关的 NK 细胞以 ZNF683 和 KRT81 为标志。肿瘤中的 Tregs 上调具有调节功能的基因如 RBPJ、CXCR3 和 ZBED2,而正常组织中的 Tregs 上调 CCR7 和 CXCR5,表明免疫细胞在肿瘤和正常组织中的机制不同 17(图 2A,B)。特别是,肿瘤浸润的巨噬细胞普遍表达免疫检查点(IL4I1)18、M2 极化相关基因(SPP1)5,19 和炎症基因(CCL7、ADAMDEC1 和 SLAMF9),而肿瘤中的树突状细胞则表现出 CCL19 和 LAMP3 的高表达,这与炎症和迁移功能有关(图 2A,B)。基因本体(GO)分析揭示,肿瘤浸润的巨噬细胞、树突状细胞和 CD8+ T 细胞中上调的基因富集在包括对病毒的防御反应、对 II 型干扰素的反应、炎症反应、淋巴细胞趋化和细胞因子介导的信号通路等相关功能和路径中(图 2C)。
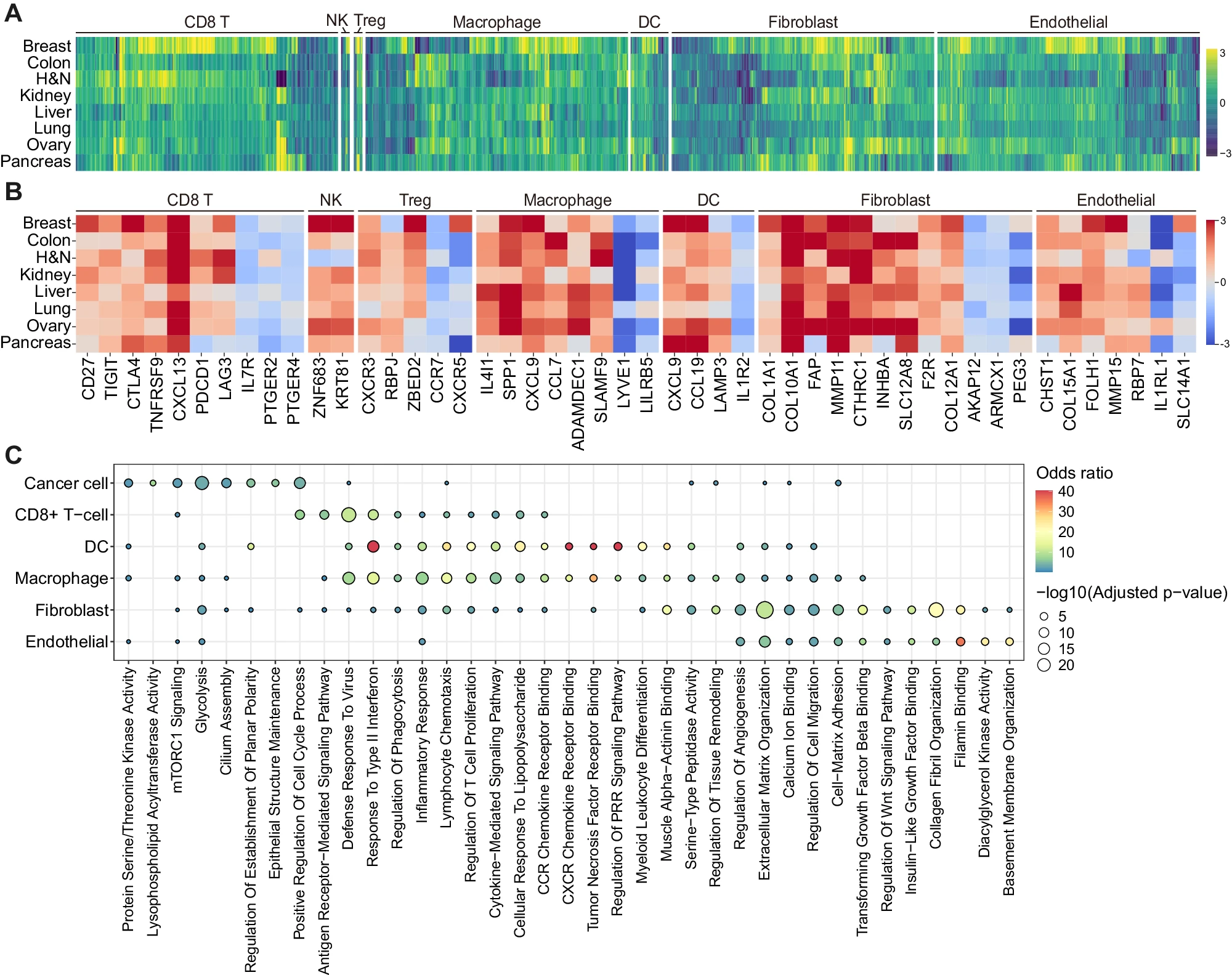
A. 各器官中肿瘤和正常生态系统的标志基因特征表征。细胞类型在热图顶部标注,仅展示在四种或更多癌症类型中上调的基因。热图中的每个框代表 log2 倍数变化值,正值表示在肿瘤中上调。H&N,头颈;DC,树突状细胞;NK,自然杀伤细胞;Treg,调节性 T 细胞。
B. 图 2A 中在各癌症和细胞类型中识别的标志基因的详细热图。热图中的每种颜色代表 log2 倍数变化值,正值表示在肿瘤中上调。H&N,头颈;DC,树突状细胞;NK,自然杀伤细胞;Treg,调节性 T 细胞。
C. 使用 Enrichr 对不同细胞类型的肿瘤标志基因进行基因本体(GO)分析。点的颜色和大小分别表示来自双侧 Fisher 精确检验的赔率比和 p 值,经过 Benjamini-Hochberg 方法调整。PRR,模式识别受体。
对于非免疫细胞类型,癌细胞普遍表现出蛋白质丝氨酸/苏氨酸激酶活性(PRKCA、GSK3B 和 CAMKK2)、糖酵解(PLOD1、EGLN3 和 P4HA1)、mTORC1 信号传导(SLC2A1、GMPS 和 PDK1)以及与细胞周期过程的正向调控相关的 GO 术语(E2F7、E2F8 和 KIF23;图 2C 和补充图 S2B)。癌相关成纤维细胞(CAF)表达众所周知的标志物,包括 FAP、COL1A1、COL10A1、MMP11 和 CTHRC120,21,以及其他基因如 INHBA、SLC12A8、F2R 和 COL12A1 在各种器官中也有表达(图 2A,B)。肿瘤内皮细胞上调了与血管生成相关的基因,包括 CHST1、FOLH1 和 MMP1522,23,24。肿瘤相关的成纤维细胞和内皮细胞在与细胞外基质组织、细胞迁移调节、细胞 - 基质黏附和 filamin 结合相关的术语中表现出富集(图 2C)。总体而言,这些结果概述了 TME 组件的失调标志特征。
将肿瘤 - 正常生态系统解构为异质性细胞状态¶
对复杂的肿瘤和正常生态系统的系统性解剖,识别出大量与先前报告的特征高度一致的细胞状态或共调控基因,以及一些在之前的全癌种分析中尚未识别的基因(图 3A 和补充图 S3, 4)。对于髓系细胞状态,我们注意到 CTSK+ 巨噬细胞(SLC9B2 和 CTSK)、CXCL9+ 巨噬细胞(CXCL9 和 ENPP2)、朗格汉斯细胞(CD1A 和 CD207)、单核吞噬细胞(DLEU2 和 FMN1)和以趋化因子(CXCL1 和 CXCL5)、迁移(SLAMF1)和免疫调节标志(ITGB8)标记的 PRR 诱导的激活状态。SLAMF1 和 ITGB8 在 TLR 诱导的 DC 成熟过程中上调,因此被命名为 PRR 诱导的激活状态。我们还识别出各种 B 细胞状态,包括生发中心 B 细胞(GCB;SUGCT 和 RGS13)和浆细胞前体(FNDC3B 和 FNDC3A)状态(补充图 S3F)。利用 Ro/e 分析评估免疫细胞状态的组织富集情况,揭示了肿瘤中衰竭的 CD8+ T 细胞(LAG3 和 TOX)、SPP1+(SPP1 和 ANGPTL4)、CTSK+ 和 CXCL9+ 巨噬细胞状态的偏好(补充图 S5A, B)。GCB 和浆细胞状态在邻近正常组织中丰富,而 PRR 诱导的激活状态在健康正常组织中富集(补充图 S5B, C)。利用细胞类型特异性细胞状态谱作为嵌入参考,我们识别出反映相应细胞状态的细胞类型(图 3B 和补充图 S6, 7):PRR 诱导的激活状态被捕获为主要来源于妇科癌症(如卵巢癌(OV)和子宫内膜癌(UCEC))患者的 PRR 诱导的 mo-DC 细胞聚类,这与 PRR 诱导的激活状态评分的分布一致(补充图 S8)。
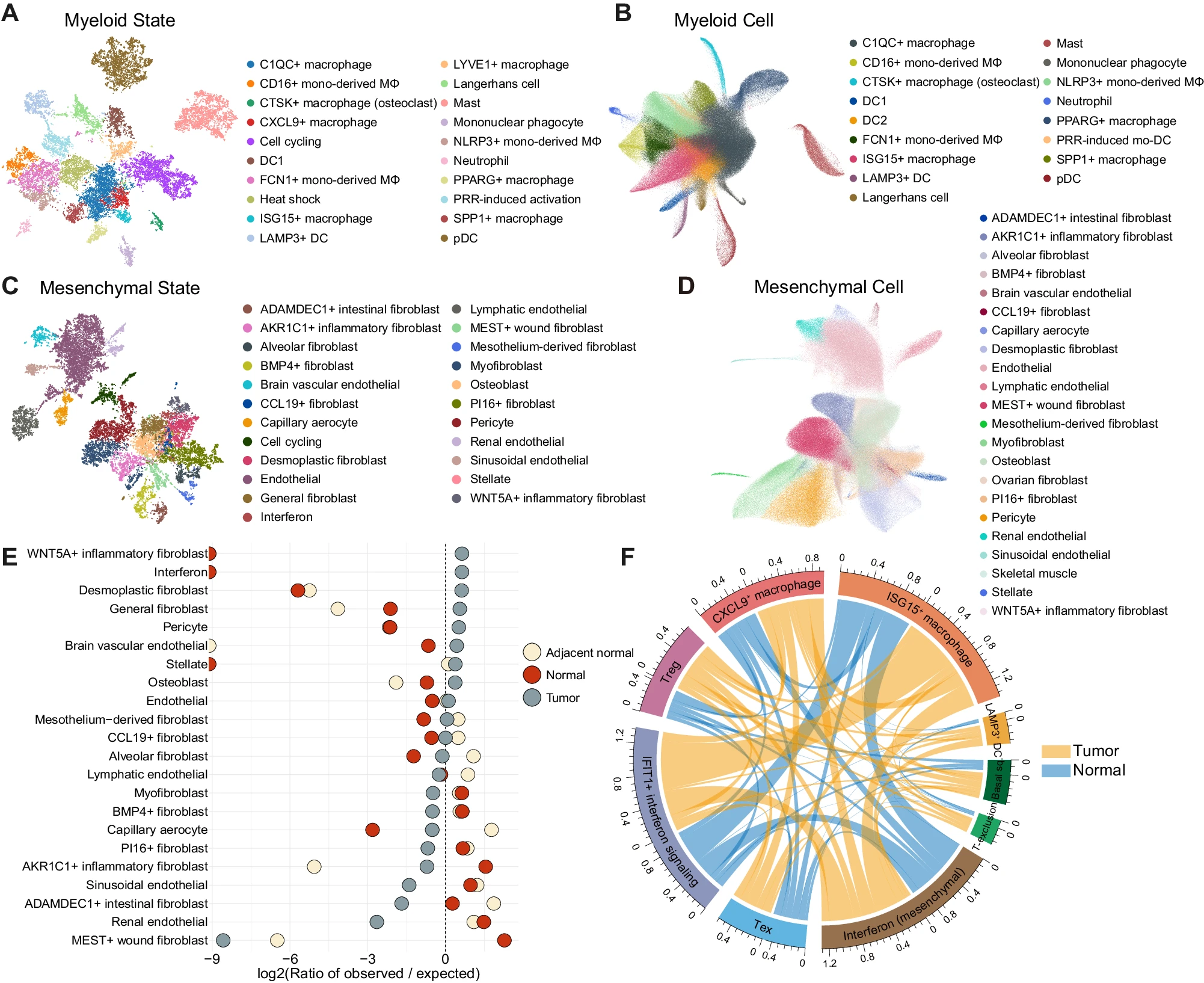
UMAP 可视化 (A) 髓系细胞状态和 (B) 髓系细胞的相应参考组件分析。NMF 模块按细胞状态进行图形聚类和着色 (A)。然后,将细胞映射到由髓系细胞状态基因组成的参考组件 (B)。DC,树突状细胞;mono-derived MΦ,单核细胞来源的巨噬细胞;mo-DC,单核细胞来源的树突状细胞;pDC,浆细胞样树突状细胞;PRR,模式识别受体。UMAP 可视化 (C) 间充质细胞状态和 (D) 间充质细胞的相应参考组件分析。NMF 模块按细胞状态进行图形聚类和着色 (C)。然后,将细胞映射到由间充质细胞状态基因组成的参考组件 (D)。E 间充质细胞状态的组织富集 Ro/e 分析。虚线垂直线表示 Ro/e 为零的位置。F Circos 图展示正常(蓝色)和肿瘤(黄色)组织中细胞状态之间的共现。弧长代表与其他细胞状态在邻近区域的共现总和。弧长越长,表示与其他细胞状态的共现越频繁。Basal sq,基底鳞状状态。DC,树突状细胞;Tex,衰竭的 CD8+ T 细胞;T-exclusion,T 细胞排斥程序;Treg,调节性 T 细胞。
上皮细胞和神经细胞被解卷为 35 种状态(补充图 S9),随后使用这些细胞状态进行的参考组件分析按其来源聚类细胞(补充图 S10A,B)。发现了上皮标志程序,如循环(TOP2A 和 BIRC5)和应激(PPP1R15A 和 KLF5)。关于先前编录的特征,肾细胞癌(RCC)和胶质母细胞瘤表现出高水平的缺氧和金属反应,而低级别胶质瘤、子宫内膜癌(UCEC)和神经母细胞瘤则分别表现出低水平的部分上皮 - 间质转化(pEMT)、氧化磷酸化和抗原呈递机制特征(补充图 S10C,D)。
特别是,我们分类和注释了多种间质来源的细胞状态,包括 CCL19+ 成纤维细胞(CCL19 和 CXCL13)、PI16+ 成纤维细胞(MFAP5 和 IGFBP6)、肌成纤维细胞(ACTA2 和 MYH11)、致密性成纤维细胞(LRRC15 和 MMP11),以及特定器官专属的成纤维细胞状态,这些状态可能反映了器官特定功能(图 3C,D 和补充图 S11)。当投影到参考组件时,这些状态也被识别为各种间质亚群。虽然致密性成纤维细胞状态高度特异于肿瘤,但肌成纤维细胞群在非恶性组织中均匀分布,这与先前报道的认为肌成纤维细胞是主要 CAF 的观点相反(图 3E)。
这些多样的细胞状态共同作用,塑造了复杂的肿瘤生态系统(图 3F)。例如,在肿瘤中,上皮细胞的 T 细胞排斥程序与 Treg 状态密切相关,促进了免疫逃逸和肿瘤进展 29,32。基底鳞状状态与炎症状态如 CXCL9+ 巨噬细胞、LAMP3+ 树突状细胞和仅在肿瘤组织中出现的间质来源的干扰素状态一致,表明基底鳞状起源的癌症类型(HNSC 和皮肤鳞状细胞癌)具有触发免疫细胞浸润的内在特征(图 3F)。特别是,涉及间质来源的干扰素状态与其他细胞类型的干扰素状态和 LAMP3+ 树突状细胞的多次共现被识别出来(图 3F)。其与 LAMP3+ 树突状细胞的肿瘤特异性共现表明,间质来源的干扰素对于启动肿瘤干扰素信号传导以随后募集免疫细胞和 T 细胞初始激活是必要的。
表征 AKR1C1+ 和 WNT5A+ 炎症成纤维细胞作为不同亚型¶
成纤维细胞是具有多种功能的高度异质性群体,如胶原沉积、血管生成和细胞因子分泌,在形成 TME 中起着核心作用 33。在癌症背景下,成纤维细胞促进炎症并协调组织微环境向免疫抑制方向发展 34,但之前的全癌种研究中对炎症成纤维细胞的多样性尚未进行广泛探索 30,31。当我们将间质细胞集合投影到定义的状态时,我们识别出多种显示免疫相关基因表达的成纤维细胞亚型(图 4A)。其中,我们注意到表达 AKR1C1+ 和 WNT5A+ 的炎症成纤维细胞的区别,这两者都与 PRR 诱导的激活状态一致,并共享类似的细胞因子基因表达(CXCL1/3/8;图 4A 和补充图 S12A)。然而,它们在标志基因(AKR1C1、FOSL1、LIF 和 THAP2 vs. WNT5A、GREM1、TNC 和 MMP1)、组织来源(正常 vs. 肿瘤)和器官偏好上显著不同(图 3E,4A,B 和补充图 S12B,C)。为了研究这两种状态是否代表真正不同的成纤维细胞亚型,我们对涵盖乳腺、结肠、头颈和卵巢的肿瘤和正常组织的 scRNA-seq 数据集进行了详细检查。除乳腺组织外,肿瘤组织表达更多的趋化因子基因(CXCL1/3/8)。同时,这两种炎症成纤维细胞中 AKR1C1 和 WNT5A 的表达模式在不同器官中有所不同(图 4C)。BRCA 和 OV 患者表达 AKR1C1 和 WNT5A,而正常乳腺组织仅表达 AKR1C1。有趣的是,结直肠癌(CRC)和 HNSC 患者表达 WNT5A 但不表达 AKR1C1,而相应的正常组织则表现出相反的表达模式(图 4C)。
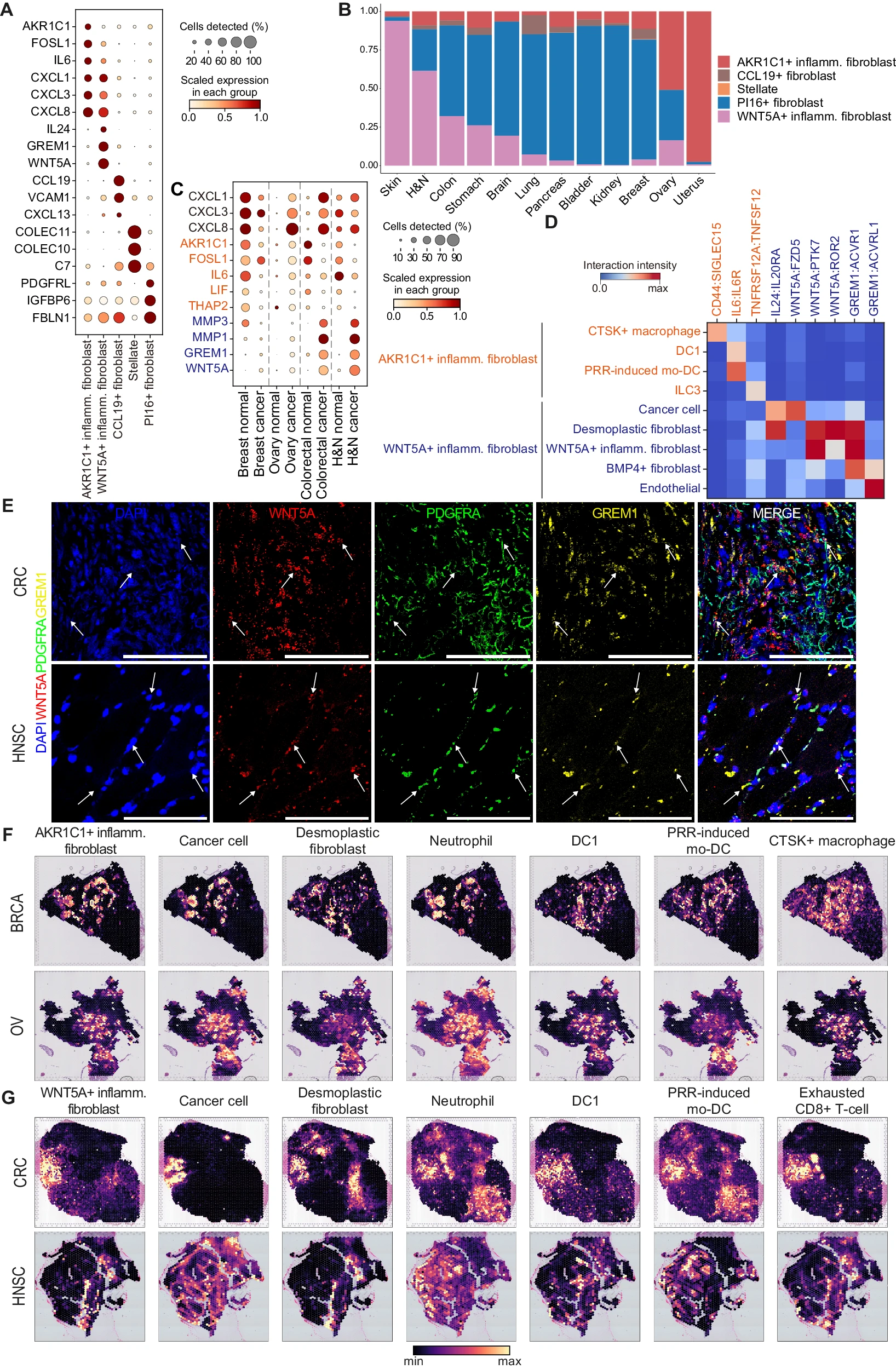
A. 炎症成纤维细胞亚型的标志基因表达的点图。Inflamm.表示炎症。 B. 各器官中炎症成纤维细胞的分布,其中 y 轴表示肿瘤组织中炎症成纤维细胞的比例。H&N 表示头颈;Inflamm.表示炎症。 C. 点图显示相关器官的正常和肿瘤组织中 AKR1C1+ 和 WNT5A+ 炎症成纤维细胞标志基因的基因表达。H&N 表示头颈。 D. AKR1C1+ 和 WNT5A+ 炎症成纤维细胞与其他细胞类型的配体 - 受体相互作用。通过乘以每对细胞的配体和受体的标准化表达值来计算相互作用强度。DC 表示树突状细胞;Inflamm.表示炎症;ILC3 表示第 3 类固有淋巴细胞;mo-DC 表示单核细胞来源的树突状细胞;PRR 表示模式识别受体。 E. CRC(上)和 HNSC(下)组织中致密基质中原位 RNA smFISH 检测 WNT5A(红色)、PDGFRA(绿色)和 GREM1(黄色)的代表性图像(n = 3)。比例尺:100 μm。放大倍数:20X。 F. AKR1C1+ 炎症成纤维细胞与相关器官中其他细胞类型的空间共定位模式,颜色表示细胞丰度。 G. WNT5A+ 炎症成纤维细胞与相关器官中其他细胞类型的空间共定位模式,颜色表示细胞丰度。DC 表示树突状细胞;Inflamm.表示炎症;mo-DC 表示单核细胞来源的树突状细胞;PRR 表示模式识别受体。
为了深入了解这些炎症成纤维细胞如何塑造 TME,我们进行了配体 - 受体分析,以揭示 AKR1C1+ 和 WNT5A+ 炎症成纤维细胞与 BRCA、CRC、HNSC 和 OV 样本中其他细胞类型的不同相互作用(图 4D 和补充图 S12D,E)。我们识别出这些炎症成纤维细胞之间的离散相互作用模式。AKR1C1+ 炎症成纤维细胞与 CTSK+ 巨噬细胞(CD44:SIGLEC15)相互作用,已知这种巨噬细胞在多种癌症类型中诱导癌症进展和转移 35(图 4D 和补充图 S12D)。此外,AKR1C1+ 炎症成纤维细胞强烈表达 IL6,这与 DC1 和 PRR 诱导的 mo-DC 表达的 IL6R 相互作用,可能促进肿瘤生长和治疗抵抗 36,37。我们还识别出 AKR1C1+ 炎症成纤维细胞的 TNFRSF12A 与 ILC3 的 TNFSF12 之间的相互作用 38。相比之下,WNT5A+ 炎症成纤维细胞通过 IL24:IL20RA 和 WNT5A:FZD5 通路与癌细胞相互作用,这可能促进癌症进展和化疗抵抗 39,40(图 4D 和补充图 S12E)。此外,WNT5A+ 炎症成纤维细胞表现出与成纤维细胞群体(包括致密性成纤维细胞)的强相互作用(WNT5A:PTK7、WNT5A:ROR2 和 GREM1:ACVR1)、自身同型相互作用(WNT5A:PTK7 和 GREM1:ACVR1)以及 BMP4+ 成纤维细胞(GREM1:ACVR1),以模仿伤口修复过程,通过自分泌和旁分泌机制增强间质细胞的增殖和迁移表型 41,42(图 4D 和补充图 S12E)。还识别出 WNT5A+ 炎症成纤维细胞的 GREM1 与内皮细胞的 ACVRL1 之间的促血管生成和促炎性相互作用 43。总之,WNT5A+ 炎症成纤维细胞分泌多种配体如 WNT5A、GREM1 和 IL24,在组织再生、炎症和癌症等多个环境中增强增殖、迁移和生存 41,44,45,46。它们与多种细胞成分如癌细胞、成纤维细胞和内皮细胞相互作用,最终形成促肿瘤和核心炎性 TME。
为了定位和验证 WNT5A+ 炎症成纤维细胞群体,我们在 CRC 和 HNSC 组织样本中进行了针对 WNT5A、GREM1 和 PDGFRA 的 RNA 单分子荧光原位杂交(smFISH)。通过癌症组织致密基质中的 WNT5A、GREM1 和 PDGFRA RNA 探针的重叠信号确认了 WNT5A+ 炎症成纤维细胞的存在(图 4E 和补充图 S13,14)。
我们假设不同的微环境导致这两种炎症成纤维细胞的不同表型。为了检查它们的共定位模式,我们分析了空间转录组学数据(BRCA、OV 和 UCEC 中的 AKR1C1+ 炎症成纤维细胞,以及 CRC 和 HNSC 中的 WNT5A+ 炎症成纤维细胞)。AKR1C1+ 炎症成纤维细胞显著与癌细胞、中性粒细胞、CTSK+ 巨噬细胞、DC1 和 PRR 诱导的 mo-DC 共定位(图 4F,G 和补充图 S15A,B)。相比之下,WNT5A+ 炎症成纤维细胞显著与致密性成纤维细胞、衰竭的 CD8+ T 细胞、Treg、DC1 和 PRR 诱导的 mo-DC 共定位,突显其在形成免疫抑制 TME 中的作用(图 4F,G 和补充图 S15A,B)。特别是,AKR1C1+ 和 WNT5A+ 炎症成纤维细胞分别与致密性成纤维细胞和中性粒细胞相互排斥,进一步突显了两种炎症成纤维细胞之间的区别(图 4F,G 和补充图 S15A,B)。WNT5A+ 炎症成纤维细胞与衰竭的 CD8+ T 细胞和 Treg 的细胞相互作用和空间邻近性促使我们调查其与免疫治疗的关联在免疫治疗前/后 HNSC 样本中的表现 47。与其他间质群体不同,免疫治疗后 WNT5A+ 炎症成纤维细胞和致密性成纤维细胞有增加的趋势(补充图 S15C)。这表明在免疫治疗过程中 WNT5A+ 炎症成纤维细胞可能受到影响。总之,尽管两种炎症成纤维细胞共同表达 CXCL1/3/8,但不同的器官/组织偏好、细胞相互作用模式和与其他细胞类型的空间邻近性表明它们在形成免疫逃逸和促肿瘤 TME 中的不同作用。
干扰素富集和促肿瘤社区的肿瘤特异性重连线¶
为了描述跨组织来源的多细胞生态系统,我们构建了一个无向网络,其中包含细胞状态的共现,并在肿瘤、邻近正常和正常组织中识别出不同的网络社区(图 5A–C)。显著的是,一个肿瘤生态系统由肿瘤中来自不同细胞类型的干扰素状态独占(干扰素富集社区;图 5A)。该社区还包含 TLS 的知名成分(LAMP3+ DC、CCL19+ 成纤维细胞和 Tfh),特定的树突状细胞和巨噬细胞(DC1、pDC 和 CXCL9+ 巨噬细胞),以及抗原呈递机制状态。相比之下,干扰素富集社区内的几个免疫细胞状态在健康正常网络中分散(图 5B)。有趣的是,在邻近正常网络中,LAMP3+ DC 与干扰素富集社区中的其他免疫细胞状态(DC1、pDC、朗格汉斯细胞和 Tfh)的邻近性被识别出来(图 5C),这表明即使在邻近正常组织中,相对于健康正常组织,也存在不同的细胞生态系统配置。
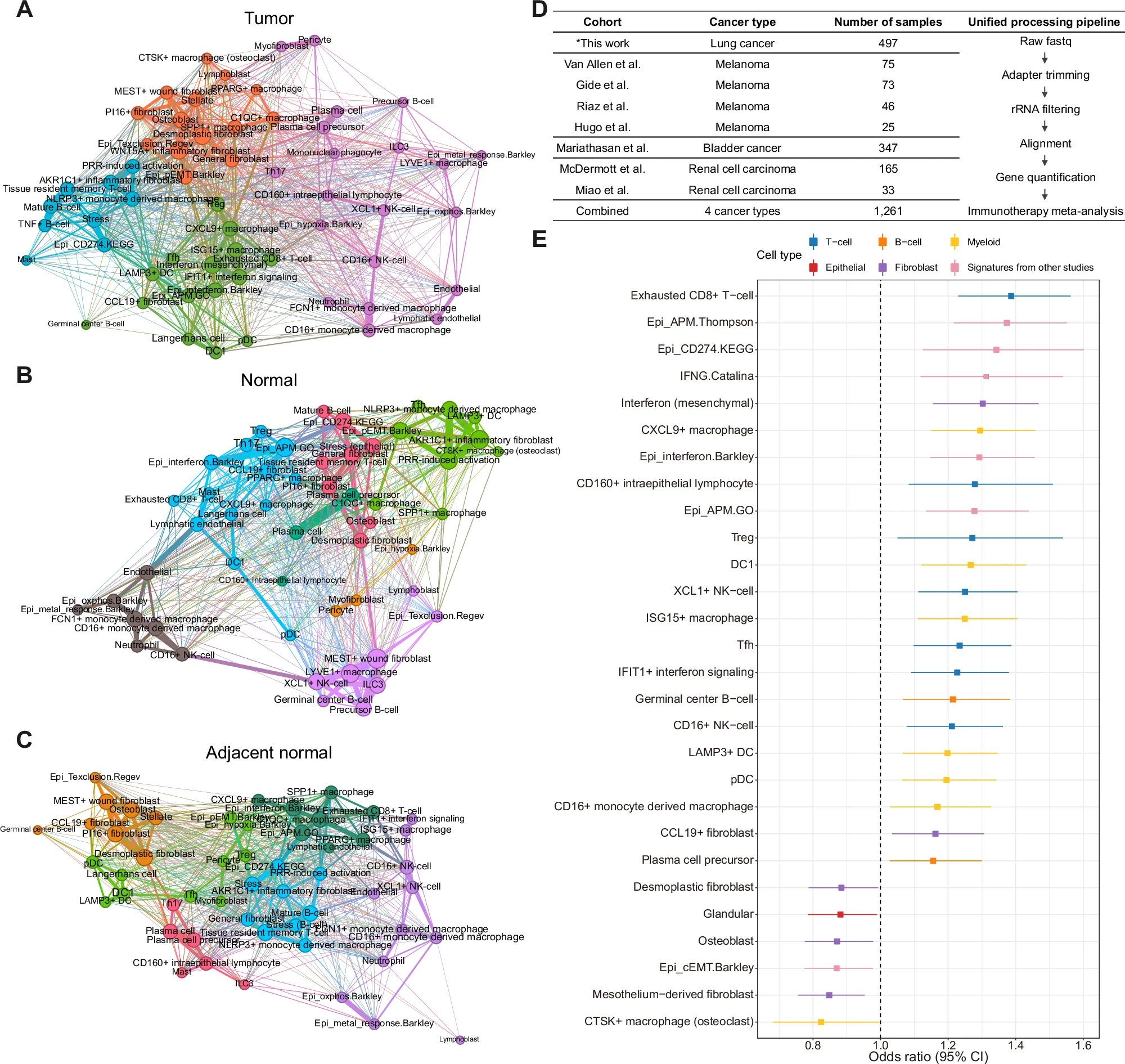
A. 肿瘤组织中的共现网络。节点和边的颜色对应于模块社区,边的厚度对应于邻接的大小。DC,树突状细胞;EMT,上皮 - 间质转化;ILC3,第 3 类固有淋巴细胞;NK,自然杀伤细胞;pDC,浆细胞样树突状细胞;PRR,模式识别受体;Texclusion,T 细胞排斥程序;Tfh,滤泡辅助性 T 细胞;Th17,辅助性 T 细胞 17;Treg,调节性 T 细胞。
B. 正常组织中的共现网络。节点和边的颜色对应于模块社区,边的厚度对应于邻接的大小。DC,树突状细胞;EMT,上皮 - 间质转化;ILC3,第 3 类固有淋巴细胞;NK,自然杀伤细胞;pDC,浆细胞样树突状细胞;PRR,模式识别受体;Texclusion,T 细胞排斥程序;Tfh,滤泡辅助性 T 细胞;Th17,辅助性 T 细胞 17;Treg,调节性 T 细胞。
C. 邻近正常组织中的共现网络。节点和边的颜色对应于模块社区,边的厚度对应于邻接的大小。DC,树突状细胞;EMT,上皮 - 间质转化;ILC3,第 3 类固有淋巴细胞;NK,自然杀伤细胞;pDC,浆细胞样树突状细胞;PRR,模式识别受体;Texclusion,T 细胞排斥程序;Tfh,滤泡辅助性 T 细胞;Th17,辅助性 T 细胞 17;Treg,调节性 T 细胞。
D. 本研究中使用的免疫治疗处理的批量 RNA-seq 队列的总结及其响应数据。* 表示新生成的数据。
E. 通过对免疫治疗处理的批量 RNA-seq 队列(n = 1261 名患者)的元分析,从其他研究中预测免疫治疗响应的细胞状态和基因特征的森林图。x 轴表示优势比,虚线垂直线表示优势比为 1,y 轴表示细胞状态和先前定义的基因特征。对于每个细胞状态,矩形和延长线分别表示通过对免疫治疗队列的临床响应进行逻辑回归元分析计算的优势比和 95% 置信区间。仅显示达到统计显著性的细胞状态。优势比大于 1 的细胞状态与免疫治疗的有利响应相关。颜色对应于每个细胞状态的细胞类型类别。APM,抗原呈递机制;DC,树突状细胞;EMT,上皮 - 间质转化;IFNG,干扰素 -γ;NK,自然杀伤细胞;pDC,浆细胞样树突状细胞;Treg,调节性 T 细胞。
讨论¶
在这项工作中,我们整合了来自 999 名个体的 4.9 百万细胞的转录组图谱,涵盖 30 种癌症类型,包括肿瘤和非肿瘤组织。我们的分析概述了标志基因特征,并系统地可视化了塑造肿瘤 - 正常生态系统的细胞状态。此外,我们阐明了 AKR1C1+ 和 WNT5A+ 成纤维细胞在形成核心炎性 TME 中的区别。干扰素富集社区的多种成分,包括 LAMP3+ 树突状细胞、CCL19+ 成纤维细胞、CXCL9+ 巨噬细胞和间质来源的干扰素状态,赋予了对免疫治疗的良好反应,并在肿瘤、邻近正常和健康正常组织之间形成不同的生态系统。结合空间转录组学,我们的全癌种单细胞图谱的高分辨率注释,使得我们能够对富含 TLS 和非 TLS 的免疫治疗有利成分中的细胞类型进行分类,并调查高度反映复杂肿瘤生物学的模式。
尽管在许多癌症类型中,组织在恶性转化过程中基因表达的差异被减弱 51,但缺乏单细胞分辨率的 TME 中共表达基因模块的证据。在此,我们旨在识别恶性组织中与正常组织相比的标志基因表达程序,跨越各种细胞类型,无论器官如何。尽管组织生态系统中嵌入了高度异质性,但我们的发现表明,在各种细胞类型中存在全器官基因表达程序。这些普遍的基因表达程序可能解释了免疫治疗(例如 PD-1/PD-L1 抑制剂和 CTLA4 抑制剂)在各种癌症中的广泛应用,相对于在特定癌症类型中具有有限适应症的靶向治疗。未来需要进行全面的研究,以揭示 TME 标志基因的免疫调节作用,这些基因可能代表各种癌症类型的 TME 中的免疫检查点或治疗靶点。
我们的研究深入调查了肿瘤和正常组织中的间质状态。特别是,我们识别了 AKR1C1+ 和 WNT5A+ 炎症成纤维细胞状态,它们表现出与 PRR 诱导的激活状态和细胞因子基因表达谱的相似共现模式。尽管这些炎症成纤维细胞群体在几种癌症类型中已被认知 40,52,53,但我们的研究提供了详细的探索,并揭示了这些群体在组织和器官偏好、细胞相互作用和空间共定位模式方面的区别,从而强调了整合图谱的必要性。AKR1C1+ 成纤维细胞状态以 AKR1C1、CXCL8、CXCL2、CXCL3 和 TNFAIP6 等基因为特征,这些基因预测对免疫治疗的不良反应 54,55,56,57,58。因此,针对 AKR1C1+ 炎症成纤维细胞可能提供新的治疗见解,特别是在 OV 和 UCEC 中。有趣的是,WNT5A+ 炎症成纤维细胞已被证明通过 WNT5A、GREM1 和 IL2441,44,45,46 与配置炎性和促肿瘤环境的各种 TME 成分相互作用。未来应进行针对 WNT5A+ 炎症成纤维细胞的治疗研究,以减轻与这些促肿瘤成纤维细胞相关的不良后果。此外,我们的研究显示肌成纤维细胞在正常组织中的富集程度相对较高,与之前的全癌种研究发现相比 30。值得注意的是,我们的研究涵盖了来自多个器官的肿瘤和正常样本,肌成纤维细胞的组织富集代表了跨所有器官的综合结果(图 3E)。器官分析揭示了乳腺、胰腺、肝脏和肾脏肿瘤组织中的肌成纤维细胞富集(补充图 S18C),这表明这些成纤维细胞在这些器官中可能具有促肿瘤作用 59,60,61。然而,结肠和胃中的肌成纤维细胞肿瘤 - 正常富集程度相当,这与先前研究记录的正常肠道中的肌成纤维细胞一致 62,63。
从不同细胞类型中产生的各种细胞状态,其中许多是干扰素富集社区的组成部分,对免疫治疗有利。虽然 TLS 以前被识别为免疫治疗反应的预测因子 12,13,但我们强调,富含 TLS 和非富含 TLS 的细胞类型都对免疫治疗反应有积极贡献。此外,我们的 TLS 特征有效地区分了 RCC 患者中 TLS 和非 TLS 区域,预测了对免疫治疗的有利反应,并成功地与多种癌症类型中的富含 TLS 细胞类型共定位。然而,尚需确认 TLS 富含细胞类型在 RCC 以外的癌症类型中是否在 TLS 定义的组织中具有可比性。此外,还描述了多种对检查点阻断具有不良反应的状态,强调了替代治疗方案的需求。需要强调的是,尽管这些免疫治疗预测细胞状态在 TCGA 队列中显示没有预后意义,但不同癌症类型中细胞状态的免疫治疗显示出不同的治疗反应,表明在免疫治疗过程中不同机制在各癌症类型中占主导地位。
我们的研究通过构建一个结合单细胞、空间和免疫治疗处理的批量数据集的广泛图谱,并系统地比较不同器官中的肿瘤和正常生态系统,提供了独特的视角。这使我们能够揭示肿瘤 - 正常生态系统的标志基因特征,概述 AKR1C1+ 和 WNT5A+ 炎症成纤维细胞之间的区别,并描述免疫治疗有利成分中的富含 TLS 和非 TLS 细胞类型。研究中提出的全癌种肿瘤 - 正常单细胞元图谱将为深入理解肿瘤 - 正常生态系统提供重要见解,并为未来的精准肿瘤学研究奠定基础。为了研究社区的利益,我们的图谱可以通过 Zenodo 存储库(DOI:10.5281/zenodo.10651059)和网络门户(https://cellatlas.kaist.ac.kr/ecosystem/)访问。