Tumour vasculature at single-cell resolution¶
摘要
肿瘤可以通过血液供应获得其进展和转移所需的营养和氧气。诱导血管生成涉及已建立的血管床的萌芽及其成熟为有组织的网络。在此,我们生成了单细胞分辨率的肿瘤血管图谱,涵盖约 20 万个细胞,来自 372 名供体,代表 31 种癌症类型。轨迹推断表明,肿瘤血管生成从静脉内皮细胞开始,并延伸至动脉内皮细胞。随着新生血管的延伸(通过血管生成阶段 SI、SII 和 SIII),在 SI 阶段的 APLN+ 尖端细胞(APLN+ TipSI)通过增加的 Notch 信号传导进展为 TipSIII 细胞。同时,跟随尖端细胞的干细胞从高趋化因子表达过渡到 TEK(也称为 Tie2)表达增加。此外,APLN+ TipSI 细胞不仅与疾病进展和预后不良相关,而且在预测抗 VEGF 疗效方面也有潜力。淋巴内皮细胞展示了两条不同的分化谱系:一条负责淋巴管生成,另一条参与抗原呈递。在周细胞中,内质网应激与促血管生成的 BASP1+ 基质生成周细胞相关。此外,细胞间通讯分析显示,新生血管内皮细胞可以形成有利于血管生成的免疫抑制微环境。本研究描绘了肿瘤血管的复杂性,并对抗血管生成疗法具有潜在的临床意义。
主要内容
肿瘤血管生成是癌症的一个关键标志。它的诱导帮助肿瘤获得维持生长所需的营养和氧气。血管生成可以在肿瘤进展的任何阶段发生,表现为从现有血管网络形成新的血管。与正常血管相比,肿瘤血管显示出更高的通透性、不规则的形态和较差的组织结构。内皮细胞(ECs)和周细胞(MCs)是直接参与肿瘤血管生成的主要血管成分。EC 亚群非常异质,具有不同的血管类型和器官特异性特征。EC 的可塑性使其成为抗血管生成疗法(AATs)的目标,旨在通过阻断血管生成信号实现肿瘤血管的正常化。在临床前研究中,结合 AAT 与肿瘤免疫疗法已显示出更强的抗肿瘤效果。利用单细胞分析技术,我们生成了覆盖约 20 万个血管细胞的全肿瘤血管景观,揭示了肿瘤血管的复杂性。
肿瘤血管单细胞图谱¶
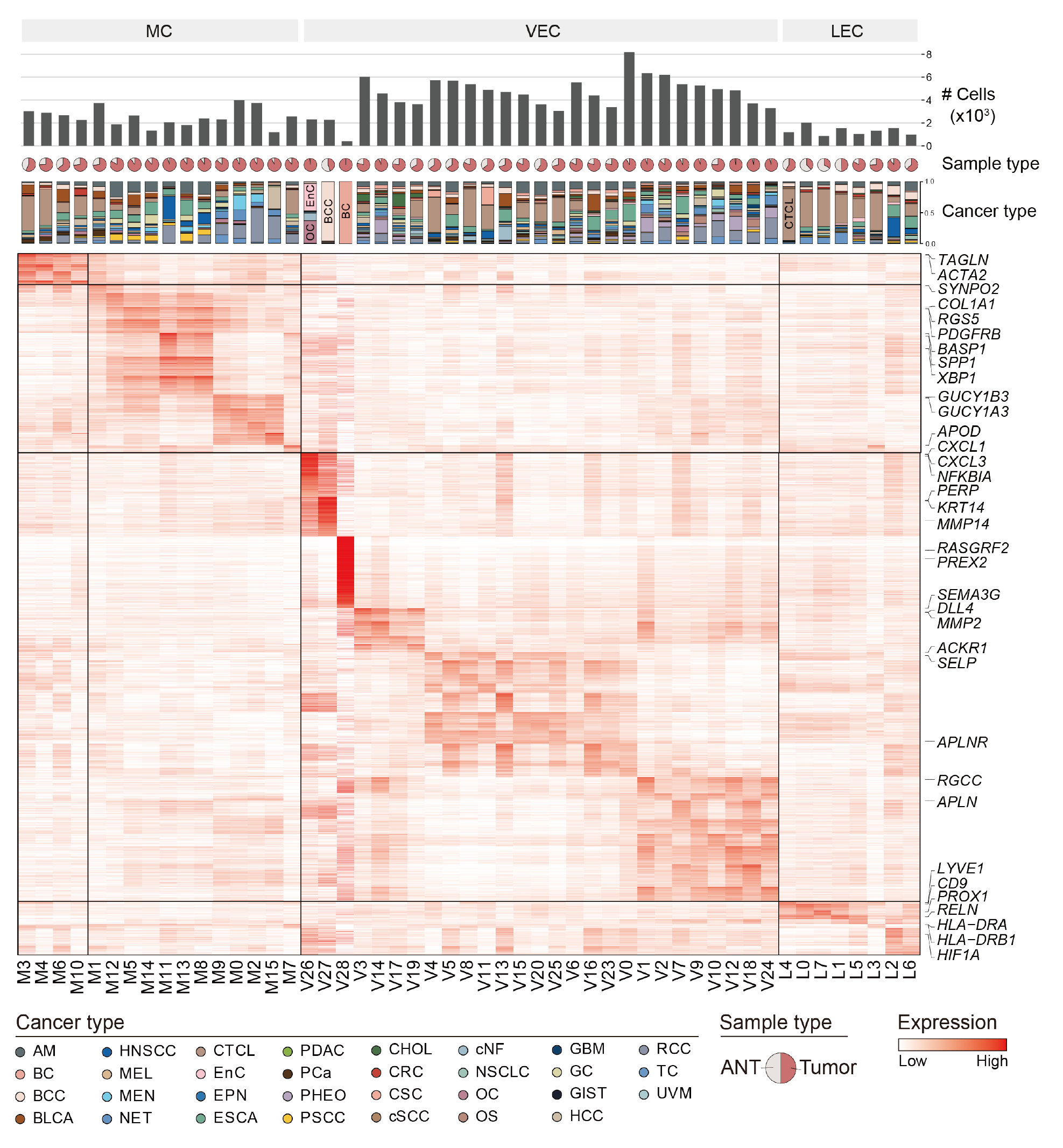
附图 1 | TVM 亚群概览。热图显示每个血管亚群标志基因的表达水平。注释面板(从上到下)分别显示每个亚群中的细胞数、样本类型百分比和癌症类型。
为了解析肿瘤血管微环境(TVM),我们对来自 372 名供体的 437 个肿瘤样本及邻近非肿瘤(ANT)样本进行了计算机辅助的内皮细胞(ECs)和周细胞(MCs)分选(图 1a,b 和补充表 1)。经过严格质量控制后,共有 183,977 个 TVM 细胞,其中 24.2% 为本研究中新测序的(图 1c)。我们整合了从多个平台生成的数据,并进行了批次效应校正(方法和扩展数据图 1)。随后,根据代表性基因特征注释了细胞类型(图 1c,d 和补充表 2)。在肿瘤组织中,ECs 主要被分类为淋巴内皮细胞(LECs)(特征为 LYVE1 和 PROX1)和血管内皮细胞(VECs),包括动脉内皮细胞(ArtECs)(特征为 SEMA3G)、静脉内皮细胞(VenECs)(特征为 ACKR1)和毛细血管样内皮细胞(CapECs)(特征为 RGCC)。此外,我们检测到正在经历内皮 - 间质转化(EndoMT)的 VECs,这些细胞共同表达 EC 和间质标志物,并进一步将这些细胞区分为 PDGFRB+ EndoMT-I 和 DCN+ EndoMT-II 细胞(扩展数据图 2a)。我们还识别了两种类型的 MCs,包括周细胞(PCs)(特征为 RGS5 和 PDGFRB)和平滑肌细胞(SMCs)(特征为 ACTA2 和 TAGLN)。值得注意的是,与 ANT 组织相比,我们在肿瘤组织中观察到更高比例的 CapECs 和 PCs(扩展数据图 2b,c)。此外,我们在相应的 ANT 组织中识别了两个特异性内皮细胞,即肾小球细胞(RGCs)(标记为 EMCN、SOST 和 PLAT)和肝窦内皮细胞(LSECs)(标记为 CLEC4G 和 CLEC1B)(图 1d)。
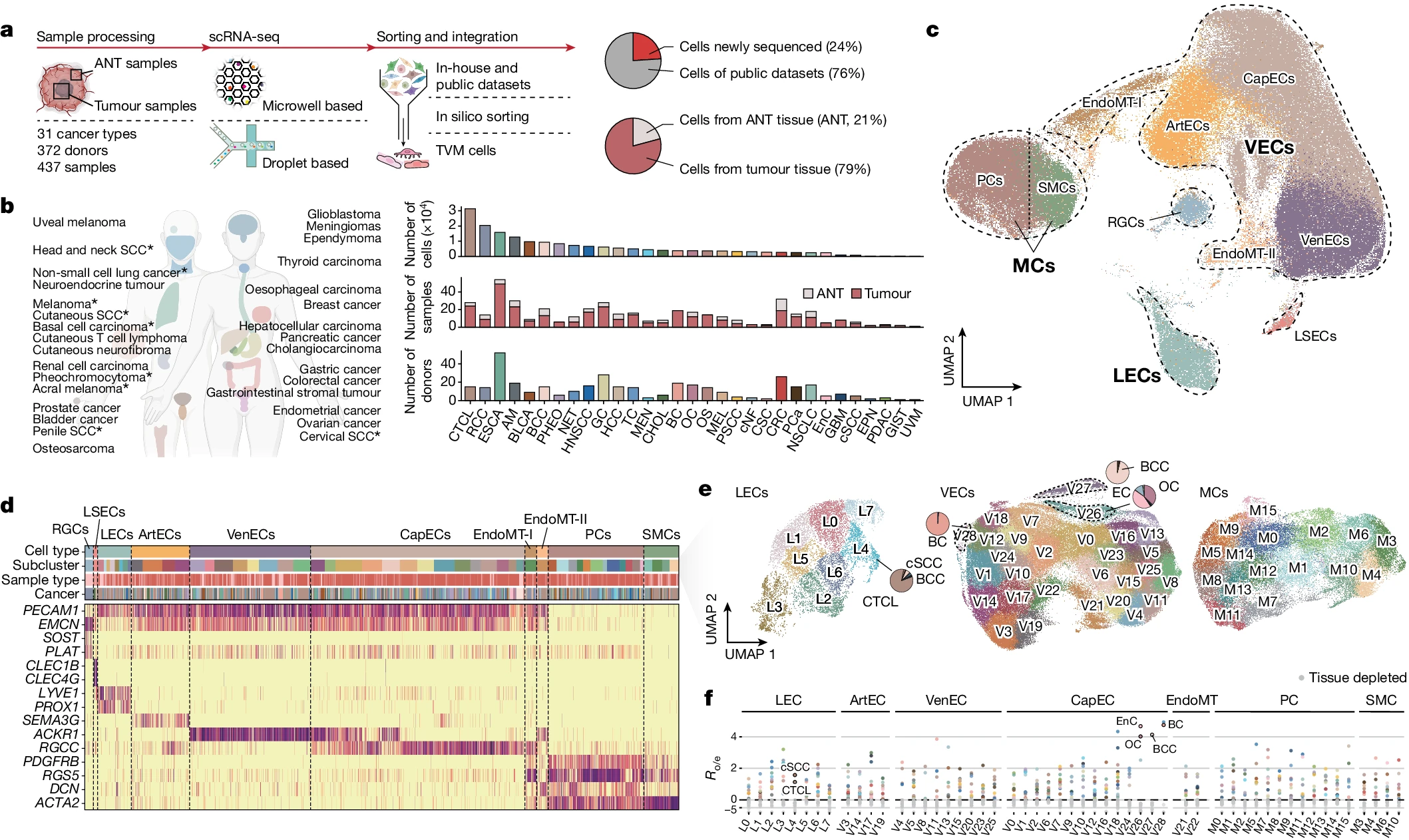
a,左,数据生成的示意图,研究设计和图谱整合。右,来自内部/公共数据集的 TVM 细胞比例(顶部)和 ANT/肿瘤组织(底部)的比例。b,本研究涵盖的癌症类型。星号表示具有新生成数据的癌症类型。条形图显示每种癌症类型的检测细胞数(顶部)、样本数(中部)和供体数(底部)。c,所有 TVM 细胞的 UMAP 分析,按细胞类型着色。d,代表性签名基因的表达水平。e,TVM 亚群的 UMAP 可视化。饼图显示类似组织来源的亚群中癌症类型的比例。f,每个 TVM 亚群中癌症类型的 Ro/e 值(对数尺度)。类似组织来源的亚群由癌症类型标记。Gynaecologic:子宫内膜癌和卵巢癌;mammary:乳腺癌;skin:基底细胞癌、皮肤 T 细胞淋巴瘤和皮肤鳞状细胞癌。AM,肢端黑色素瘤;BC,乳腺癌;BCC,基底细胞癌;BLCA,膀胱癌;CHOL,胆管癌;cNF,皮肤神经纤维瘤;CRC,结肠直肠癌;CSC,宫颈鳞状细胞癌;cSCC,皮肤鳞状细胞癌;CTCL,皮肤 T 细胞淋巴瘤;EnC,子宫内膜癌;EPN,室管膜瘤;ESCA,食管癌;GBM,胶质母细胞瘤;GIST,胃肠道间质瘤;GC,胃癌;HCC,肝细胞癌;HNSCC,非小细胞肺癌;MEL,黑色素瘤;MEN,脑膜瘤;NET,神经内分泌肿瘤;NSCLC,非小细胞肺癌;OC,卵巢癌;OS,骨肉瘤;PCa,前列腺癌;PDAC,胰腺癌;PHEO,嗜铬细胞瘤;PSCC,阴茎鳞状细胞癌;RCC,肾细胞癌;TC,甲状腺癌;UVM,葡萄膜黑色素瘤。图 a 和 b 中的图表是使用 BioRender 创建的。
我们接下来对 VECs、LECs 和 MCs 进行了无监督图聚类分析,并将这些细胞分成了 29 个 VECs 亚群(V0-V28)、8 个 LECs 亚群(L0-L7)和 16 个 MCs 亚群(M0-M15)(图 1e 和补充图 1)。我们进一步通过计算观察到的细胞数量与预期细胞数量的相对得分(Ro/e 分数;图 1f 和方法),估计了这些亚群的癌症类型富集情况。大多数亚群展示了全肿瘤的来源。然而,某些 VECs 和 LECs 亚群,位于均匀流形近似和投影(UMAP)表示的边缘位置,展示了组织来源的相似性,包括妇科癌症来源的 CapECs(V26)、乳腺癌(BC)来源的 CapECs(V28)以及皮肤肿瘤来源的 CapECs 和 LECs(V27 和 L4)(图 1e,f)。
对这些边缘亚群的标记基因分析显示,妇科癌症来源的 CapECs 特别表达 CXCL1 和 CXCL3,与子宫内膜癌和卵巢癌的进展相关 11。BC 来源的 CapECs 表现出 PREX2 和 RASGRF2 的高表达,可能在对 VEGF 的响应中得到增强 12。此外,皮肤肿瘤来源的 CapECs 和 LECs 表达与皮肤相关的标志物 PERP 和 KRT1413(扩展数据图 2d)。这些亚群未包括在随后的全肿瘤分析中。
肿瘤血管生成从 VenECs 发芽¶
肿瘤血管生成涉及从已建立的血管床发展新生血管 2,3。为了探索其分子基础,我们基于对 116,958 个全肿瘤 VECs 的转录组分析,包括 18,048 个 ArtECs、61,400 个 CapECs 和 37,510 个 VenECs(图 2a 和补充图 2)。与 ANT 组织相比,肿瘤组织中 CapECs 的浸润多于 VenECs 和 ArtECs(图 2b)。为捕捉肿瘤血管生成的轨迹,我们进行了扩散成分分析 14,推断了从 VenECs 经过 CapECs 到 ArtECs 的分化轨迹(图 2c)。这一结果通过不同算法原理的轨迹推断方法再现(方法和扩展数据图 3a,b)。进一步评估细胞分化能力 15 显示,VenECs 在其转化为 CapECs 过程中,最初经历了去分化过程(扩展数据图 3c)。接下来,我们通过将肿瘤细胞移植到斑马鱼中构建了体内肿瘤血管生成模型(方法)。我们观察到,肿瘤诱导的新生血管逐渐从亚肠静脉(SIV)向肿瘤位置延伸(图 2d)。然而,当正常细胞注入斑马鱼时,这种现象并未发生。这些结果表明,肿瘤血管生成是由 VenECs 启动的。
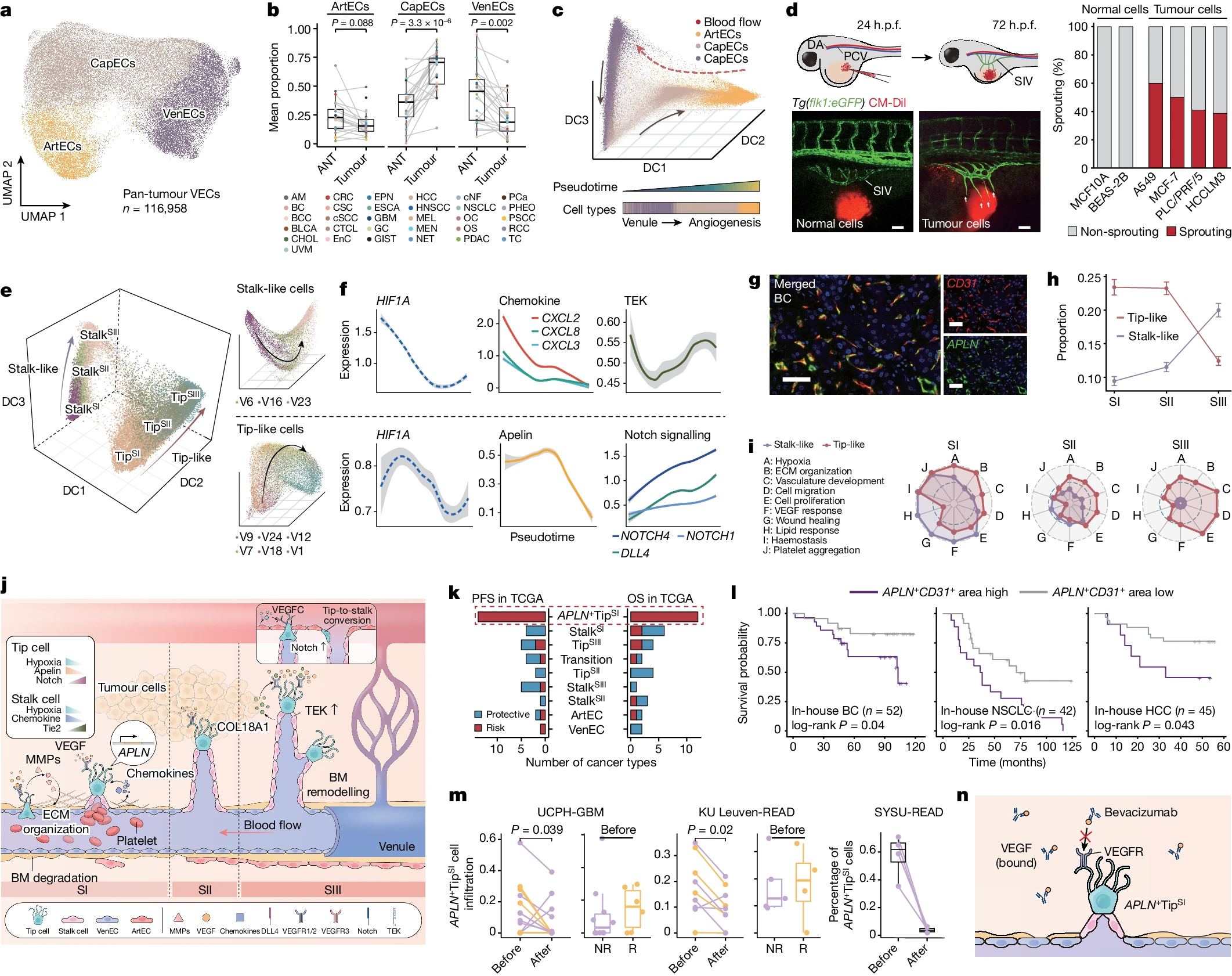
a,全肿瘤 VECs 的 UMAP 分析,按推断的细胞类型着色。b,VECs 亚型在 ANT 和肿瘤组织中每种癌症类型的平均比例。点表示癌症类型(n=31)。c,扩散映射显示肿瘤血管生成的轨迹。d,转基因 Tg(flk1:eGFP) 斑马鱼胚胎中的肿瘤血管生成。代表性图像显示用红色荧光染料(CM-Dil)标记的正常细胞(BEAS-2B)和肿瘤细胞(A549)移植到斑马鱼胚胎中后 SIV 的形态。条形图显示斑马鱼胚胎发育中 SIV 的异位分枝率。h.p.f.表示受精后小时。e,扩散映射中尖型和茎型细胞的轨迹。f,伪时间中指定基因的 Loess 回归平滑表达(y 轴)。阴影区域表示 95% 置信区间。g,BC 中 APLN+ TipSI 细胞标志物的代表性 mIHC 分析,来自 n=10 个独立生物复制品。h,尖型和茎型细胞中血管生成阶段的比例(尖型细胞:n=9,063(SI)、n=13,390(SII)、n=6,351(SIII);茎型细胞:n=4,399(SI)、n=3,361(SII)、n=5,517(SIII))。数据为均值±标准误差。i,显示了 TAPFs 的比例活动的雷达图。j,肿瘤血管生成可能过程的示意图。k,在 TCGA 队列中,VECs 与总生存期/无进展生存期相关的癌症类型数量。l,根据低和高 APLN+CD31+ 面积对内部队列的 Kaplan-Meier 曲线进行分层。m,贝伐单抗治疗后 APLN+ TipSI 细胞浸润的变化。显示了 UCPH-GBM(n=6(N)、n=10(NR))、KU Leuven-READ(n=6(N)、n=5(NR))和 SYSU-READ(n=4)中治疗前反应者(R)和非反应者(NR)之间的差异。n,说明贝伐单抗通过抑制 VEGF 介导的血管生成的图示。对于箱线图,中心线表示中位数,箱限表示上下四分位数,触须延伸到 1.5 倍的四分位距。统计分析使用双侧 Mann-Whitney U 检验(b 和 m)和 log-rank 检验(l)进行。图 n 是使用 BioRender 创建的。对于 d 和 g,比例尺为 50µm。
新生血管缺乏母体血管的形态特征,可以解释为具有毛细血管样特征。对细胞纯度的评估还表明,与 VenECs 和 ArtECs 相比,CapECs 表现出更高的异质性(扩展数据图 3d)。在血管生成过程中,尖端细胞感知并响应引导信号,带路,而柄细胞则跟随其后,延长芽的柄部并形成管腔。因此,我们对 CapECs 的表型进行了评分(方法),并鉴定出类似尖端的 CapECs、类似柄的 CapECs 以及处于尖端和柄细胞过渡状态中的 CapECs(扩展数据图 3e)。我们观察到肿瘤组织中显著富集了类似尖端的 CapECs(扩展数据图 3f,g)。接下来,我们追踪了类似尖端和类似柄细胞的分化轨迹,将血管生成阶段定义为 SI、SII 和 SIII(扩展数据图 4a–c)。总体而言,类似尖端和类似柄细胞的发育轨迹是独立的,并在三维扩散组分空间中呈正交趋势(图 2e)。在 SI 阶段,类似尖端和类似柄的 CapECs 均表现出 HIF-1α(由 HIF1A 编码)活性增强(扩展数据图 4d),随后在 SII 和 SIII 阶段逐渐减弱(图 2f),这支持低氧环境在触发肿瘤芽生成中起着关键作用。
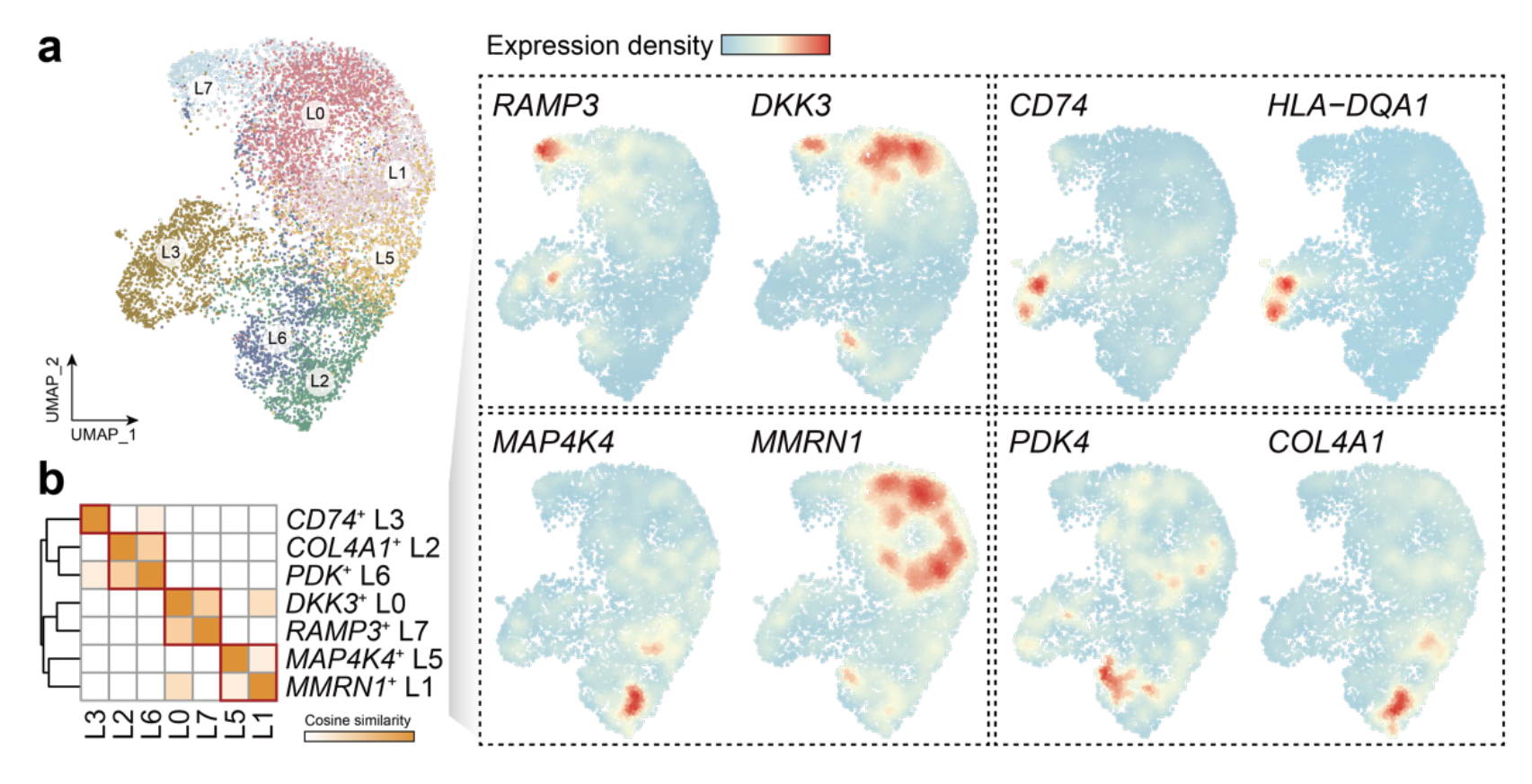
附图 3 | LEC 亚群概览。a. 显示 LEC 细胞亚群的 UMAP 图。b. 热图显示 LEC 亚群之间的余弦相似性。基于前 100 个标记基因集计算了 LEC 亚群之间的成对余弦相似性。每个亚群中顶级标记基因的表达密度图显示在右侧。
在血管生成的起始阶段,APLN(编码 apelin)被确认为 TipSI 细胞的特异性标记(下称为 APLN+ TipSI 细胞)(图 2f 和扩展数据图 4e)。对肿瘤切片的多色免疫组化染色进一步确认了 APLN+ TipSI 细胞在各种癌症中的普遍存在(图 2g 和扩展数据图 4f)。此外,新生血管的动态分析显示,尖端细胞主要存在于 SI 阶段,而柄细胞则更多见于 SIII 阶段(图 2h)。我们接着根据自组织图(SOM)分析功能过表达的代基因,总结了肿瘤血管生成过程(TAPFs)的特征,其中包括低氧、细胞外基质(ECM)组织、血管发育、细胞迁移、细胞增殖、VEGF 反应、伤口愈合、脂质反应、止血和血小板聚集(扩展数据图 4h)。我们利用 AUCell 进一步评估了活跃的 TAPFs,并将它们分配给新生血管细胞(图 2i)。通过进一步整合细胞间通信的建模(方法、扩展数据图 4i,j 和补充表 3),我们重建了动态肿瘤血管生成的推测过程(图 2j)。在 SI 阶段,APLN 和趋化因子(CXCL2、CXCL3 和 CXCL8)被确认为尖端和柄细胞中的活跃配体(扩展数据图 4i)。TipSI 细胞分泌 apelin,促进柄细胞增殖和芽伸长。同时,StalkSI 细胞释放的趋化因子可以促进尖端细胞的丝状伪足形成和迁移。APLN+ TipSI 和 StalkSI 细胞均表达 ECM 调节因子,重新组织 ECM,并表现出高的血小板聚集、止血和伤口愈合活动(图 2i)。到了 SIII 阶段,柄细胞增殖减弱,而尖端细胞增殖增强(图 2i)。此外,我们观察到 StalkSIII 细胞中血管生成素受体 TEK 的表达升高(扩展数据图 4e),这可能有助于稳定新血管生成。COL18A1 也被鉴定为 StalkSIII 细胞的特异性配体(扩展数据图 4i)。COL18A1 是血管基底膜的成分之一,还能结合 VEGF 受体(FLT1 和 KDR),竞争性抑制 VEGF 信号,暗示基底膜在 SIII 阶段的重塑和血管生成完成。此外,Notch 信号在 TipSIII 细胞中被激活(图 2f),Notch 信号的激活促进 TipSIII 细胞通过高表达 VEGFR3(由 FLT4 编码)进行尖端到柄细胞的表型转换,使尖端细胞整合到动脉中,限制血管生成。
为了研究 VECs 的临床意义,我们在癌症基因组图谱(TCGA)队列中使用解卷积分析评估了细胞浸润情况(方法和补充表 4)。在大多数 ANT 组织中,APLN+ TipSI 细胞几乎检测不到(扩展数据图 5a),但在 TCGA 肿瘤样本中,随着肿瘤分期的升高而增加(扩展数据图 5b)。Cox 比例风险模型表明,高浸润的 APLN+ TipSI 细胞预测了较差的无进展生存和总生存(图 2k 和扩展数据图 5c)。此外,在我们的 BC、非小细胞肺癌(NSCLC)和肝细胞癌(HCC)的内部队列中,具有高比例 APLN+ TipSI 细胞的肿瘤患者具有较差的总生存结果(图 2l 和补充表 5–7)。接着我们考察了 VEC 亚型对抗血管生成治疗(AAT)的反应。贝伐单抗——一种抗 VEGF 抗体——被开发用于靶向肿瘤血管生成。在三个接受贝伐单抗联合治疗的独立队列中(补充表 1),贝伐单抗治疗后,APLN+ TipSI 细胞的浸润显著减少。治疗反应者在治疗前具有比非反应者更高水平的 APLN+ TipSI 细胞浸润(图 2m 和扩展数据图 5d,e),这暗示 APLN+ TipSI 细胞可能成为贝伐单抗诱导肿瘤血管正常化的靶点(图 2n)。这些发现进一步表明,具有高 APLN+ TipSI 细胞肿瘤浸润的患者可能会受益于 AAT,尽管其预后较差。
肿瘤 LECs 的分化谱系¶
总共检测到 9,283 个全肿瘤 LEC(L0–L3 和 L5–L7)(补充图 3)。我们发现 L0 和 L7 亚群主要来自 ANT 样本(扩展数据图 6a,b),因此被注释为正常样 LEC(NLECs)。进一步的轨迹推断显示,从 NLECs 出发存在两个明显的分化谱系(图 3a 和扩展数据图 6c)。轨迹 1(T1)通过 L1/L5 导向 L2/L6,而轨迹 2(T2)则直接分化为 L3 通过 L1(扩展数据图 6d)。我们因此表征了 L2/L6 和 L3 谱系的特性。比较 L2 和 L6 细胞之间尖端和柄样 CapECs 的标记表达水平(补充表 2)表明,L2 细胞表现出尖端样表型,而 L6 细胞显示出柄样表型(图 3b)。基因表达分析进一步显示,尖端样(L2)和柄样(L6)LEC 高表达 VEGFR3 和趋化因子(CCL2、CXCL1 和 CXCL2),而 L3 细胞则表达多种 MHC-II 分子(HLA-DP、HLA-DQ 和 HLA-DR)和 MHC-II 相关的不变链(CD74)(图 3c)。我们因此将 L3 标注为抗原呈递 LEC(apLEC)。功能富集分析还显示,尖端样和柄样 LEC 与 VEGF 信号通路和炎症反应相关,而 apLECs 在抗原呈递、免疫过程和 ECM 组织中显著富集(扩展数据图 6e)。我们推测 T1 代表了肿瘤淋巴生成过程(下称 T1- 淋巴生成),而 T2 代表了 apLECs 的分化(下称 T2-apLEC)。值得注意的是,T1- 淋巴生成显示比 T2-apLEC 更高的 HIF1A 活性(图 3c 和扩展数据图 6f),这暗示低氧可能是肿瘤淋巴生成的诱导因素。
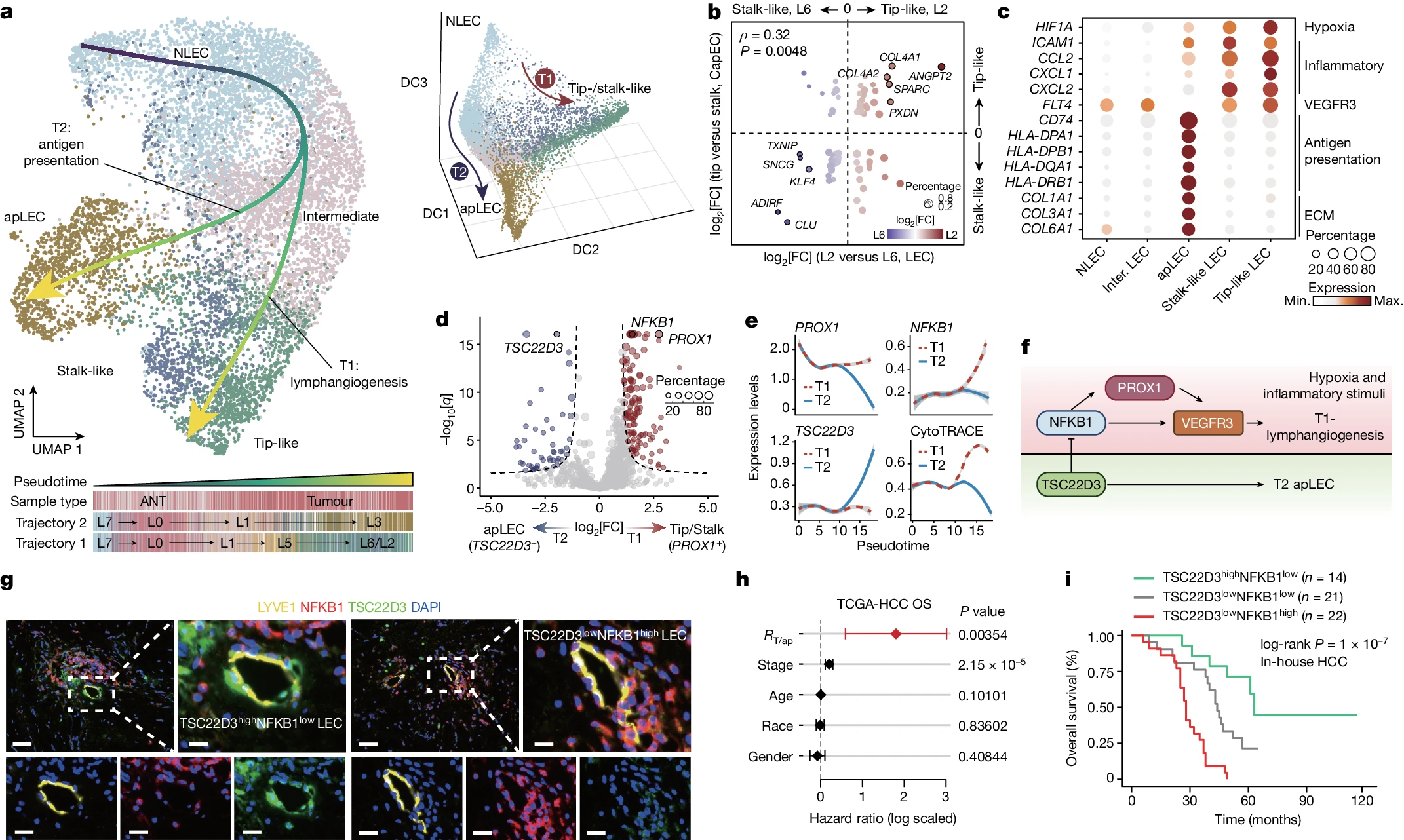
a,LEC 分化轨迹。左侧显示了 Slingshot 推导的 LEC 轨迹的 UMAP 图。右侧显示了扩散成分图。轨迹 1(T1)和轨迹 2(T2)分别从 NLECs 起始,通向类似尖端/柄状 LECs 或 apLECs。b,L2 高度表达类似尖端 CapEC 标记物,而 L6 高度表达类似柄状 CapEC 标记物。显示了 Spearman 相关系数及其 P 值。c,选择的尖端/柄状 LECs 和 apLECs 标志基因的表达水平。d,T1 和 T2 之间差异表达的转录因子(TFs)。e,指定基因和 CytoTRACE 评分在伪时间中的 Loess 回归平滑表达(y 轴)。阴影区域表示 95% 的置信区间。f,LEC 谱系中提议的 TF 调控。g,BC 中类似尖端 LECs(左)和 apLECs(右)标志物的代表性 mIHC 分析,来自 n=6 个独立生物复制品。比例尺,50μm(顶部,第 1 和第 3 列),20μm(底部)和 12.5μm(顶部,第 2 和第 4 列)。h,在 TCGA-HCC 队列(n=363 例)中,通过调整肿瘤分期、年龄、种族和性别的多种 Cox 比例风险模型识别 RT/ap 相关的风险比(x 轴,误差条的中心)和 95% 置信区间(误差条)(y 轴)。显示了 Cox 比例风险模型的双侧 P 值。i,在内部 HCC 队列中,将患者分为指定组别的 Kaplan-Meier 生存曲线。对于 i,统计分析使用 log-rank 检验进行。
接下来,我们识别了与谱系相关的转录因子(TFs)(方法)。PROX1 的表达在 T1- 淋巴生成中显著高于 T2-apLEC(图 3d,e)。此外,PROX1 是 TF NFKB1 的下游基因,NFKB1 在 T1- 淋巴生成中也富集。功能上,PROX1 和 NFKB1 都可以被炎症刺激激活,并调节 LEC 中 VEGFR3 的表达,导致淋巴生成。与 T1- 淋巴生成相反,抗炎 TF TSC22D3 在 apLEC 中特异性表达(图 3d,e 和扩展数据图 6g)。先前的研究表明,TSC22D3 可以抑制 NF-κB(由 NFKB1 编码)的核转位和 DNA 结合。这些证据表明,炎症反应在 LEC 分化中起着关键作用(图 3f)。此外,我们确定了细胞分化状态(方法),显示 T2-apLEC 沿着分化水平逐步增加,而 T1- 淋巴生成沿着分化水平逐步降低(图 3e)。
值得注意的是,我们观察到 apLECs 与类似尖端和柄状 LECs 的比例之间存在显著的负相关关系(扩展数据图 6h)。在 TCGA 队列中也观察到了相同的趋势。我们使用 mIHC 染色法在 BC(图 3g)和 HCC(扩展数据图 6i)中确认了 apLECs(TSC22D3 高 NFKB1 低)和类似尖端的 LECs(TSC22D3 低 NFKB1 高)的存在。在 TCGA-HCC 中,高肿瘤浸润的类似尖端 LECs 与较差的预后结果相关,而 apLEC 浸润可以作为保护因素(扩展数据图 6j)。在经过年龄、性别、种族和肿瘤分期校正的 Cox 回归模型中,类似尖端 LEC 与 apLEC 浸润水平的比率(RT/ap)也成为独立的风险因素(图 3h)。在我们的内部 HCC 队列中,与具有高比例类似尖端 LECs(TSC22D3 低 NFKB1 高)的患者相比,具有高比例 apLECs(TSC22D3 高 NFKB1 低)的患者具有更长的总生存期(图 3i 和补充表 8)。这种现象也出现在多种癌症中(扩展数据图 6k),暗示不同的 LEC 分化谱系可能与不同的预后结果相关。
ER 应激驱动促血管生成的 matPCs¶
我们在 TVM 中检测到 40,391 个 MCs,包括 29,572 个 PCs 和 10,819 个 SMCs(补充图 4)。与 ANT 组织相比,肿瘤组织中 PCs 的比例显著高于 SMCs(扩展数据图 7a)。我们还注意到 PCs 的细胞纯度低于 SMCs(扩展数据图 7b),这促成了 PCs 的异质性表型。在全肿瘤组织中识别出四种 PCs 亚型(扩展数据图 7c):(1)与血管舒张相关的 PCs(vdPCs);(2)脂肪细胞样 PCs;(3)肌样 PCs;(4)基质生成 PCs(matPCs)(图 4a)。值得注意的是,vdPCs 表达的鸟苷酸环化酶亚基(GUCY1A3 和 GUCY1B3)也与静息 PCs 标记相关。因此,我们根据静息 PCs 标记对 PCs 的静息状态进行了评分(方法)。vdPCs 的静息评分高于其他 PCs 亚型(扩展数据图 7d)。我们还分析了静息高(Qhigh)和静息低(Qlow)细胞之间的差异表达基因(DEGs),显示 Qhigh 细胞高度表达氧化磷酸化相关基因,而 Qlow 细胞高度表达 ECM 和糖酵解调节因子(图 4b),暗示代谢重编程与 PCs 活性密切相关。
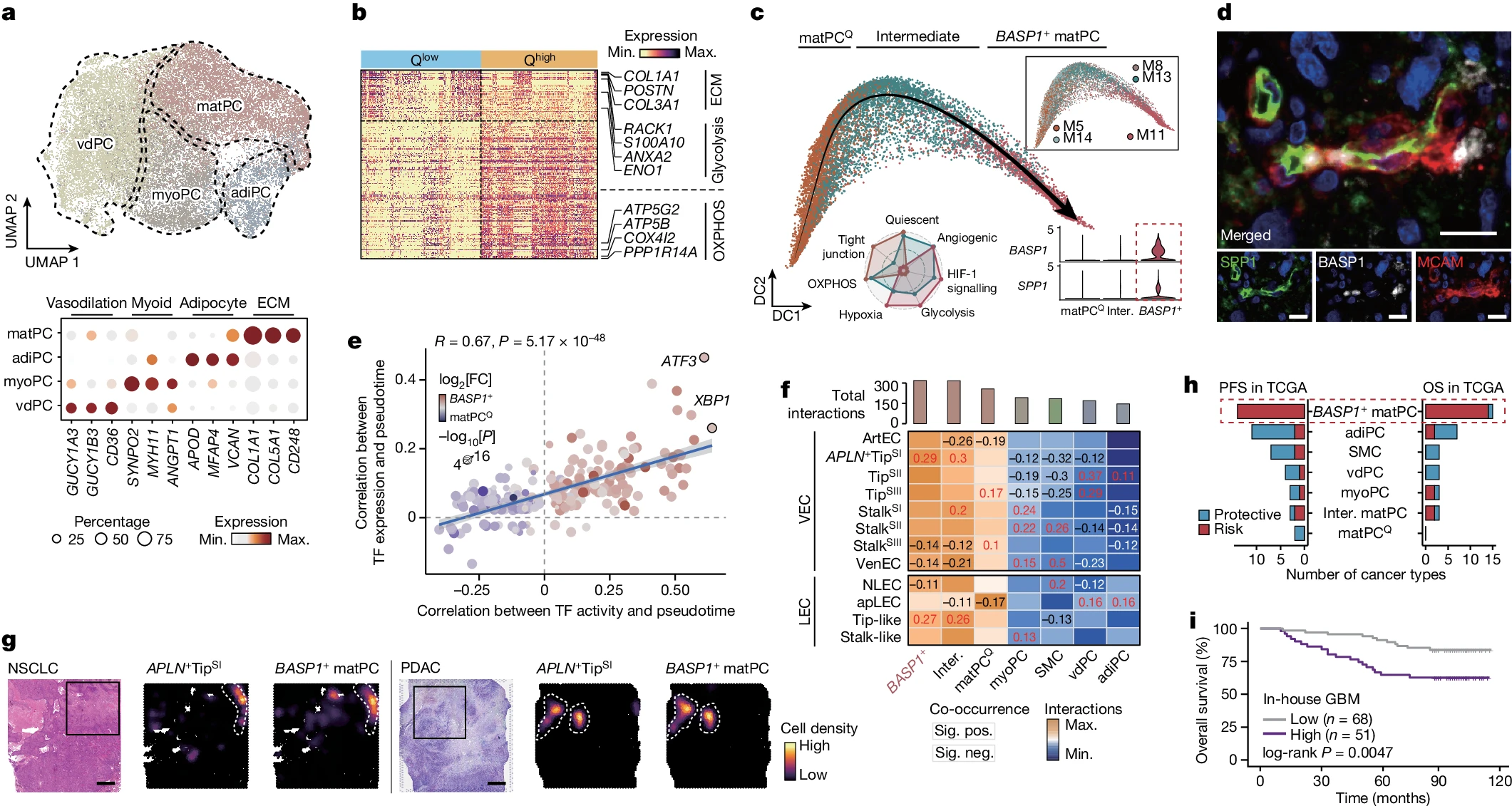
a,PCs 的 UMAP 分析,按 PC 表型着色。底部显示了 PC 表型的选定基因的表达。b,Qhigh 和 Qlow PCs 之间的差异表达基因(DEGs)。c,matPC 分化的轨迹。雷达图显示了 PC 属性的缩放活动。小提琴图显示了 BASP1 和 SPP1 在 matPC 亚型中的表达。d,GBM 中 BASP1+ matPC 标志物的代表性 mIHC 分析,来自 n=6 个独立生物复制品。比例尺,10μm。e,伪时间相关的转录因子(TFs)。计算了伪时间与 TF 转录(y 轴)和 TF SCENIC 评分(x 轴)之间的 Spearman 相关系数。显示了 x 轴和 y 轴之间 Spearman 相关系数及其 P 值。点代表 TFs,按倍数变化着色,并根据 matPCQ 和 BASP1+ matPC 之间双侧 Mann-Whitney U 检验的−log[P] 大小调整大小。f,MCs 和 ECs 之间的细胞 - 细胞相互作用。条形图显示了 MCs 的总 LR 相互作用数。热图显示了 MC 亚型和 EC 亚型之间的相互作用数量。热图上显示了显著的共现 Pearson 相关系数;正相关(红色)和负相关(黑色)被指示。g,来自 NSCLC(n=1 例)和胰腺癌(n=1 例)的空间转录组学数据中 APLN+ TipSI 细胞和 BASP1+ matPCs 在空间位置上的共定位。比例尺,1mm。h,在 TCGA 队列中 MCs 与总生存期和无进展生存期显著相关的癌症类型数量。i,在内部 GBM 队列中低比例和高比例 BASP1+ matPCs 患者的 Kaplan-Meier 生存曲线。对于 i,统计分析使用 log-rank 检验进行。
值得注意的是,matPCs 表现出静息和代谢状态的转换(扩展数据图 7d-f)。我们接着在伪时间上排列 matPCs,观察到处于 Qhigh 状态的 matPCs(matPCQ)可以分化为 BASP1+ matPCs(图 4c 和扩展数据图 7g,h)。进一步的活性分析表明,与 matPCQ 细胞相比,BASP1+ matPCs 表现出较低的紧密连接、氧化磷酸化和静息水平,以及较高的血管生成、低氧和糖酵解水平(图 4c 和扩展数据图 7i)。值得注意的是,BASP1+ matPCs 还表达骨桥蛋白(由 SPP1 编码)(图 4c)。血管周围的骨桥蛋白被报道具有促血管生成作用。我们进一步使用 mIHC 染色验证了胶质母细胞瘤(GBM)中 BASP1+ matPCs 的存在(图 4d)。接下来,我们评估了 matPCs 的 TF 活性。内质网(ER)应激相关的 TFs ATF3 和 XBP1 在转录和 TF 活性水平上均与伪时间明显相关(图 4e 和扩展数据图 7j)。对 ER 应激相关基因的分析还表明,在 matPC 分化过程中引发了 ER 应激(扩展数据图 7k)。此外,几个 ER 应激相关的 TFs 在 BASP1+ matPCs 中表现出高活性(扩展数据图 7l,m)。这些结果表明 ER 应激可能是 matPCs 演化的关键因素。
随后,我们分析了 MCs 和 ECs 之间的细胞间相互作用(方法和补充表 9)。BASP1+ matPCs 在所有 MC 亚型中表现出最高的相互作用频率,突显了它们在 TVM 中的高活性(图 4f)。值得注意的是,BASP1+ matPCs 的比例与 APLN+ TipSI 细胞的比例之间存在显著的正相关关系(扩展数据图 8a)。我们接着对 NSCLC 的苏木精和伊红切片进行了空间转录组测序(方法)。在内部和外部的空间转录组数据(补充表 1)中,我们进行了空间转录组解卷分析。结果显示 BASP1+ matPCs 和 APLN+ TipSI 细胞在空间上是近邻的(图 4g 和扩展数据图 8b)。配体 - 受体(LR)相互作用表明,BASP1+ matPCs 可以分泌 VEGF,促进血管生成(扩展数据图 8c,d)。染色质免疫沉淀后测序(ChIP-seq)数据表明,VEGFA 和 VEGFB 也是 TFs XBP1 和 ATF3 的潜在下游基因,支持 ER 应激在肿瘤血管生成中的关键作用。此外,肿瘤浸润的 BASP1+ matPCs 不仅在更高肿瘤分期的样本中增加(扩展数据图 8f),还与 TCGA 队列中的疾病进展和预后不良相关(图 4h 和扩展数据图 8g)。我们还验证了高比例 BASP1+ matPCs 与内部 GBM 队列中较差的总生存期相关(图 4i 和补充表 10)。
血管与肿瘤微环境(TME)之间的对话¶
为了解析与血管相关的细胞间通信,我们分选了 160,366 个髓系细胞、261,751 个 T 细胞和自然杀伤细胞(T/NK 细胞)、42,342 个 B 细胞和 92,297 个成纤维细胞,并确定了它们的亚型(方法和补充图 5)。我们进一步在肿瘤微环境(TME)中识别了同质的 meta 细胞(图 5a),并在血管细胞和 TME 细胞之间进行无偏 LR 相互作用分析(方法和补充表 11)。髓系细胞和成纤维细胞显示出比 T/NK 和 B 细胞更多的相互作用,包括关键的 VEGF 信号通路。对 VEGF-VEGFR 信号通路(VEGFA, VEGFB-FLT1, KDR)的分析表明,SPP1+ 肿瘤相关巨噬细胞(TAMs)、髓系抑制细胞(MDSCs)、单核细胞、2 型常规树突细胞(cDC2s)、癌症相关成肌纤维细胞(myoCAFs)、基质相关 CAFs(matCAFs)和 BASP1+ matPCs 是新生血管生成的主要 VEGF 来源(扩展数据图 8c 和 9a,b)。我们观察到 SPP1+ TAMs 与血管细胞的相互作用数量最多(图 5b)。SPP1+ TAMs 还显著与尖端细胞共现,并且与 APLN+ TipSI 细胞的相互作用数量高于与 SII 和 SIII 尖端细胞的相互作用数量。空间转录组数据表明 APLN+ TipSI 细胞和 SPP1+ TAMs 在空间上共定位。此外,mIHC 分析提供了 SPP1+CD68+ TAMs 和 APLN+CD31+ ECs 在原位接近的额外证据。
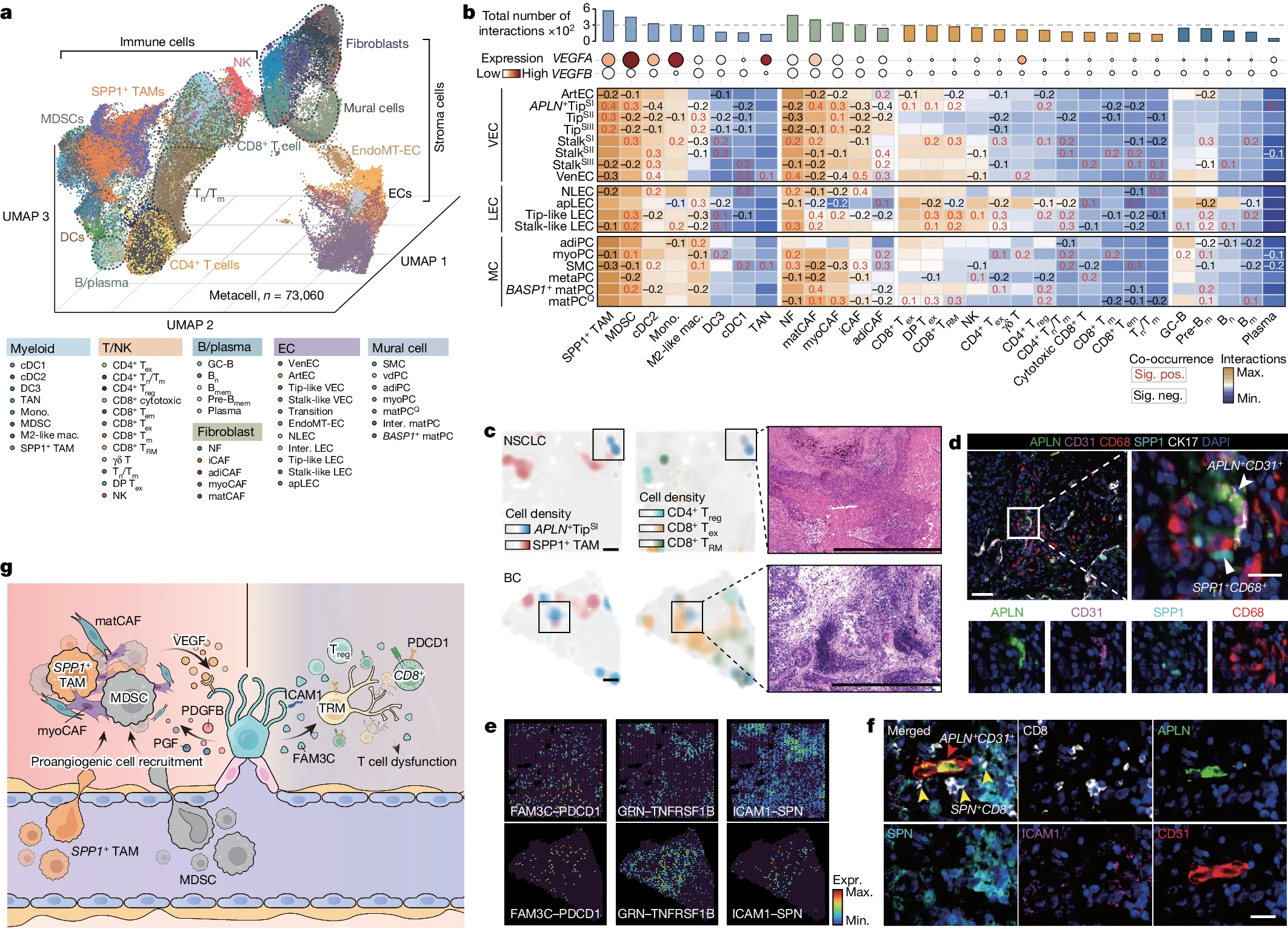
a,泛肿瘤 TME 和血管 meta 细胞的 UMAP 图,按注释的细胞身份着色。点代表由 SuperCell 生成的 meta 细胞。Bmem,记忆 B 细胞;DP,CD4+CD8+ 双阳性细胞;GC-B,生发中心 B 细胞;Mac.,巨噬细胞;Mono.,单核细胞;Pre-Bmem,前体记忆 B 细胞;TEM,效应记忆 T 细胞;Texh,耗竭 T 细胞;Tn/Tm,初始和记忆 T 细胞;Treg,调节性 T 细胞;TRM,驻留记忆 T 细胞;TAN,肿瘤相关中性粒细胞;NF,正常样成纤维细胞。b,TME 细胞与血管细胞之间的细胞 - 细胞相互作用。条形图显示了 TME 细胞的总 LR 相互作用数。热图显示了血管细胞与 TME 细胞之间的 LR 相互作用数量。热图上显示了显著的共现 Pearson 相关系数;显著正相关(sig. pos.,红色)和显著负相关(sig. neg.,黑色)。c,来自 NSCLC(n=1 例)和 BC(n=1 例)的空间转录组学数据中 APLN+ TipSI 细胞、SPP1+ TAMs 和耗竭 T 细胞的共定位。局部放大的苏木精和伊红染色图像。比例尺,1mm。d,来自 BC 的 APLN+ 尖端细胞和 SPP1+ TAMs 的代表性 mIHC 分析,来自 n=6 个独立生物复制品。箭头指示特定细胞类型。比例尺,左图为 50μm,右图为 15μm。e,空间图显示了免疫抑制相互作用 LR 对的表达强度。表达强度通过 LR 对的几何平均值计算。f,来自 NSCLC 的表达 ICAM1 的 APLN+ 尖端细胞和表达 SPN 的 CD8+ T 细胞的代表性 mIHC 分析,来自 n=5 个独立生物复制品。箭头指示特定细胞类型。比例尺,20μm。g,示意图显示了 APLN+ TipSI 细胞如何形成促血管生成的免疫抑制微环境。图 g 由 BioRender 创建。
除了 SPP1+ TAMs 和 BASP1+ matPCs 外,我们还发现其他与 APLN+ TipSI 细胞显著共存的促血管生成细胞,包括 MDSCs、myoCAFs 和 matCAFs。LR 分析表明,APLN+ TipSI 细胞不仅可以释放趋化因子 PDGFB 和 PGF 来招募基质促血管生成细胞(BASP1+ matPCs、myoCAFs 和 matCAFs),还可以通过 CD99-PILRA 促进髓系促血管生成细胞(SPP1+ TAMs 和 MDSCs)向肿瘤组织的渗出(扩展数据图 8c、9a 和 10a)。此外,mIHC 染色揭示了表达 PDGFB 的 APLN+ 尖端细胞和表达 PDGFRB 的 myoCAFs(aSMA+)的共定位(扩展数据图 10b)。这些促血管生成细胞在物理上也彼此接近,形成了一个支持性环境(扩展数据图 9e)。值得注意的是,APLN+ TipSI 细胞显示出对 T 细胞的免疫调节潜力。调节性 T 细胞(Treg)、驻留记忆 T 细胞和耗竭 T 细胞与 APLN+ TipSI 细胞共存(图 5c 和扩展数据图 9e)。APLN+ TipSI 细胞表达的配体 PODXL 和 CD34 可以与 Treg 细胞上的归巢受体 L-selectin(由 SELL 编码)相互作用,促进 Treg 细胞的肿瘤浸润(扩展数据图 10c)。此外,APLN+ TipSI 细胞可以通过 FAM3C-PDCD1、GRN-TNFRSF1B 和 ICAM1-SPN 配对诱导 T 细胞功能障碍和耗竭(图 5e)。mIHC 染色确认了在 NSCLC 中表达 ICAM1 的 APLN+ 尖端细胞和表达 SPN 的 CD8+ T 细胞的共定位(图 5f),这可能与细胞毒性 T 细胞的抑制有关。这种免疫抑制作用在 SII/SIII 新生血管和淋巴管中也持续存在(扩展数据图 10d)。这些结果表明,APLN+ TipSI 细胞有助于形成促血管生成的免疫抑制肿瘤微环境(TME)(图 5g)。
讨论¶
肿瘤血管生成是肿瘤的重要过程,但推动这一动态过程的内皮细胞的起源仍然是一个未解决的重要问题。在此,我们使用计算机模拟和体内模型证明了 VenECs 是血管生成起始的起点,这进一步提供了令人信服的证据,表明肿瘤血管生成起源于小静脉。在肿瘤血管生成开关启动后,APLN+ TipSI 细胞芽在低氧和 VEGF 信号的影响下从静脉内皮中出现。此外,虽然 APLN+ TipSI 细胞在肿瘤组织中的存在可以用作多种癌症类型中预后不良和疾病进展的指标,但它也为贝伐单抗介导的血管生成破坏提供了一个潜在的靶点。这一证据也提出了一个问题,即在未来贝伐单抗前瞻性随机试验中,肿瘤活检中增加的 APLN+ TipSI 细胞血管计数是否可以被认为是临床获益的预测生物标志物。炎症和低氧 TVM 激活了淋巴管生成。值得注意的是,我们发现了一种特定的 LEC 谱系,apLEC,其特异性表达抗炎 TF TSC22D3。最近的研究表明,TSC22D3 可以抑制 NF-κB 的 DNA 结合,并可以介导 HIF-1α的泛素依赖性降解。我们还分析了 MC 分类法,强调了异质的 matPCs。其中一种促血管生成的 matPC,以 BASP1 表达为特征,是由 ER 和氧化应激驱动的。这种 matPC 表现出降低的紧密连接水平,促进尖端细胞的萌芽。肿瘤浸润的 BASP1+ matPCs 与多种癌症的不良预后相关。总之,我们的综合血管手册将有助于肿瘤血管生物学的探索,并为潜在的临床意义和抗血管生成治疗的靶点提供计算资源。
数据可用性 本文报告的原始测序数据、处理过的单细胞 RNA 和空间转录组数据已存储在中国北京国家基因组数据中心的基因组序列档案中,BioProject 编号为 PRJCA018695。所存储和公开的数据符合中华人民共和国科学技术部的规定。整合的基因表达数据可以通过在线数据浏览器访问(http://resource.yin-lab.com/Panvascular/)。本研究中使用的公共单细胞 RNA 数据集和空间转录组数据见补充表 1,TCGA 数据集来自 https://portal.gdc.cancer.gov/。
代码可用性 自定义代码已存储在 GitHub(https://github.com/bio-Pixel/panVC)和 Zenodo91(https://zenodo.org/records/11188740)。