6.2 Cell-cell communication¶
摘要:本章简要概述了从单细胞转录组学数据推断细胞间通讯的基本概念和假设。我们提供了两种最常见的CCC推断方法的示例:一种是专注于配体-受体相互作用的,另一种是包括下游反应的。
Motivation¶
细胞通讯是细胞对其环境和自身刺激作出反应的过程。在多细胞生物中,细胞的动态协调,也称为细胞间通讯(CCC),参与许多生物过程,如凋亡和细胞迁移,因此在维持体内平衡和疾病过程中至关重要。CCC通常关注蛋白质介导的相互作用,最常见的是分泌的配体与其相应的质膜受体结合。然而,这一概念可以扩展到包括分泌酶、细胞外基质蛋白、转运蛋白以及需要细胞之间物理接触的相互作用,如细胞间粘附蛋白和间隙连接 {cite}armingol_2021。细胞通讯并非独立于其他过程,相反,外部刺激通常会引发下游反应。在CCC的情况下,这通常表现为接受信号的细胞,即受体细胞中经典通路和下游转录因子的诱导。最终,这些外部刺激会改变受体细胞的功能,并通过这些细胞与其微环境的后续相互作用进一步传播。
传统上,研究CCC需要专门的原位生化检测方法,如邻近标记蛋白质组学、共免疫沉淀和酵母双杂交筛选 {cite}armingol_2021。然而,转录组数据生成的快速发展和成本下降使研究重点从关注存在的细胞类型转向研究它们之间的关系 {cite}almet_2021。因此,从单细胞数据中推断CCC现在已成为一种常规方法,能够提供体内细胞间交叉对话的系统级假设。
Approaches¶
由于这种兴趣的增加,许多用于从单细胞转录组学推断CCC的计算工具应运而生,这些工具可以分为两类:一种是仅预测CCC相互作用的方法,通常称为配体-受体推断方法(例如 {cite}efremova_2020,jin_2021,raredon_2022,hou2020predicting),另一种是额外估计由CCC引起的细胞内活动的方法(例如 {cite}wang_2019,browaeys_2020,hu_2021)。这两类工具都使用基因表达信息作为蛋白质丰度的替代指标,通常需要将细胞聚类成具有生物学意义的组(参见注释教程)。这些CCC工具推断细胞群体间的细胞间交互,一组是CCC事件的源细胞群,另一组是受体细胞群。因此,CCC事件通常表示为源细胞群和受体细胞群表达的蛋白质之间的相互作用。
关于相互作用蛋白质的信息通常从先前的知识资源中提取。对于配体-受体方法,相互作用也可以由异源蛋白复合物表示,因为不同的亚基组合可以引发不同的反应,并且包括蛋白复合物信息已被证明可以降低假阳性率 {cite}efremova_2020,jin_2021,liu_2022。另一方面,建模细胞内信号传导的方法也利用了受体细胞类型的功能信息,因此需要额外的信息,如细胞内蛋白质-蛋白质相互作用网络和/或基因调控相互作用。
最近的研究强调了在使用不同工具时,方法和/或资源的选择导致推断预测的共识有限 {cite}dimitrov_2022,wang_2022,liu_2022,因此在解释其输出时需要谨慎。CCC领域还受到缺乏能够捕捉大量细胞和分子之间复杂且动态相互作用的真实数据的困扰 {cite}armingol_2021,almet_2021。尽管如此,独立评估表明,CCC方法对噪音的引入具有相当的鲁棒性 {cite}dimitrov_2022,wang_2022,liu_2022,并且在很大程度上与细胞内信号传导和空间信息等替代数据模式一致 {cite}dimitrov_2022,liu_2022。
在本章中,我们首先介绍并举例说明最常见和最简单的CCC方法,即使用CellPhoneDB {cite}efremova_2020 和 LIANA {cite}dimitrov_2022进行的配体-受体推断。然后,我们展示NicheNet作为一个重点关注CCC事件下游细胞内活动的CCC推断方法的示例 {cite}browaeys_2020。最后,我们强调了从单细胞转录组学数据推断CCC的常见假设和限制,并提出了提高细胞间通讯预测置信度的建议。需要注意的是,为了简单起见,我们在此进行了概括,然而,有许多不同的和新兴的CCC方法。在展望部分,我们强调了其中的一些方法。
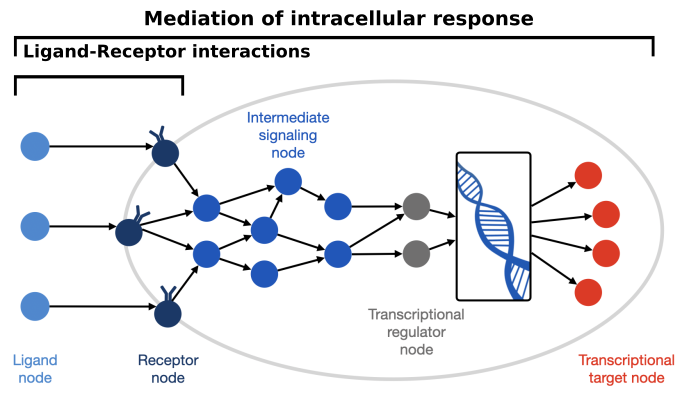
FIGURE 22.1. Cell-Cell Communication Overview
Environment setup¶
# python libs
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
import scanpy as sc
import liana as li
import decoupler as dc
import session_info
# Setting up R dependencies
import anndata2ri
import rpy2
from rpy2.robjects import r
import random
anndata2ri.activate()
%load_ext rpy2.ipython
%%R
suppressPackageStartupMessages({
library(reticulate)
library(ggplot2)
library(tidyr)
library(dplyr)
library(purrr)
library(tibble)
})
# figure settings
sc.settings.set_figure_params(dpi=200, frameon=False)
sc.set_figure_params(dpi=200, facecolor="white")
sc.set_figure_params(figsize=(5, 5))
Case Study¶
作为一个简单的示例,我们将研究来自8位狼疮患者的约25k个PBMCs(外周血单个核细胞),分别在IFN-β刺激前和刺激后 {cite}kang2018multiplexed。请注意,在本教程中,通过聚焦于PBMCs,我们假设在它们之间发生协调事件。
那么,首先让我们下载预处理数据。
# Read in
adata = sc.read(
"kang_counts_25k.h5ad", backup_url="https://figshare.com/ndownloader/files/34464122"
)
adata
# Store the counts for later use
adata.layers["counts"] = adata.X.copy()
应用基本的质量控制步骤,以去除任何低质量的细胞和低表达的基因。我们建议用户参阅 {ref}质量控制 章节,以获取更全面的质量控制步骤。
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
# Store the counts for later use
adata.layers["counts"] = adata.X.copy()
# Rename label to condition, replicate to patient
adata.obs = adata.obs.rename({"label": "condition", "replicate": "patient"}, axis=1)
# assign sample
adata.obs["sample"] = (
adata.obs["condition"].astype("str") + "&" + adata.obs["patient"].astype("str")
)
我们还将对数据进行标准化,以确保所有细胞的计数深度相等,因为我们需要基因表达值在不同细胞类型之间具有可比性。我们建议用户参阅 {ref}标准化 章节,以获取更多信息和可能更适合其数据的其他标准化方法。
# log1p normalize the data
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
在这个案例研究中,我们假设B细胞和CD4 T细胞扮演信号介质的角色,而其他细胞类型,如CD8 T细胞和自然杀伤细胞,则由执行响应的细胞组成。换句话说,我们将B细胞和CD4 T细胞视为CCC信号的来源,而后者则是CCC刺激的接收者。当然,这是一个过度简化的假设,因为信号源和接收者预期是动态和多方向的,因此我们将哪些细胞类型视为哪一类取决于我们所持的假设。
adata.obs["cell_type"].cat.categories
Index(['CD4 T cells', 'CD14+ Monocytes', 'B cells', 'NK cells', 'CD8 T cells',
'FCGR3A+ Monocytes', 'Dendritic cells', 'Megakaryocytes'],
dtype='object')
Show pre-computed UMAP, just to showcase the data
sc.pl.umap(adata, color=["condition", "cell_type"], frameon=False)
/home/dbdimitrov/anaconda3/envs/cellcell/lib/python3.10/site-packages/scanpy/plotting/_tools/scatterplots.py:364: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored /home/dbdimitrov/anaconda3/envs/cellcell/lib/python3.10/site-packages/scanpy/plotting/_tools/scatterplots.py:364: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored

Ligand-receptor inference¶
首先,让我们使用CellPhoneDB(v2)配体-受体方法 {cite}efremova_2020。
我们将在IFN-β刺激后的数据上运行CellPhoneDB,因为这些方法最初是为了在“稳态”数据中推断CCC事件而设计的,换句话说,它们旨在用于单一条件或样本,而不是跨样本或条件。尽管如此,某些方法确实存在可以跨条件应用配体-受体方法,但这些方法不在本教程的讨论范围内,我们将在展望部分提及这些方法。
adata_stim = adata[adata.obs["condition"] == "stim"].copy()
adata_stim
AnnData object with n_obs × n_vars = 12301 × 15701
obs: 'nCount_RNA', 'nFeature_RNA', 'tsne1', 'tsne2', 'condition', 'cluster', 'cell_type', 'patient', 'nCount_SCT', 'nFeature_SCT', 'integrated_snn_res.0.4', 'seurat_clusters', 'n_genes', 'sample'
var: 'name', 'n_cells'
uns: 'log1p', 'condition_colors', 'cell_type_colors'
obsm: 'X_pca', 'X_umap'
layers: 'counts'
# import cellphonedb method via liana
from liana.method import cellphonedb
CellPhoneDB是最常用的细胞间通讯(CCC)工具之一,它通过参与相互作用的蛋白质的平均基因表达来表示细胞间的通信事件。参与的蛋白质也可以形成异源复合物,在这种情况下,考虑的是亚基的最小基因表达。除了表达平均值之外,还通过打乱细胞群标签生成的零分布来确定相互作用的显著性。
请注意,我们是按细胞类型分组的,因此我们获得的CCC统计数据将反映先前预定义的细胞类型。
cellphonedb(
adata_stim, groupby="cell_type", use_raw=False, return_all_lrs=True, verbose=True
)
Using `.X`! Converting mat to CSR format 227 features of mat are empty, they will be removed.
/home/dbdimitrov/anaconda3/envs/cellcell/lib/python3.10/site-packages/pandas/core/indexing.py:1728: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
0.46 of entities in the resource are missing from the data. Generating ligand-receptor stats for 12301 samples and 15474 features
100%|██████████| 1000/1000 [00:22<00:00, 45.07it/s]
默认情况下,结果会写入anndata对象内的特定位置,更具体地说,是在 .uns['liana_res'] 中。
让我们检查一下CellPhoneDB方法的输出结果:
adata_stim.uns["liana_res"].head()
| ligand | ligand_complex | ligand_means | ligand_props | receptor | receptor_complex | receptor_means | receptor_props | source | target | lrs_to_keep | lr_means | cellphone_pvals | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | LGALS9 | LGALS9 | 0.072739 | 0.101973 | PTPRC | PTPRC | 0.457824 | 0.501761 | CD4 T cells | CD4 T cells | True | 0.265281 | 1.0 |
| 9 | LGALS9 | LGALS9 | 0.072739 | 0.101973 | CD44 | CD44 | 0.304042 | 0.364741 | CD4 T cells | CD4 T cells | True | 0.188391 | 1.0 |
| 37 | VIM | VIM | 0.507059 | 0.519725 | CD44 | CD44 | 0.304042 | 0.364741 | CD4 T cells | CD4 T cells | True | 0.405550 | 1.0 |
| 38 | PKM | PKM | 0.230863 | 0.278267 | CD44 | CD44 | 0.304042 | 0.364741 | CD4 T cells | CD4 T cells | True | 0.267453 | 1.0 |
| 66 | LGALS9 | LGALS9 | 0.072739 | 0.101973 | CD47 | CD47 | 0.213795 | 0.272807 | CD4 T cells | CD4 T cells | True | 0.143267 | 1.0 |
在这里,我们看到统计数据同时提供了配体和受体实体的详细信息,具体来说:
ligand和receptor通常是相互作用的两个实体。需要提醒的是,CCC事件不限于分泌信号,为简化起见,我们称其为ligand和receptor。在异源复合物的情况下,
ligand和receptor列表示最小表达的亚基,而*_complex则对应实际复合物,亚基由_分隔。source和target列分别表示每次相互作用的源/发送细胞和目标/接收细胞的身份。*_props: 表示表达该实体的细胞比例。默认情况下,在CellPhoneDB和LIANA中,任何实体在每种细胞类型中表达不超过10%的相互作用都被认为是假阳性,因为假设CCC发生在细胞类型之间,细胞内的足够比例应表达这些基因。
*_means: 每种细胞类型的实体表达均值。lr_means: 配体-受体表达均值,作为配体-受体相互作用的 强度 的度量。cellphone_pvals: 基于置换的p值,作为相互作用 特异性 的度量。
请注意,ligand、receptor、source 和 target 列由每个配体-受体方法返回,而其余列则因配体-受体方法而异,因为每种方法依赖于不同的假设和评分函数,因此每种方法返回不同的配体-受体评分。不过,通常大多数方法使用一对评分函数 - 其中一个通常对应于相互作用的 强度 (magnitude),另一个反映特定相互作用对某对细胞身份的 特异性 (specificity)。
Visual exploration¶
现在,我们可以将刚刚获得的结果可视化为点图,其中行表示源/发送(顶部)和目标/接收(底部)细胞类型之间的优先相互作用。
li.pl.dotplot(
adata=adata_stim,
colour="lr_means",
size="cellphone_pvals",
inverse_size=True, # we inverse sign since we want small p-values to have large sizes
# We choose only the cell types which we wish to plot
source_labels=["CD4 T cells", "B cells", "FCGR3A+ Monocytes"],
target_labels=["CD8 T cells", "CD14+ Monocytes", "NK cells"],
# since cpdbv2 suggests using a filter to FPs
# we can filter the interactions according to p-values <= 0.01
filterby="cellphone_pvals",
filter_lambda=lambda x: x <= 0.01,
# as this type of methods tends to result in large numbers
# of predictions, we can also further order according to
# expression magnitude
orderby="lr_means",
orderby_ascending=False, # we want to prioritize those with highest expression
top_n=20, # and we want to keep only the top 20 interactions
figure_size=(9, 5),
size_range=(1, 6),
)

<ggplot: (8783502753003)>
很好,我们得到了一些可能与IFN-β刺激相关的相互作用。
我们还可以看到,相互作用的强度(表达强度)和特异性都依赖于细胞类型。例如,HLA-B与CD8A/B的潜在结合显然只在接收细胞是CD8 T细胞时发生。
Generating a Ligand-Receptor consensus with LIANA¶
鉴于不同配体-受体方法推断的相互作用之间报告的有限一致性,为了进一步增加对潜在感兴趣相互作用的信心,可以检查该相互作用是否被多个方法预测为相关。以同样的方式,也可以使用多种方法并关注它们的共识,换句话说,关注一致预测为相关的相互作用。为此,我们将运行liana的 rank_aggregate 方法 {cite}dimitrov_2022,该方法生成跨方法高度排序相互作用的概率分布。
Let's first examine the ligand-receptor methods in LIANA:
li.method.show_methods()
| Method Name | Magnitude Score | Specificity Score | Reference | |
|---|---|---|---|---|
| 0 | CellPhoneDB | lr_means | cellphone_pvals | Efremova, M., Vento-Tormo, M., Teichmann, S.A.... |
| 0 | Connectome | expr_prod | scaled_weight | Raredon, M.S.B., Yang, J., Garritano, J., Wang... |
| 0 | log2FC | None | lr_logfc | Dimitrov, D., Türei, D., Garrido-Rodriguez, M.... |
| 0 | NATMI | expr_prod | spec_weight | Hou, R., Denisenko, E., Ong, H.T., Ramilowski,... |
| 0 | SingleCellSignalR | lrscore | None | Cabello-Aguilar, S., Alame, M., Kon-Sun-Tack, ... |
| 0 | CellChat | lr_probs | cellchat_pvals | Jin, S., Guerrero-Juarez, C.F., Zhang, L., Cha... |
| 0 | Rank_Aggregate | magnitude_rank | specificity_rank | Dimitrov, D., Türei, D., Garrido-Rodriguez, M.... |
| 0 | Geometric Mean | lr_gmeans | gmean_pvals | CellPhoneDBv2's permutation approach applied t... |
现在让我们运行Rank_Aggregate方法,该方法本质上将在后台运行其他方法,然后生成共识结果。
from liana.method import rank_aggregate
rank_aggregate(
adata_stim, groupby="cell_type", return_all_lrs=True, use_raw=False, verbose=True
)
Using `.X`! Converting mat to CSR format 227 features of mat are empty, they will be removed.
/home/dbdimitrov/anaconda3/envs/cellcell/lib/python3.10/site-packages/pandas/core/indexing.py:1728: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
0.46 of entities in the resource are missing from the data. Generating ligand-receptor stats for 12301 samples and 15474 features Assuming that counts were `natural` log-normalized! Running CellPhoneDB
100%|██████████| 1000/1000 [00:05<00:00, 171.04it/s]
Running Connectome Running log2FC Running NATMI Running SingleCellSignalR Running CellChat
100%|██████████| 1000/1000 [01:59<00:00, 8.35it/s]
Let's now check how the output of liana's rank_aggregate:
adata_stim.uns["liana_res"].drop_duplicates(
["ligand_complex", "receptor_complex"]
).head()
| source | target | ligand_complex | receptor_complex | lr_means | cellphone_pvals | expr_prod | scaled_weight | lr_logfc | spec_weight | lrscore | lr_probs | cellchat_pvals | steady_rank | specificity_rank | magnitude_rank | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1129 | CD8 T cells | CD8 T cells | B2M | CD3D | 2.562213 | 0.0 | 3.070147 | 1.070524 | 0.690730 | 0.062383 | 0.982125 | 0.123504 | 0.0 | 2.092188e-11 | 1.415831e-09 | 9.840017e-13 |
| 831 | CD8 T cells | NK cells | B2M | KLRD1 | 2.511733 | 0.0 | 2.622731 | 1.541763 | 0.858930 | 0.080626 | 0.980689 | 0.110393 | 0.0 | 2.092188e-11 | 1.415831e-09 | 8.965786e-11 |
| 468 | FCGR3A+ Monocytes | CD14+ Monocytes | TIMP1 | CD63 | 1.877567 | 0.0 | 3.374182 | 1.478816 | 1.808744 | 0.123245 | 0.982935 | 0.136754 | 0.0 | 2.092188e-11 | 1.415831e-09 | 9.348292e-09 |
| 1513 | CD8 T cells | FCGR3A+ Monocytes | B2M | LILRB2 | 2.402974 | 0.0 | 1.658766 | 1.466587 | 0.604138 | 0.078486 | 0.975838 | 0.063762 | 0.0 | 2.092188e-11 | 1.415831e-09 | 1.935320e-08 |
| 836 | CD8 T cells | NK cells | B2M | CD247 | 2.414776 | 0.0 | 1.763377 | 1.156763 | 0.644404 | 0.074642 | 0.976548 | 0.041130 | 0.0 | 2.092188e-11 | 1.415831e-09 | 1.228218e-07 |
在这里,我们可以看到所有方法的评分函数的输出(如果有兴趣,可以参考上表了解每个评分对应的方法)。更重要的是,我们还获得了每个相互作用的 magnitude_rank 和 specificity_rank,它们分别代表共识相互作用的 强度(表达强度)和 特异性(在所有细胞类型对之间)。例如,回到CellPhoneDB,lr_mean 和 cellphone_pvals 将相应地聚合到这些共识排名中。
Let's generate the same plot, but now using the aggregate of the methods:
li.pl.dotplot(
adata=adata_stim,
colour="magnitude_rank",
size="specificity_rank",
inverse_colour=True, # we inverse sign since we want small p-values to have large sizes
inverse_size=True,
# We choose only the cell types which we wish to plot
source_labels=["CD4 T cells", "B cells", "FCGR3A+ Monocytes"],
target_labels=["CD8 T cells", "CD14+ Monocytes", "NK cells"],
# since the rank_aggregate can also be interpreted as a probability distribution
# we can again filter them according to their specificity significance
# yet here the interactions are filtered according to
# how consistently highly-ranked is their specificity across the methods
filterby="specificity_rank",
filter_lambda=lambda x: x <= 0.05,
# again, we can also further order according to magnitude
orderby="magnitude_rank",
orderby_ascending=True, # prioritize those with lowest values
top_n=20, # and we want to keep only the top 20 interactions
figure_size=(9, 5),
size_range=(1, 6),
)

<ggplot: (8783489520283)>
尽管CellPhoneDB和LIANA优先选择的相互作用在生物学上似乎合理且可能与治疗相关,但要确定其相关性仍然具有挑战性。特别是,这些方法以无假设的方式生成系统级洞见的优势恰好也是它们的主要缺点之一。具体来说,配体-受体工具返回每对细胞类型的所有合理的配体-受体相互作用,因此我们最终会得到大量的相互作用列表,选择目标进行后续实验验证可能会很困难。
因此,在进行实验验证之前,我们建议任何潜在的相互作用假设都应得到额外的先验知识支持。这可能包括特定条件的领域知识(例如,感兴趣的细胞类型或受体),以及其他模式,如蛋白质丰度、空间共定位或下游信号传导。为此,我们还建议读者参考 {ref}富集分析 和 空间CCC(待添加)章节。
Modelling differential intercellular signalling with NicheNet¶
NicheNet是一种考虑细胞间相互作用引发的细胞内信号效应的CCC方法 {cite}browaeys_2020。
简而言之,NicheNet推断配体和它们可能调节的下游靶标之间的关联 {cite}browaeys_2020。换句话说,NicheNet假设某种发送/来源细胞类型产生配体,该配体与特定接收/目标细胞类型结合后,导致信号传播,影响主基因调控因子或转录因子,进而影响它们的靶标。因此,关于哪些靶基因受哪些配体-受体对调节的预测可能提供有关正在进行的CCC事件的有趣假设。此外,特定配体-受体对的靶基因在接收细胞类型中的富集也可以表明该配体-受体对在功能上是活跃的。总之,NicheNet分析可以通过以下方式进行:1)推断表达的配体-受体相互作用的潜在靶基因;2)根据它们在接收者中的靶基因富集优先排序配体-受体相互作用。这种富集被称为“配体活性”,类似于转录因子活性 {ref}enrichment-analysis。
为了执行这些任务,NicheNet利用了关于配体-靶标关联的先验知识。然而,与配体-受体数据库不同的是,全面的配体-靶标数据库并不存在。因此,NicheNet通过整合覆盖配体-受体、细胞内信号传导和基因调控相互作用的三层先验知识来预测配体-靶标关联。使用这三层信息,NicheNet为每个配体-靶标链接计算一个调控潜力分数。这种调控潜力表示先验知识支持配体可能调节靶基因表达的程度。为了计算调控潜力,NicheNet首先在综合信号网络上应用一种称为个性化PageRank(PPR)的网络扩散算法,以估计给定配体可能向特定调控因子传递信号的概率。具体来说,NicheNet中的PPR实现将给定配体视为感兴趣的种子节点。在此假设下,信号网络中离配体较近的节点比较远的节点得分更高,因为假设在配体附近的网络调控因子更可能受配体调节。对数据库中所有配体应用PPR的结果是一个配体-调控因子的信号概率矩阵,然后将其与调控因子到靶基因的权重矩阵相乘,以获得配体-靶标调控潜力分数 {cite}browaeys_2020。概念上,这意味着如果配体能够向靶基因的调控因子传递信号,则配体-靶标对会获得较高的调控潜力分数。这些先验知识衍生的分数与交互细胞的表达数据结合使用,以优先排序配体-受体相互作用并预测其靶基因。
因为NicheNet专注于配体如何影响潜在交互细胞的基因表达,所以需要能够定义哪些基因表达变化可能(部分)由CCC过程引起。为了根据配体在接收细胞类型中的潜在配体活性对配体进行排序,需要一组假设受接收细胞类型中的CCC事件影响的基因。NicheNet的作者建议在处理不同条件(例如治疗与对照)时理想地定义这些基因集。为此,我们将以这种方式应用NicheNet,并比较数据中患者在IFN-β刺激前后的表达。
有关NicheNet方法的更详细描述,我们特别建议读者参考 {ref}enrichment-analysis 章节的足迹部分以及NicheNet的教程和论文 {cite}browaeys_2020。
Load NicheNet Prior-Knowledge¶
如上所述,NicheNet需要关于配体-受体相互作用和配体-靶标链接的先验知识:
ligand_target_matrix - 表示配体可能调控靶基因表达的潜力。该矩阵中的权重基于先验知识,并且需要用于优先排序可能的配体-受体相互作用和受影响的靶基因。
lr_network - 配体-受体相互作用的数据库,用于定义表达的配体、受体及其相互作用。
我们将从Zenodo加载这些数据 ![]() (可能需要几分钟)。
(可能需要几分钟)。
%%R
# load NicheNet (NicheNet is only available on GitHub)
suppressPackageStartupMessages({
if(!require(nichenetr)) remotes::install_github("saeyslab/nichenetr", upgrade = "never")
})
%%R
# Increase timeout threshold
options(timeout=600)
# Load PK
ligand_target_matrix <- readRDS(url("https://zenodo.org/record/7074291/files/ligand_target_matrix_nsga2r_final.rds"))
lr_network <- readRDS(url("https://zenodo.org/record/7074291/files/lr_network_human_21122021.rds"))
此外,可以使用 OmnipathR 包根据手头的数据量身定制NicheNet的先验知识,并对网络进行上下文化处理。
步骤 1. 定义感兴趣的细胞类型,作为CCC相互作用的发送方/源和接收方/靶¶
在这里,我们假设多个细胞类型能够影响接收细胞类型
sender_celltypes = ["CD4 T cells", "B cells", "FCGR3A+ Monocytes"]
receiver_celltypes = ["CD8 T cells"]
步骤 2. 定义一组可能影响接收细胞类型的配体¶
与上述的配体-受体方法类似,这里我们只关注涉及每种细胞类型中充分表达基因的潜在相互作用。因此,我们假设例如10%的细胞是一个良好的阈值,反映出细胞类型内部基因的表达情况。
# Helper function to obtain sufficiently expressed genes
from functools import reduce
def get_expressed_genes(adata, cell_type, expr_prop):
# calculate proportions
temp = adata[adata.obs["cell_type"] == cell_type, :]
a = temp.X.getnnz(axis=0) / temp.X.shape[0]
stats = (
pd.DataFrame({"genes": temp.var_names, "props": a})
.assign(cell_type=cell_type)
.sort_values("genes")
)
# obtain expressed genes
stats = stats[stats["props"] >= expr_prop]
expressed_genes = stats["genes"].values
return expressed_genes
sender_expressed = reduce(
np.union1d,
[
get_expressed_genes(adata, cell_type=cell_type, expr_prop=0.1)
for cell_type in sender_celltypes
],
)
receiver_expressed = reduce(
np.union1d,
[
get_expressed_genes(adata, cell_type=cell_type, expr_prop=0.1)
for cell_type in receiver_celltypes
],
)
Then use this information to keep only ligand-receptor pairs in the NicheNet network that are expressed.
%%R -i sender_expressed -i receiver_expressed
# get ligands and receptors in the resource
ligands <- lr_network %>% pull(from) %>% unique()
receptors <- lr_network %>% pull(to) %>% unique()
# only keep the intersect between the resource and the data
expressed_ligands <- intersect(ligands, sender_expressed)
expressed_receptors <- intersect(receptors, receiver_expressed)
# filter the network to only include ligands for which both the ligand and receptor are expressed
potential_ligands <- lr_network %>%
filter(from %in% expressed_ligands & to %in% expressed_receptors) %>%
pull(from) %>% unique()
步骤 3. 在接收细胞类型中定义感兴趣的基因集¶
这一步是NicheNet分析中最关键的一步。在这里,需要定义哪些基因可能受细胞间通信的调节,即受配体信号传导影响的基因。例如,可以假设在接收细胞类型之间的不同条件下表达差异的基因是由一个或多个发送细胞群体的配体驱动的。另一个例子可能是在分化细胞群体和祖细胞群体之间的基因差异表达,如果分化可能是通过与其他细胞类型的相互作用诱导的。
So, 我们现在将使用 decoupler 为每个细胞类型和样本生成伪批量(pseudobulk)文件,并对其进行差异表达分析。伪批量的一个关键步骤是过滤掉大部分细胞和样本中未表达的基因,因为这些基因非常嘈杂,可能导致不稳定的对数倍变化。为了获得稳健的文件,基因会被过滤掉,如果它们在每个样本中的表达不足(min_prop),以及在不足的样本中表达。我们建议读者参考{ref}normalization 和 {ref}differential-analysis章节,获取更多关于差异表达分析和伪批量文件的信息。
# Get pseudo-bulk profile
pdata = dc.get_pseudobulk(
adata,
sample_col="sample",
groups_col="cell_type",
min_prop=0.1,
min_smpls=3,
layer="counts",
)
Normalize the pseudobulk counts
# Storing the raw counts
pdata.layers["counts"] = pdata.X.copy()
# Does PC1 captures a meaningful biological or technical fact?
pdata.obs["lib_size"] = pdata.X.sum(1)
# Normalize
sc.pp.normalize_total(pdata, target_sum=1e4)
sc.pp.log1p(pdata)
# check how this looks like
pdata
AnnData object with n_obs × n_vars = 108 × 4412
obs: 'condition', 'cell_type', 'patient', 'sample', 'lib_size'
uns: 'log1p'
layers: 'counts'
然后我们进行非常简单的差异分析对比。在这个例子中,我们将使用在 scanpy 中实现的 t-test,但我们也可以使用其他任何方法。
logFCs, pvals = dc.get_contrast(
pdata,
group_col="cell_type",
condition_col="condition",
condition="stim",
reference="ctrl",
method="t-test",
)
然后保留在接收细胞类型中显著上调的差异表达基因。
# Visualize those for e.g. CD14+ Monocytes
dc.plot_volcano(logFCs, pvals, "CD14+ Monocytes", top=15, sign_thr=0.05, lFCs_thr=1)

# format results
deg = dc.format_contrast_results(logFCs, pvals)
# only keep the receiver cell type(s)
deg = deg[np.isin(deg["contrast"], receiver_celltypes)]
deg.head()
| contrast | name | logFCs | pvals | adj_pvals | |
|---|---|---|---|---|---|
| 13236 | CD8 T cells | SAMD9L | 3.592979 | 1.818029e-10 | 0.000001 |
| 13237 | CD8 T cells | IFIT3 | 6.486142 | 6.843872e-10 | 0.000002 |
| 13238 | CD8 T cells | IFI6 | 5.555549 | 1.835813e-09 | 0.000003 |
| 13239 | CD8 T cells | OAS1 | 5.479982 | 7.165866e-09 | 0.000008 |
| 13240 | CD8 T cells | IFIT1 | 6.617856 | 1.149598e-08 | 0.00001 |
现在我们已经获得了接收细胞类型的差异表达统计数据,我们可以使用这些数据来定义NicheNet中配体活性分析的背景和感兴趣的基因集。
# define background of sufficiently expressed genes
background_genes = deg["name"].values
# only keep significant and positive DE genes
deg = deg[(deg["pvals"] <= 0.05) & (deg["logFCs"] > 1)]
# get geneset of interest
geneset_oi = deg["name"].values
步骤 4. NicheNet 配体活性估计¶
为了估计配体活性,NicheNet利用基因的调控潜力(基于先验知识)来预测哪种配体最能预测我们为接收细胞类型定义的感兴趣基因集。
换句话说,它评估具有特定配体高调控潜力的基因,是否更可能属于我们为接收细胞类型推导出的感兴趣基因集。
从概念上讲,这与标准的{ref}enrichment-analysis并不完全不同,NicheNet提出了不同的方法来估计配体活性,例如接收操作特征曲线下面积或Pearson相关性 {cite}browaeys_2020。
%%R -i geneset_oi -i background_genes -o ligand_activities
ligand_activities <- predict_ligand_activities(geneset = geneset_oi,
background_expressed_genes = background_genes,
ligand_target_matrix = ligand_target_matrix,
potential_ligands = potential_ligands)
ligand_activities <- ligand_activities %>%
arrange(-aupr) %>%
mutate(rank = rank(desc(aupr)))
# show top10 ligand activities
head(ligand_activities, n=10)
# A tibble: 10 × 6 test_ligand auroc aupr aupr_corrected pearson rank <chr> <dbl> <dbl> <dbl> <dbl> <dbl> 1 PTPRC 0.801 0.116 0.0882 0.168 1 2 HLA-F 0.781 0.104 0.0765 0.188 2 3 HLA-A 0.776 0.0941 0.0662 0.180 3 4 HLA-B 0.766 0.0864 0.0586 0.162 4 5 HLA-E 0.767 0.0856 0.0578 0.129 5 6 CCL5 0.762 0.0812 0.0534 0.0527 6 7 CD48 0.762 0.0786 0.0508 0.106 7 8 B2M 0.753 0.0755 0.0477 0.121 8 9 HLA-DRA 0.730 0.0694 0.0416 0.111 9 10 CXCL10 0.736 0.0678 0.0400 0.0803 10
Step 5. Infer & Visualize top-predicted target genes for top ligands¶
%%R -o vis_ligand_target
top_ligands <- ligand_activities %>%
top_n(15, aupr) %>%
arrange(-aupr) %>%
pull(test_ligand) %>%
unique()
# get regulatory potentials
ligand_target_potential <- map(top_ligands,
~get_weighted_ligand_target_links(.x,
geneset = geneset_oi,
ligand_target_matrix = ligand_target_matrix,
n = 500)
) %>%
bind_rows() %>%
drop_na()
# prep for visualization
active_ligand_target_links <-
prepare_ligand_target_visualization(ligand_target_df = ligand_target_potential,
ligand_target_matrix = ligand_target_matrix)
# order ligands & targets
order_ligands <- intersect(top_ligands,
colnames(active_ligand_target_links)) %>% rev() %>% make.names()
order_targets <- ligand_target_potential$target %>%
unique() %>%
intersect(rownames(active_ligand_target_links)) %>%
make.names()
rownames(active_ligand_target_links) <- rownames(active_ligand_target_links) %>%
make.names() # make.names() for heatmap visualization of genes like H2-T23
colnames(active_ligand_target_links) <- colnames(active_ligand_target_links) %>%
make.names() # make.names() for heatmap visualization of genes like H2-T23
vis_ligand_target <- active_ligand_target_links[order_targets, order_ligands] %>%
t()
# convert to dataframe, and then it's returned to py
vis_ligand_target <- vis_ligand_target %>%
as.data.frame() %>%
rownames_to_column("ligand") %>%
as_tibble()
# convert dot to underscore and set ligand as index
vis_ligand_target["ligand"] = vis_ligand_target["ligand"].replace("\.", "_", regex=True)
vis_ligand_target.set_index("ligand", inplace=True)
# keep only columns where at least one gene has a regulatory potential >= 0.05
vis_ligand_target = vis_ligand_target.loc[
:, vis_ligand_target[vis_ligand_target >= 0.05].any()
]
vis_ligand_target.head()
| IFI16 | IFIH1 | IFIT2 | IRF7 | MT2A | MX1 | NMI | PARP14 | PARP9 | SOCS1 | TNFSF10 | EPSTI1 | HERC6 | LAP3 | PARP12 | TMEM140 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ligand | ||||||||||||||||
| ICAM1 | 0.010111 | 0.008649 | 0.010065 | 0.013067 | 0.007855 | 0.007761 | 0.000000 | 0.007268 | 0.0000 | 0.008907 | 0.012580 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.00000 |
| CXCL11 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0000 | 0.000000 | 0.007285 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.00000 |
| HLA_DQB1 | 0.000000 | 0.000000 | 0.000000 | 0.007094 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0000 | 0.000000 | 0.007733 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.00000 |
| HMGB1 | 0.158681 | 0.077152 | 0.151255 | 0.091884 | 0.075502 | 0.074791 | 0.147717 | 0.075350 | 0.0745 | 0.078945 | 0.082315 | 0.074556 | 0.073812 | 0.145847 | 0.147686 | 0.07226 |
| ITGB2 | 0.007564 | 0.000000 | 0.006784 | 0.009015 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0000 | 0.000000 | 0.008748 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.00000 |
Visualize top ligands & regulatory targets¶
fig, ax = plt.subplots(1, 1, figsize=(15, 5))
sns.heatmap(vis_ligand_target, xticklabels=True, ax=ax)
plt.show()

Perfect, we end up with ligands that are most probable to affect downstream signalling in the receiver cell type(s), as well as their most likely targets.
Combining NicheNet output with ligand-receptor inference¶
NicheNet和配体-受体方法并不是互斥的,而是互补的,因为它们以不同的方式解决不同的问题。配体-受体方法,例如我们上面展示的方法,通过分析配体和受体的表达来推断配体-受体相互作用,通常适用于“稳态”数据。相反,NicheNet基于目标细胞类型中诱导的基因表达变化,预测这些推断的配体-受体链接可能是最功能性的,这一过程主要依赖于先验知识。因此,虽然工具选择取决于研究问题,但人们也可以将配体-受体方法(例如CellPhoneDB或LIANA)和细胞内信号CCC方法(例如NicheNet)视为互补的。
例如,鉴于我们优先考虑的前三个配体,我们可以集中关注包含这些配体和我们用来推断活跃配体的接收细胞类型的配体-受体对。
因此,让我们看看哪些配体-受体相互作用涉及NicheNet中排名前五的潜在活跃配体,并且被LIANA中不同方法一致地优先考虑为相关的。
ligand_oi = ligand_activities.head(3)["test_ligand"].values
ligand_oi
array(['PTPRC', 'HLA-F', 'HLA-A'], dtype=object)
li.pl.dotplot(
adata=adata_stim,
colour="lr_means",
size="cellphone_pvals",
inverse_size=True, # we inverse sign since we want small p-values to have large sizes
# We choose only the cell types which we wish to plot
source_labels=sender_celltypes,
target_labels=receiver_celltypes,
# keep only those ligands
filterby="ligand_complex",
filter_lambda=lambda x: np.isin(x, ligand_oi),
# as this type of methods tends to result in large numbers
# of predictions, we can also further order according to
# expression magnitude
orderby="magnitude_rank",
orderby_ascending=False, # we want to prioritize those with highest expression
top_n=25, # and we want to keep only the top 25 interactions
figure_size=(9, 9),
size_range=(1, 6),
)

<ggplot: (8783461218238)>
从上面的结果中,我们现在可以看到与NicheNet推断的配体活性相关的具体配体-受体相互作用。此外,我们可以假设,NicheNet推断的配体活性可能通过特定的细胞类型对之间的配体-受体相互作用(例如 HLA-A -> CD3G)影响细胞类型。换句话说,虽然相同的配体可能是由于IFN-β刺激的结果而活跃的,但它们可能引起特定细胞类型的信号传导变化。
虽然在这个案例中我们使用了NicheNet和LIANA,但一些最近的细胞间通信工具开发结合了这两类细胞间通信推断方法中的各种思想和假设(例如 {cite}zhang2021cellcall,zhang2021cellinker,baruzzo2022identify)。
Key takeaways¶
假设与限制¶
本文考虑的方法的共同目的是利用单细胞转录组数据预测不同细胞类型之间最相关的细胞间相互作用。因此,所有方法都假设不同细胞类型的基因表达是采样组织内发生的细胞间通信事件的信息代理。尽管单细胞转录组学使得以前无法预料地推断细胞间通信事件成为可能,但仍需考虑一些假设和限制。
首先,这些方法假设蛋白共表达反映细胞间相互作用,并因此反映任何先于相互作用的事件,包括蛋白翻译和加工、分泌和扩散 {cite}armingol_2021,dimitrov_2022(图22.2)。此外,如果将生物体内的细胞间通信概念化为发生在不同“长度尺度”或范围内的细胞间通信事件的组合 {cite}palla2022spatial,那么从单细胞转录组学中推断出的细胞间通信事件可能仅限于发生在“局部”范围内的蛋白介导事件,即采样的细胞类型之间的事件。因此,长距离信号传导和由其他分子介导的细胞间通信,如内分泌信号和系统梯度(如钙和氧浓度),可能无法被捕获 {cite}dimitrov_2022。
此外,尽管按其谱系将细胞类型分组是结构化数据的常见做法,但考虑到组织是通信发生的地方,细胞间的相互作用不一定发生在细胞类型之间,而更可能是单个细胞之间 {cite}wilk_2022。此外,细胞间相互作用通常被呈现为细胞类型和/或蛋白质之间的一对一事件。因此,跨细胞类型的潜在共表达事件不一定反映真正的信号传递,因此,为了基本上捕捉社区内事件,我们必须确定细胞之间传递的消息的幅度、方向性和生物学相关性 {cite}armingol_2021。
总结与展望:¶
在本章中,我们介绍了两种细胞间通信推断方法的应用。具体来说,我们使用了CellPhoneDB和LIANA来预测从单一上下文(或稳态)数据中得出的相关配体-受体相互作用,同时使用NicheNet在差异表达的背景下推断潜在的活跃配体及其靶标。
随着单细胞领域的关注点进一步从谱系定义转向在不同条件下细胞类型内部变化的表征,解开不同上下文中细胞间通信洞见的方法变得至关重要。因此,除了NicheNet之外,我们还建议用户参考其他能够进行跨条件比较的方法,如NATMI的差异细胞连接分析 {cite}hou2020predicting,Crosstalker的网络拓扑测量 {cite}nagai2021crosstalker,CellChat的路径集中的流形学习 {cite}jin_2021,以及Tensor-cell2cell的非靶向因子分解方法来推断不同上下文中的细胞间通信模式 {cite}armingol_2022。
由于单细胞和细胞间通信领域的持续发展,涌现了越来越多的方法,其中一些提出了替代方法来预测细胞间通信事件,如那些在单细胞分辨率下工作的方法 {cite}raredon_2023,wang_2019,wilk_2022。而其他方法则试图解决上述的一些限制,例如通过包括推断由代谢物或小分子介导的相互作用 {cite}zheng_2022,garciaalonso_2022,zhang_2021。
尽管本章未涵盖分离的单细胞数据中细胞间通信领域的多样性和所有当前的发展,但它旨在突出一些一般的思想和限制。因此,我们希望它作为一个起点,读者可以通过结合其他章节的知识以及深入阅读细胞间通信领域本身的文献来进一步扩展。
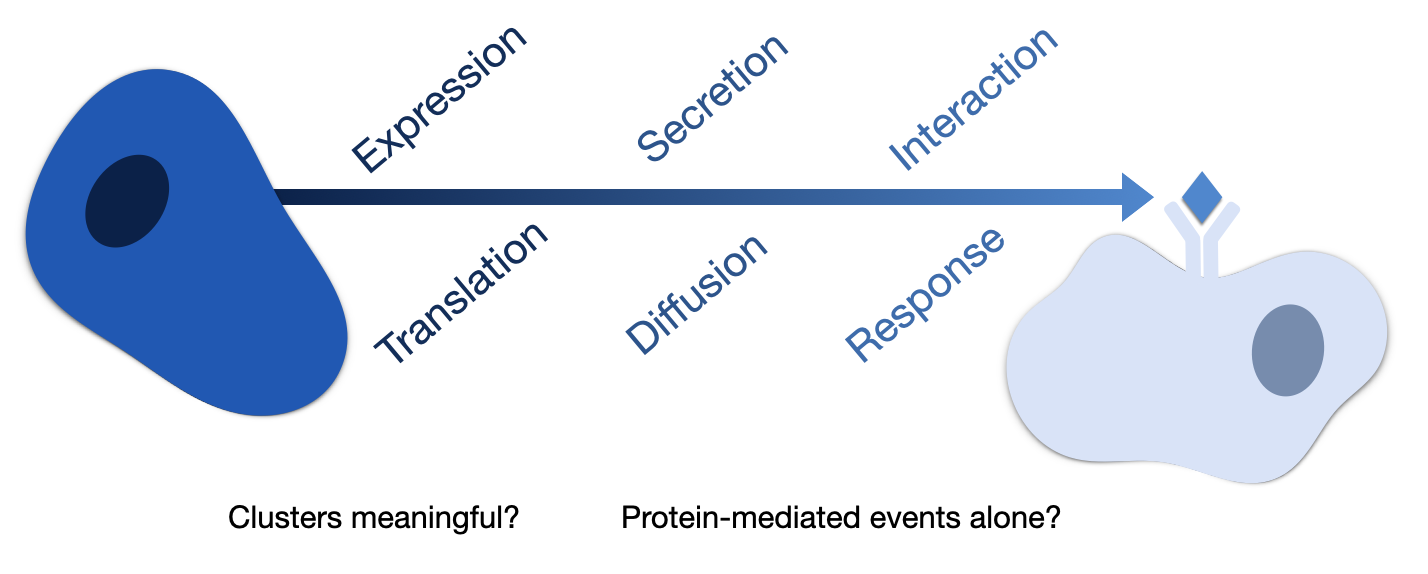
FIGURE 22.2. Assumptions and Limitations of Cell-Cell Communication from single-cell transcriptomics data
Quiz¶
- Q1. What are the first three limitations that come to mind when inferring CCC from single-cell transcriptomics data?
- Q2. What is the role of heteromeric complexes in ligand-receptor inference?
- Q3. Can you think of an advantage and a disadvantage of inference based on prior-knowledge?
- Q4: Can you think of an important limitation we should consider here regarding the definition of the geneset of interest for the NicheNet analysis on the IFN-beta stimulated PBMC dataset?
Session Info¶
%%R
sessionInfo()
R version 4.2.2 (2022-10-31) Platform: x86_64-conda-linux-gnu (64-bit) Running under: Ubuntu 20.04.5 LTS Matrix products: default BLAS/LAPACK: /home/dbdimitrov/anaconda3/envs/cellcell/lib/libopenblasp-r0.3.21.so locale: [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C [3] LC_TIME=de_DE.UTF-8 LC_COLLATE=en_US.UTF-8 [5] LC_MONETARY=de_DE.UTF-8 LC_MESSAGES=en_US.UTF-8 [7] LC_PAPER=de_DE.UTF-8 LC_NAME=C [9] LC_ADDRESS=C LC_TELEPHONE=C [11] LC_MEASUREMENT=de_DE.UTF-8 LC_IDENTIFICATION=C attached base packages: [1] tools stats graphics grDevices utils datasets methods [8] base other attached packages: [1] nichenetr_1.1.1 tibble_3.1.8 purrr_1.0.1 dplyr_1.0.9 [5] tidyr_1.2.0 ggplot2_3.4.0 reticulate_1.25 loaded via a namespace (and not attached): [1] backports_1.4.1 circlize_0.4.15 Hmisc_4.7-2 [4] plyr_1.8.8 igraph_1.3.5 lazyeval_0.2.2 [7] sp_1.6-0 splines_4.2.2 listenv_0.9.0 [10] scattermore_0.8 digest_0.6.31 foreach_1.5.2 [13] htmltools_0.5.4 fansi_1.0.3 checkmate_2.1.0 [16] magrittr_2.0.3 tensor_1.5 cluster_2.1.4 [19] doParallel_1.0.17 ROCR_1.0-11 limma_3.54.0 [22] tzdb_0.3.0 recipes_1.0.4 ComplexHeatmap_2.14.0 [25] globals_0.16.2 readr_2.1.2 gower_1.0.1 [28] matrixStats_0.63.0 hardhat_1.2.0 timechange_0.2.0 [31] spatstat.sparse_3.0-0 jpeg_0.1-10 colorspace_2.0-3 [34] ggrepel_0.9.2 xfun_0.36 crayon_1.5.2 [37] jsonlite_1.8.4 progressr_0.13.0 spatstat.data_3.0-0 [40] survival_3.5-0 zoo_1.8-11 iterators_1.0.14 [43] glue_1.6.2 polyclip_1.10-4 gtable_0.3.1 [46] ipred_0.9-13 leiden_0.4.3 GetoptLong_1.0.5 [49] future.apply_1.10.0 shape_1.4.6 BiocGenerics_0.44.0 [52] abind_1.4-5 scales_1.2.1 spatstat.random_3.0-1 [55] miniUI_0.1.1.1 Rcpp_1.0.9 htmlTable_2.4.1 [58] viridisLite_0.4.1 xtable_1.8-4 clue_0.3-63 [61] foreign_0.8-84 proxy_0.4-27 Formula_1.2-4 [64] stats4_4.2.2 lava_1.7.1 prodlim_2019.11.13 [67] htmlwidgets_1.6.1 httr_1.4.4 DiagrammeR_1.0.9 [70] RColorBrewer_1.1-3 ellipsis_0.3.2 Seurat_4.3.0 [73] ica_1.0-3 pkgconfig_2.0.3 nnet_7.3-18 [76] uwot_0.1.14 deldir_1.0-6 utf8_1.2.2 [79] caret_6.0-93 tidyselect_1.2.0 rlang_1.0.6 [82] reshape2_1.4.4 later_1.3.0 visNetwork_2.1.0 [85] munsell_0.5.0 cli_3.6.0 generics_0.1.3 [88] ggridges_0.5.4 fdrtool_1.2.17 stringr_1.5.0 [91] fastmap_1.1.0 goftest_1.2-3 knitr_1.41 [94] ModelMetrics_1.2.2.2 fitdistrplus_1.1-8 caTools_1.18.2 [97] randomForest_4.7-1.1 RANN_2.6.1 pbapply_1.7-0 [100] future_1.30.0 nlme_3.1-161 mime_0.12 [103] rstudioapi_0.13 compiler_4.2.2 plotly_4.10.1 [106] png_0.1-8 e1071_1.7-12 spatstat.utils_3.0-1 [109] stringi_1.7.12 lattice_0.20-45 Matrix_1.5-3 [112] vctrs_0.5.1 pillar_1.8.1 lifecycle_1.0.3 [115] spatstat.geom_3.0-3 lmtest_0.9-40 GlobalOptions_0.1.2 [118] RcppAnnoy_0.0.20 bitops_1.0-7 data.table_1.14.6 [121] cowplot_1.1.1 irlba_2.3.5.1 httpuv_1.6.8 [124] patchwork_1.1.2 latticeExtra_0.6-30 R6_2.5.1 [127] promises_1.2.0.1 KernSmooth_2.23-20 gridExtra_2.3 [130] IRanges_2.32.0 parallelly_1.34.0 codetools_0.2-18 [133] MASS_7.3-58.1 rjson_0.2.21 withr_2.5.0 [136] SeuratObject_4.1.3 sctransform_0.3.5 S4Vectors_0.36.0 [139] parallel_4.2.2 hms_1.1.1 grid_4.2.2 [142] rpart_4.1.19 timeDate_4022.108 class_7.3-20 [145] Rtsne_0.16 spatstat.explore_3.0-5 pROC_1.18.0 [148] base64enc_0.1-3 shiny_1.7.4 lubridate_1.9.0 [151] interp_1.1-3
session_info.show()
Click to view session information
----- anndata 0.8.0 anndata2ri 1.1 decoupler 1.3.3 liana 0.1.6 matplotlib 3.6.3 numpy 1.23.5 pandas 1.5.3 plotnine 0.10.1 rpy2 3.5.7 scanpy 1.7.2 seaborn 0.12.2 session_info 1.0.0 -----
Click to view modules imported as dependencies
PIL 9.4.0 adjustText NA asttokens NA backcall 0.2.0 beta_ufunc NA binom_ufunc NA cffi 1.15.1 colorama 0.4.6 cycler 0.10.0 cython_runtime NA dateutil 2.8.2 debugpy 1.5.1 decorator 5.1.1 defusedxml 0.7.1 dunamai 1.15.0 entrypoints 0.4 executing 0.8.3 fontTools 4.38.0 get_version 3.5.4 h5py 3.7.0 hypergeom_ufunc NA invgauss_ufunc NA ipykernel 6.9.1 ipython_genutils 0.2.0 ipywidgets 7.6.5 jedi 0.18.1 jinja2 3.1.2 joblib 1.2.0 kiwisolver 1.4.4 legacy_api_wrap 0.0.0 llvmlite 0.39.1 markupsafe 2.1.2 matplotlib_inline NA mizani 0.8.1 mpl_toolkits NA natsort 8.2.0 nbinom_ufunc NA ncf_ufunc NA nct_ufunc NA ncx2_ufunc NA numba 0.56.4 numexpr 2.8.3 packaging 23.0 palettable 3.3.0 parso 0.8.3 patsy 0.5.3 pexpect 4.8.0 pickleshare 0.7.5 pkg_resources NA prompt_toolkit 3.0.20 ptyprocess 0.7.0 pure_eval 0.2.2 pycparser 2.21 pydev_ipython NA pydevconsole NA pydevd 2.6.0 pydevd_concurrency_analyser NA pydevd_file_utils NA pydevd_plugins NA pydevd_tracing NA pygments 2.11.2 pyparsing 3.0.9 pytz 2022.7.1 pytz_deprecation_shim NA scipy 1.10.0 setuptools 66.1.1 setuptools_scm NA sinfo 0.3.1 six 1.16.0 skewnorm_ufunc NA sklearn 1.2.0 stack_data 0.2.0 statsmodels 0.13.5 tables 3.7.0 threadpoolctl 3.1.0 tornado 6.1 tqdm 4.64.1 traitlets 5.1.1 typing_extensions NA tzlocal NA wcwidth 0.2.5 zmq 23.2.0 zoneinfo NA
----- IPython 8.4.0 jupyter_client 7.2.2 jupyter_core 4.10.0 notebook 6.4.12 ----- Python 3.10.8 | packaged by conda-forge | (main, Nov 22 2022, 08:26:04) [GCC 10.4.0] Linux-5.15.0-60-generic-x86_64-with-glibc2.31 ----- Session information updated at 2023-02-19 13:46