5.1 Differential gene expression analysis¶
Motivation¶
本章是“注释”子章节的更详细延续,已经介绍了差异基因表达(DGE)作为注释簇与细胞类型工具的方法。 在这里,我们将重点讨论差异基因表达测试在更复杂的实验设计中的高级用例,这些设计涉及一种或多种条件,如疾病、基因敲除或药物。 在这种情况下,我们通常对目标条件与参考条件之间基因表达模式差异的大小和显著性感兴趣。这个参考可以是任何东西,但通常是健康样本。这个统计测试可以应用于任意群体,但在单细胞RNA测序的情况下,通常在细胞类型水平上应用。
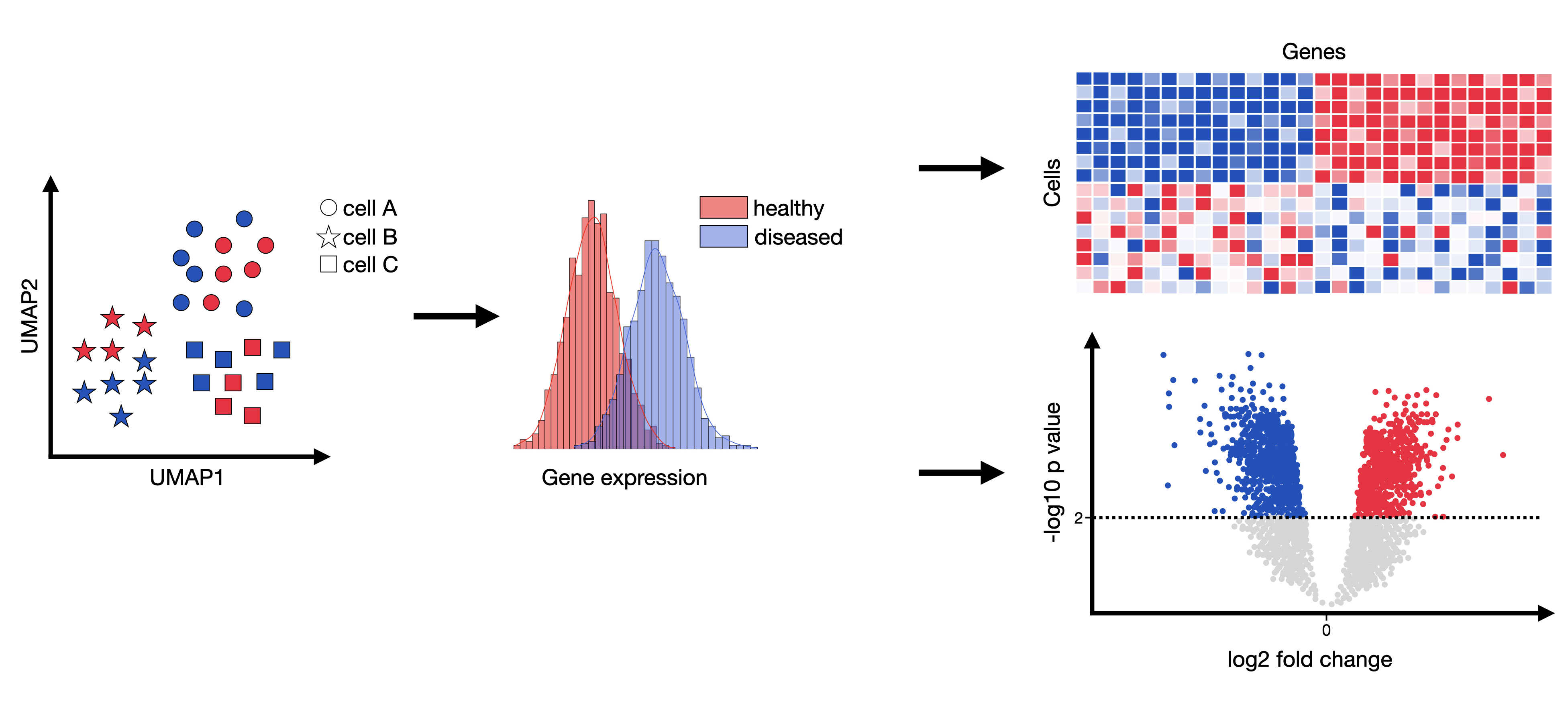
差异基因表达分析试图推断在任何比较组之间(通常在每种细胞类型之间的健康和条件之间)统计显著的过表达或下表达的基因。
这种分析的结果可能是影响并潜在解释任何观察到的表型的基因集。这些基因集可以更仔细地检查,例如,受影响的通路或诱导的细胞-细胞通信变化。
差异基因表达测试通常返回每个比较基因在每个比较条件下的log2倍变和调整后的p值。然后可以按p值对该列表进行排序,并进行更详细的调查。
流行的学生t检验是一种进行这种测试的方法。然而,它未能考虑到几种单细胞RNA测序的特性,如由丢失现象引起的过多零值或复杂实验设计的需求。更具体地说,很少有人有足够的样本数量来在不跨基因汇集信息的情况下准确估计方差。此外,原始计数永远不是给定样本中特定基因表达的绝对测量值。每个基因的实际读取数量取决于文库制备的效率、非编码转录本的污染量和测序深度。因此,它在单细胞RNA测序中的灵敏度和特异性都不足,更不用说实验设计的灵活性了。
结果,差异基因表达测试是一个经典的生物信息学问题,已经被许多工具解决。一般来说,目前这个问题从两个视角来处理:样本级视角和细胞级视角。在样本级视角中,表达被聚合以创建“伪批量”并使用最初为批量表达样本设计的方法进行分析,如edgeR {cite}de:Robinson2010 或 DEseq2 {cite}Love2014。在细胞级视角中,细胞使用广义混合效应模型单独建模,如MAST {cite}Finak2015 或 glmmTMB {cite}Brooks2017。DGE工具在数据集之间的一致性和稳健性较低 {cite}de:Wang2019, Das2021。如前所述,尽管单细胞数据包含技术噪声伪影,如丢失、零膨胀和高细胞间变异性 {cite}Hicks2017, Vallejos2017, de:Lücken2019,为批量RNA-seq数据设计的方法与为scRNA-seq数据专门设计的方法相比表现出色 {cite}Das2021, Soneson2018, Jaakkola2016, de:Squair2021。发现单细胞特定方法特别容易错误地将高表达基因标记为差异表达基因。
一项最近的研究强调了伪重复的问题,即将推断统计应用于不具有统计独立性的生物重复样本。未能考虑重复样本(来自同一个体的细胞)的内在相关性会导致假阳性发现率(FDR)的增加 {cite}de:Squair2021, Zimmerman2021, Junttila2022。因此,在进行DGE分析之前,应进行批次效应校正或通过对每个个体的细胞类型特异性表达值进行汇总、平均或随机效应聚合(即生成伪批量),以考虑样本内相关性 {cite}Zimmerman2021。通常,汇总聚合的伪批量方法如edgeR、DESeq2或Limma {cite}Ritchie2015 以及具有随机效应设置的混合模型如MAST,被发现优于不考虑这些因素的简单方法,如流行的Wilcoxon秩和检验或Seurat的潜在模型 {cite}de:Hao2021 {cite}Junttila2022。
在Zimmerman论文提出的问题中,Murphy等人对Zimmerman的基准测试策略进行了严格审查并进行了改进 {cite}Murphy_2022。他们得出的结论是,伪批量方法表现最佳,但汇总或平均聚合哪个效果更好仍需进一步研究。
因此,在本笔记本中,我们展示了如何使用两种工具进行差异表达分析:edgeR(使用准似然检验)和MAST(使用随机效应)。由于edgeR和MAST都是在R中实现的,我们使用anndata2ri包,以便能够同时处理Python中的AnnData对象和R中的SingleCellExperiment对象。
对于这两种方法,我们展示了两个用例:如何在全数据上运行分析并对多种细胞类型进行测试,以及如何对一种特定的细胞类型进行测试。
Environment setup¶
import warnings
warnings.filterwarnings("ignore")
import matplotlib.pyplot as plt
import seaborn as sns
import scanpy as sc
import pandas as pd
import numpy as np
import random
import sc_toolbox
import pertpy
import rpy2.rinterface_lib.callbacks
import anndata2ri
import logging
from rpy2.robjects import pandas2ri
from rpy2.robjects import r
sc.settings.verbosity = 0
rpy2.rinterface_lib.callbacks.logger.setLevel(logging.ERROR)
pandas2ri.activate()
anndata2ri.activate()
%load_ext rpy2.ipython
To use sccoda or tasccoda please install ete3 with pip install ete3
%%R
library(edgeR)
library(MAST)
Preparing the dataset¶
我们将使用Kang数据集,该数据集是基于10x微滴的scRNA-seq外周血单核细胞(PBMC)数据,来自8位狼疮患者在使用INF-β治疗前后6小时的样本(共16个样本){cite}de:kang2018。干扰素β以天然成纤维细胞或重组制剂(干扰素β-1a和干扰素β-1b)的形式使用,具有类似于干扰素α的抗病毒和抗增殖特性。干扰素β已被批准用于治疗复发缓解型多发性硬化症和继发进行型多发性硬化症。
First, we load the full dataset.
adata = pertpy.data.kang_2018()
adata
AnnData object with n_obs × n_vars = 24673 × 15706
obs: 'nCount_RNA', 'nFeature_RNA', 'tsne1', 'tsne2', 'label', 'cluster', 'cell_type', 'replicate', 'nCount_SCT', 'nFeature_SCT', 'integrated_snn_res.0.4', 'seurat_clusters'
var: 'name'
obsm: 'X_pca', 'X_umap'
We will need label (which contains the condition label), replicate and cell_type columns of the .obs.
adata.obs[:5]
| nCount_RNA | nFeature_RNA | tsne1 | tsne2 | label | cluster | cell_type | replicate | nCount_SCT | nFeature_SCT | integrated_snn_res.0.4 | seurat_clusters | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| index | ||||||||||||
| AAACATACATTTCC-1 | 3017.0 | 877 | -27.640373 | 14.966629 | ctrl | 9 | CD14+ Monocytes | patient_1016 | 1704.0 | 711 | 1 | 1 |
| AAACATACCAGAAA-1 | 2481.0 | 713 | -27.493646 | 28.924885 | ctrl | 9 | CD14+ Monocytes | patient_1256 | 1614.0 | 662 | 1 | 1 |
| AAACATACCATGCA-1 | 703.0 | 337 | -10.468194 | -5.984389 | ctrl | 3 | CD4 T cells | patient_1488 | 908.0 | 337 | 6 | 6 |
| AAACATACCTCGCT-1 | 3420.0 | 850 | -24.367997 | 20.429285 | ctrl | 9 | CD14+ Monocytes | patient_1256 | 1738.0 | 653 | 1 | 1 |
| AAACATACCTGGTA-1 | 3158.0 | 1111 | 27.952170 | 24.159738 | ctrl | 4 | Dendritic cells | patient_1039 | 1857.0 | 928 | 12 | 12 |
我们需要处理原始计数数据,因此我们要检查 .X 是否确实包含原始计数,并将其放入我们的 AnnData 对象的 counts 层。
np.max(adata.X)
3828.0
adata.layers["counts"] = adata.X.copy()
We have 8 control and 8 disease patients.
print(len(adata[adata.obs["label"] == "ctrl"].obs["replicate"].cat.categories))
print(len(adata[adata.obs["label"] == "stim"].obs["replicate"].cat.categories))
8 8
我们对细胞进行初步质量控制,过滤掉基因数量少于200的细胞以及在少于3个细胞中检测到的基因。
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
adata
AnnData object with n_obs × n_vars = 24562 × 15701
obs: 'nCount_RNA', 'nFeature_RNA', 'tsne1', 'tsne2', 'label', 'cluster', 'cell_type', 'replicate', 'nCount_SCT', 'nFeature_SCT', 'integrated_snn_res.0.4', 'seurat_clusters', 'n_genes'
var: 'name', 'n_cells'
obsm: 'X_pca', 'X_umap'
layers: 'counts'
Pseudobulk¶
edgeR 是一个在 R 中实现的差异基因表达测试工具,最初设计用于批量基因表达数据。它基于负二项分布、经验贝叶斯估计、精确检验、广义线性模型 (GLM) 和准似然检验,实施了广泛的统计方法。
更多详细信息请阅读原始出版物{cite}de:Robinson2010。
在这里,我们将使用准似然检验,因为它考虑到了离散估计的不确定性。相比之下,精确检验假设估计的离散度是实际值,这可能会导致一些不准确性。此外,准似然GLM在实验设计方面更加灵活。
由于edgeR是作为批量数据的差异表达分析方法引入的,我们首先需要从我们的单细胞数据集中创建伪批量样本。对于每个患者,我们通过聚合每个亚群中的细胞并取该亚群内的平均基因表达来创建每种细胞类型的1个伪批量样本。
如果您处理的数据没有重复样本,则创建多个(例如2-3个)伪批量样本可能是有益的,以考虑患者变异性。在这里,我们选择为每个患者创建1个伪批量样本,因为每个患者我们有2个重复样本,一个是对照组,一个是刺激组。
我们强烈建议阅读这个关于设计矩阵的指南 https://f1000research.com/articles/9-1444。 无论我们是仅对几个细胞亚群运行分析并为每个细胞单独拟合模型,还是为所有细胞拟合一个模型,我们首先需要准备数据,定义一个函数来创建伪批量样本并运行edgeR流程。首先,让我们准备数据。
由于我们需要为每个患者-条件组合创建伪批量样本,因此我们首先需要通过连接replicate和label来创建这样一列。
adata.obs["sample"] = [
f"{rep}_{l}" for rep, l in zip(adata.obs["replicate"], adata.obs["label"])
]
我们需要清理细胞类型名称,即将空格替换为下划线并移除加号,以避免 Python 到 R 转换问题。
adata.obs["cell_type"] = [ct.replace(" ", "_") for ct in adata.obs["cell_type"]]
adata.obs["cell_type"] = [ct.replace("+", "") for ct in adata.obs["cell_type"]]
我们需要将分类元数据确实设置为分类类型,以创建伪批量样本。
adata.obs["replicate"] = adata.obs["replicate"].astype("category")
adata.obs["label"] = adata.obs["label"].astype("category")
adata.obs["sample"] = adata.obs["sample"].astype("category")
adata.obs["cell_type"] = adata.obs["cell_type"].astype("category")
aggregate_and_filter 函数用于将单细胞数据聚合成伪重复样本:
- 首先,从原始单细胞 AnnData 对象中选择指定的细胞亚群。
- 对于每个供体(患者),创建一个包含指定亚群的伪重复样本。
- 过滤掉细胞数量少于30的供体,以确保数据的质量。
- 通过更改
replicates_per_patient参数,可以为每个样本创建多个伪重复样本。细胞将被分成大小大致相等的子集。
NUM_OF_CELL_PER_DONOR = 30
def aggregate_and_filter(
adata,
cell_identity,
donor_key="sample",
condition_key="label",
cell_identity_key="cell_type",
obs_to_keep=[], # which additional metadata to keep, e.g. gender, age, etc.
replicates_per_patient=1,
):
# subset adata to the given cell identity
adata_cell_pop = adata[adata.obs[cell_identity_key] == cell_identity].copy()
# check which donors to keep according to the number of cells specified with NUM_OF_CELL_PER_DONOR
size_by_donor = adata_cell_pop.obs.groupby([donor_key]).size()
donors_to_drop = [
donor
for donor in size_by_donor.index
if size_by_donor[donor] <= NUM_OF_CELL_PER_DONOR
]
if len(donors_to_drop) > 0:
print("Dropping the following samples:")
print(donors_to_drop)
df = pd.DataFrame(columns=[*adata_cell_pop.var_names, *obs_to_keep])
adata_cell_pop.obs[donor_key] = adata_cell_pop.obs[donor_key].astype("category")
for i, donor in enumerate(donors := adata_cell_pop.obs[donor_key].cat.categories):
print(f"\tProcessing donor {i+1} out of {len(donors)}...", end="\r")
if donor not in donors_to_drop:
adata_donor = adata_cell_pop[adata_cell_pop.obs[donor_key] == donor]
# create replicates for each donor
indices = list(adata_donor.obs_names)
random.shuffle(indices)
indices = np.array_split(np.array(indices), replicates_per_patient)
for i, rep_idx in enumerate(indices):
adata_replicate = adata_donor[rep_idx]
# specify how to aggregate: sum gene expression for each gene for each donor and also keep the condition information
agg_dict = {gene: "sum" for gene in adata_replicate.var_names}
for obs in obs_to_keep:
agg_dict[obs] = "first"
# create a df with all genes, donor and condition info
df_donor = pd.DataFrame(adata_replicate.X.A)
df_donor.index = adata_replicate.obs_names
df_donor.columns = adata_replicate.var_names
df_donor = df_donor.join(adata_replicate.obs[obs_to_keep])
# aggregate
df_donor = df_donor.groupby(donor_key).agg(agg_dict)
df_donor[donor_key] = donor
df.loc[f"donor_{donor}_{i}"] = df_donor.loc[donor]
print("\n")
# create AnnData object from the df
adata_cell_pop = sc.AnnData(
df[adata_cell_pop.var_names], obs=df.drop(columns=adata_cell_pop.var_names)
)
return adata_cell_pop
我们还需要定义一个单独的函数来拟合 edgeR GLM:
fit_model函数接受 SingleCellExperiment 对象作为输入,创建设计矩阵并输出拟合的 GLM。我们还输出 edgeR 类的 DGEList 对象,以便进行一些探索性数据分析 (EDA)。
%%R
fit_model <- function(adata_){
# create an edgeR object with counts and grouping factor
y <- DGEList(assay(adata_, "X"), group = colData(adata_)$label)
# filter out genes with low counts
print("Dimensions before subsetting:")
print(dim(y))
print("")
keep <- filterByExpr(y)
y <- y[keep, , keep.lib.sizes=FALSE]
print("Dimensions after subsetting:")
print(dim(y))
print("")
# normalize
y <- calcNormFactors(y)
# create a vector that is a concatenation of condition and cell type that we will later use with contrasts
group <- paste0(colData(adata_)$label, ".", colData(adata_)$cell_type)
replicate <- colData(adata_)$replicate
# create a design matrix: here we have multiple donors so also consider that in the design matrix
design <- model.matrix(~ 0 + group + replicate)
# estimate dispersion
y <- estimateDisp(y, design = design)
# fit the model
fit <- glmQLFit(y, design)
return(list("fit"=fit, "design"=design, "y"=y))
}
现在我们已经定义了所有必需的函数,因此我们可以继续创建假批量(pseudobulks)。以后我们可能需要查看可用元数据,因此将其保留在 AnnData 对象中。
obs_to_keep = ["label", "cell_type", "replicate", "sample"]
我们需要将原始计数传递给 edgeR。因此,我们将 .X 设置为 counts 层,以确保为原始计数创建伪重复项。
adata.X = adata.layers["counts"].copy()
Next, we create the AnnData object with pseudobulks.
# process first cell type separately...
cell_type = adata.obs["cell_type"].cat.categories[0]
print(
f'Processing {cell_type} (1 out of {len(adata.obs["cell_type"].cat.categories)})...'
)
adata_pb = aggregate_and_filter(adata, cell_type, obs_to_keep=obs_to_keep)
for i, cell_type in enumerate(adata.obs["cell_type"].cat.categories[1:]):
print(
f'Processing {cell_type} ({i+2} out of {len(adata.obs["cell_type"].cat.categories)})...'
)
adata_cell_type = aggregate_and_filter(adata, cell_type, obs_to_keep=obs_to_keep)
adata_pb = adata_pb.concatenate(adata_cell_type)
Processing B_cells (1 out of 8)... Dropping the following samples: ['patient_1039_ctrl'] Processing donor 16 out of 16... Processing CD14_Monocytes (2 out of 8)... Processing donor 16 out of 16... Processing CD4_T_cells (3 out of 8)... Processing donor 16 out of 16... Processing CD8_T_cells (4 out of 8)... Dropping the following samples: ['patient_101_ctrl', 'patient_1039_ctrl', 'patient_1039_stim', 'patient_107_ctrl', 'patient_107_stim', 'patient_1244_ctrl', 'patient_1244_stim'] Processing donor 16 out of 16... Processing Dendritic_cells (5 out of 8)... Dropping the following samples: ['patient_1016_ctrl', 'patient_1016_stim', 'patient_101_ctrl', 'patient_1039_ctrl', 'patient_1039_stim', 'patient_107_ctrl', 'patient_107_stim'] Processing donor 16 out of 16... Processing FCGR3A_Monocytes (6 out of 8)... Dropping the following samples: ['patient_1039_ctrl', 'patient_1039_stim', 'patient_107_ctrl', 'patient_107_stim', 'patient_1244_stim'] Processing donor 16 out of 16... Processing Megakaryocytes (7 out of 8)... Dropping the following samples: ['patient_1015_ctrl', 'patient_1015_stim', 'patient_1016_ctrl', 'patient_101_ctrl', 'patient_1039_stim', 'patient_107_stim', 'patient_1244_ctrl', 'patient_1244_stim', 'patient_1256_ctrl', 'patient_1256_stim', 'patient_1488_ctrl'] Processing donor 11 out of 11... Processing NK_cells (8 out of 8)... Dropping the following samples: ['patient_1039_ctrl', 'patient_1039_stim'] Processing donor 16 out of 16...
差异基因表达结果的有效性在很大程度上取决于统计模型中主要变异方向的捕获。中间数据探索步骤,例如对伪批量样本进行主成分分析 (PCA) 或多维尺度缩放 (MDS),可以识别变异来源,从而指导构建相应的模型数据的的设计矩阵和对比矩阵 [引用来源: Law2020]。
对于包含生物重复样本的实验,未能解释多个生物变异来源会夸大错误发现率 (FDR) [引用来源: Thurman2021],[引用来源: Lähnemann2020]。虽然增加每个个体的细胞数量会提高精确度,但它对检测个体间差异的能力影响有限。因此,提高统计功效的最佳方法是增加独立实验样本的数量 [引用来源: Zimmerman2021]。
由于我们的数据已经生成,我们无法进一步增加独立实验样本的数量。然而,我们现在将探索我们的数据以确定主要变异方向,以便正确生成我们的设计矩阵。
我们对创建的伪重复样本进行非常基本的探索性数据分析 (EDA),以检查是否存在需要排除的异常患者/伪批量样本,以免对差异表达 (DE) 结果产生偏差。我们将原始计数保存 فى (zài) 'counts' 层,然后对计数进行归一化并计算归一化伪批量计数的 PCA 坐标。
adata_pb.layers['counts'] = adata_pb.X.copy()
sc.pp.normalize_total(adata_pb, target_sum=1e6)
sc.pp.log1p(adata_pb)
sc.pp.pca(adata_pb)
接下来,我们将创建的伪重复样本绘制在 PCA 图上,并根据所有可用元数据进行染色,以查看是否存在我们可能想要包含在设计矩阵中的混杂因素。我们还添加了 lib_size 和 log_lib_size 列来检查文库大小和 PC 组件之间是否存在相关性。
adata_pb.obs["lib_size"] = np.sum(adata_pb.layers["counts"], axis=1)
adata_pb.obs["log_lib_size"] = np.log(adata_pb.obs["lib_size"])
sc.pl.pca(adata_pb, color=adata_pb.obs, ncols=1, size=300)

从 PCA 图上我们可以观察到细胞类型的分离以及刺激和未刺激细胞的分离。其他协变量(批次)似乎与 PCA 组件没有明显相关性,因此我们不会将任何这些信息纳入设计矩阵。
正如上文所述,edgeR 需要原始计数作为输入,因此在继续之前,我们将计数放回 .X 字段。
adata_pb.X = adata_pb.layers['counts'].copy()
One group¶
首先,我们将展示如何针对一种特定细胞类型准备数据、构建设计矩阵并进行差异表达分析。
由于论文表明在 CD14+ 单核细胞亚群中发现了数量最多的差异表达基因,因此我们将管道应用于数据的 CD14+ 单核细胞子集。
adata_mono = adata_pb[adata_pb.obs["cell_type"] == "CD14_Monocytes"]
adata_mono
View of AnnData object with n_obs × n_vars = 16 × 15701
obs: 'label', 'cell_type', 'replicate', 'sample', 'batch', 'lib_size', 'log_lib_size'
uns: 'log1p', 'pca', 'label_colors', 'cell_type_colors', 'replicate_colors', 'sample_colors', 'batch_colors'
obsm: 'X_pca'
varm: 'PCs'
layers: 'counts'
Clean the sample names to make plots less crowded.
adata_mono.obs_names = [
name.split("_")[2] + "_" + name.split("_")[3] for name in adata_mono.obs_names
]
%%time
%%R -i adata_mono
outs <-fit_model(adata_mono)
[1] "Dimensions before subsetting:" [1] 15701 16 [1] "" [1] "Dimensions after subsetting:" [1] 3709 16 [1] "" CPU times: user 2.95 s, sys: 224 ms, total: 3.18 s Wall time: 4.27 s
%%R
fit <- outs$fit
y <- outs$y
由于我们没有预先假设某个特定基因的上调或下调,因此我们需要可视化来理解 DGE 结果。MDS(多维尺度缩放)绘图允许进行高级概述。通常,我们期望来自不同条件的样本之间会存在分离,正如以下针对我们结果的绘图所示。
%%R
plotMDS(y, col=ifelse(y$samples$group == "stim", "red", "blue"))
Fontconfig warning: ignoring UTF-8: not a valid region tag

生物变异系数 (BCV) 图展示了每个基因在生物分组间的平均变异性,并将其作为一个平均表达的函数进行可视化。例如,BCV 为 0.3 表示基因表达在不同分组间的平均变异性为 30%。
通常,低丰度基因的 BCV 值会更高,因为低丰度基因的读计数测量存在更大不确定性。另一方面,高平均表达的基因定量更加可靠,因此它们通常具有更低的变异性,从而导致更低的 BCV 值。我们可以利用 BCV 图检测异常基因或识别其他可能需要反映在设计矩阵中的实验因素。例如,如果图中右上角出现一组平均表达高、BCV 值也高的基因,则可能表明实验压力、污染等问题,尤其是在这些基因属于相似基因家族的情况下。
BCV 是离散度的平方根。标签分散 (tagwise dispersion) 和通用 BCV 趋势之间的距离表明标签分散估计是否高度可变(即异质基因表达)。如果标签分散值非常异质,则应用于捕获异质性的调节就更少。曲线上的任意两点之间的距离反映了基因的离散度向公共趋势收缩的程度。也就是说,它捕获了应用的离散度调节量。
在下方的 BCV 图中,我们观察到一些低丰度基因具有高 BCV 值,但没有高丰度基因具有高 BCV 值,这表明不需要将任何进一步的实验因素纳入设计矩阵建模。
%%R
plotBCV(y)

接下来,我们将执行拟似逼近检验 (quasi-likelihood test) 以寻找控制和刺激条件之间差异表达的基因。让我们检查设计矩阵的列,以便指定用于测试的正确列。
%%R
colnames(y$design)
[1] "groupctrl.CD14_Monocytes" "groupstim.CD14_Monocytes" [3] "replicatepatient_107" "replicatepatient_1015" [5] "replicatepatient_1016" "replicatepatient_1039" [7] "replicatepatient_1244" "replicatepatient_1256" [9] "replicatepatient_1488"
%%R -o tt
myContrast <- makeContrasts('groupstim.CD14_Monocytes-groupctrl.CD14_Monocytes', levels = y$design)
qlf <- glmQLFTest(fit, contrast=myContrast)
# get all of the DE genes and calculate Benjamini-Hochberg adjusted FDR
tt <- topTags(qlf, n = Inf)
tt <- tt$table
让我们检查表格:对于 edgeR 未过滤掉的每个基因(在本例中为 3709 个),表格都包含差异表达测试的结果。 我们可以将此表格保存为 .csv 文件,以便稍后用于可视化和其他分析。在本节末尾运行完整数据分析后,我们将展示如何使用火山图进行可视化。
tt.shape
(3709, 5)
tt[:5]
| logFC | logCPM | F | PValue | FDR | |
|---|---|---|---|---|---|
| HESX1 | 8.345536 | 6.773420 | 1281.013295 | 1.837373e-15 | 2.766927e-12 |
| CD38 | 7.126846 | 7.420668 | 1243.793133 | 2.266164e-15 | 2.766927e-12 |
| NT5C3A | 5.657050 | 8.327003 | 1218.102628 | 2.628780e-15 | 2.766927e-12 |
| SOCS1 | 4.388247 | 6.943768 | 1191.289806 | 3.079524e-15 | 2.766927e-12 |
| GMPR | 6.943484 | 7.031832 | 1159.601183 | 3.730018e-15 | 2.766927e-12 |
值得一提的是,还可以使用 glmTreat 函数来测试基因相对于指定阈值折叠变化的差异表达,该函数可以针对提供的系数或对比进行测试。
%%R
tr <- glmTreat(fit, contrast=myContrast, lfc=1.5)
print(head(topTags(tr)))
Coefficient: -1*groupctrl.CD14_Monocytes 1*groupstim.CD14_Monocytes
logFC unshrunk.logFC logCPM PValue FDR
HESX1 8.345536 8.702486 6.773420 2.108737e-14 3.535749e-11
CD38 7.126846 7.219184 7.420668 2.292459e-14 3.535749e-11
NT5C3A 5.657050 5.674640 8.327003 3.149544e-14 3.535749e-11
IL1RN 6.588583 6.596787 10.358946 4.249159e-14 3.535749e-11
GMPR 6.943484 7.047504 7.031832 4.766446e-14 3.535749e-11
DEFB1 6.654201 6.696759 8.067159 7.670147e-14 4.741429e-11
最后,我们可以查看 FDR 校正值小于 0.01 的基因数量。
涂抹图(也称为 MA 图,M 值 vs A 值图)显示了基因的对数 fold 变化与其平均丰度之间的关系。我们通常会观察到低丰度范围内的较高 logFC,因为低丰度读计数的变异性更大,从而导致较大的 logFC 估计值。如果我们将一条 Loess 曲线拟合到 logFC 和平均 logCPM 值,则趋势应该围绕零为中心。任何偏离都可能表明数据标准化不正确。具有大平均表达和绝对值大的 logFC 的基因可能标志着具有生物学意义的基因,值得进一步研究和追踪。
%%R
plotSmear(qlf, de.tags = rownames(tt)[which(tt$FDR<0.01)])

Multiple groups¶
接下来,我们将展示如何使用对比分析方法,针对所有可用细胞类型的完整数据集准备数据、构建设计矩阵并执行差异表达分析
%%time
%%R -i adata_pb
outs <-fit_model(adata_pb)
[1] "Dimensions before subsetting:" [1] 15701 90 [1] "" [1] "Dimensions after subsetting:" [1] 2358 90 [1] "" CPU times: user 11.9 s, sys: 39.7 ms, total: 12 s Wall time: 12 s
%%R
fit <- outs$fit
y <- outs$y
现在,我们将使用对比分析方法针对每个细胞类型执行拟似逼近检验。由于 R 循环内部无法直接将表格从 R 传递到 Python,因此我们将手动获取每个细胞类型的结果。
%%R -i adata_pb -o de_per_cell_type
de_per_cell_type <- list()
for (cell_type in unique(colData(adata_pb)$cell_type)) {
print(cell_type)
# create contrast for this cell type
myContrast <- makeContrasts(paste0("groupstim.", cell_type, "-groupctrl.", cell_type), levels = y$design)
# perform QLF test
qlf <- glmQLFTest(fit, contrast=myContrast)
# get all of the DE genes and calculate Benjamini-Hochberg adjusted FDR
tt <- topTags(qlf, n = Inf)
# save in the list with the results for all the cell types
de_per_cell_type[[cell_type]] <- tt$table
}
[1] "B_cells" [1] "CD14_Monocytes" [1] "CD4_T_cells" [1] "CD8_T_cells" [1] "Dendritic_cells" [1] "FCGR3A_Monocytes" [1] "NK_cells"
现在让我们使用来自 sc_toolbox 软件包 (https://github.com/schillerlab/sc-toolbox) 的 sc_toolbox.tools.de_res_to_anndata 函数,将结果保存到我们原始 adata 对象的 .uns 层中。这个函数会将结果保存成类似于使用 scanpy 的 rank_genes_groups() 函数创建的格式。这样做的优点是,我们现在可以使用标准的 scanpy 绘图函数进行可视化。
此外,我们还将每个细胞类型的差异表达基因 (DEG) 结果分别保存为单独的 .csv 文件,因为我们稍后在细胞间通讯章节中会用到它们。
# get cell types that we ran the analysis for
cell_types = de_per_cell_type.keys()
# add the table to .uns for each cell type
for cell_type in cell_types:
df = de_per_cell_type[cell_type]
df["gene_symbol"] = df.index
df["cell_type"] = cell_type
sc_toolbox.tools.de_res_to_anndata(
adata,
df,
groupby="cell_type",
score_col="logCPM",
pval_col="PValue",
pval_adj_col="FDR",
lfc_col="logFC",
key_added="edgeR_" + cell_type,
)
df.to_csv(f"de_edgeR_{cell_type}.csv")
为了再次将表格转换为 pandas 数据框,我们使用 sc.get.rank_genes_groups_df 函数。
sc.get.rank_genes_groups_df(adata, group="CD14_Monocytes", key="edgeR_CD14_Monocytes")[
:5
]
| names | scores | logfoldchanges | pvals | pvals_adj | |
|---|---|---|---|---|---|
| 0 | FTH1 | 16.186098 | -0.572272 | 0.001589 | 0.002712 |
| 1 | MALAT1 | 16.025682 | 0.010049 | 0.877078 | 0.897245 |
| 2 | B2M | 15.468524 | 0.677266 | 0.0 | 0.0 |
| 3 | TMSB4X | 15.054697 | -0.217131 | 0.004663 | 0.007331 |
| 4 | FTL | 14.984937 | -0.041832 | 0.669951 | 0.710956 |
edgeR 笔记¶
- 需要原始计数作为输入
- 需要来自单细胞实验的伪批量
- 如果单细胞实验有多个供体,并且用户想要解释患者的可变性,我们建议为每个患者创建 2 或 3 个伪重复,并将患者信息包含到设计矩阵中
Single-cell specific¶
MAST 框架使用二元广义线性模型对单细胞基因表达进行建模。MAST 的一个部分用于模拟每个基因在细胞间的离散表达率,另一个部分用于模拟条件连续表达水平(以基因表达为条件)。
有关详细信息,请阅读原始论文 {cite}Finak2015。
MAST 需要归一化计数作为输入,因此我们首先需要处理单细胞 RNA 测序数据的“计数”层,然后进行归一化步骤。
adata.X = adata.layers["counts"].copy()
sc.pp.normalize_total(adata, target_sum=1e6)
sc.pp.log1p(adata)
为了帮助处理 R 和 Python 之间的一些对象类型转换问题,我们定义了一个小型辅助函数。 另外,由于模型需要估计每个基因的方差,因此我们需要过滤掉每个子群中表达于少量细胞(例如少于 3 个细胞)的基因。
def prep_anndata(adata_):
def fix_dtypes(adata_):
df = pd.DataFrame(adata_.X.A, index=adata_.obs_names, columns=adata_.var_names)
df = df.join(adata_.obs)
return sc.AnnData(df[adata_.var_names], obs=df.drop(columns=adata_.var_names))
adata_ = fix_dtypes(adata_)
sc.pp.filter_genes(adata_, min_cells=3)
return adata_
One group¶
与 edgeR 一样,可以使用完整数据集拟合模型,然后使用对比度测试每个感兴趣的细胞类型,但是为了缩短运行时间,本例程仅展示了针对一种细胞类型(即 CD14 单核细胞)的 MAST-RE 管道。
adata_mono = adata[adata.obs["cell_type"] == "CD14_Monocytes"].copy()
adata_mono
AnnData object with n_obs × n_vars = 5696 × 15701
obs: 'nCount_RNA', 'nFeature_RNA', 'tsne1', 'tsne2', 'label', 'cluster', 'cell_type', 'replicate', 'nCount_SCT', 'nFeature_SCT', 'integrated_snn_res.0.4', 'seurat_clusters', 'n_genes', 'sample'
var: 'name', 'n_cells'
uns: 'edgeR_B_cells', 'edgeR_CD14_Monocytes', 'edgeR_CD4_T_cells', 'edgeR_CD8_T_cells', 'edgeR_Dendritic_cells', 'edgeR_FCGR3A_Monocytes', 'edgeR_NK_cells', 'log1p'
obsm: 'X_pca', 'X_umap'
layers: 'counts'
sc.pp.filter_genes(adata_mono, min_cells=3)
adata_mono
AnnData object with n_obs × n_vars = 5696 × 12268
obs: 'nCount_RNA', 'nFeature_RNA', 'tsne1', 'tsne2', 'label', 'cluster', 'cell_type', 'replicate', 'nCount_SCT', 'nFeature_SCT', 'integrated_snn_res.0.4', 'seurat_clusters', 'n_genes', 'sample'
var: 'name', 'n_cells'
uns: 'edgeR_B_cells', 'edgeR_CD14_Monocytes', 'edgeR_CD4_T_cells', 'edgeR_CD8_T_cells', 'edgeR_Dendritic_cells', 'edgeR_FCGR3A_Monocytes', 'edgeR_NK_cells', 'log1p'
obsm: 'X_pca', 'X_umap'
layers: 'counts'
Next we filter both objects as mentioned above.
adata_mono = prep_anndata(adata_mono)
adata_mono
AnnData object with n_obs × n_vars = 5696 × 12268
obs: 'nCount_RNA', 'nFeature_RNA', 'tsne1', 'tsne2', 'label', 'cluster', 'cell_type', 'replicate', 'nCount_SCT', 'nFeature_SCT', 'integrated_snn_res.0.4', 'seurat_clusters', 'n_genes', 'sample'
var: 'n_cells'
adata_mono.obs["cell_type"] = [
ct.replace(" ", "_") for ct in adata_mono.obs["cell_type"]
]
adata_mono.obs["cell_type"] = [
ct.replace("+", "") for ct in adata_mono.obs["cell_type"]
]
%%R
find_de_MAST_RE <- function(adata_){
# create a MAST object
sca <- SceToSingleCellAssay(adata_, class = "SingleCellAssay")
print("Dimensions before subsetting:")
print(dim(sca))
print("")
# keep genes that are expressed in more than 10% of all cells
sca <- sca[freq(sca)>0.1,]
print("Dimensions after subsetting:")
print(dim(sca))
print("")
# add a column to the data which contains scaled number of genes that are expressed in each cell
cdr2 <- colSums(assay(sca)>0)
colData(sca)$ngeneson <- scale(cdr2)
# store the columns that we are interested in as factors
label <- factor(colData(sca)$label)
# set the reference level
label <- relevel(label,"ctrl")
colData(sca)$label <- label
celltype <- factor(colData(sca)$cell_type)
colData(sca)$celltype <- celltype
# same for donors (which we need to model random effects)
replicate <- factor(colData(sca)$replicate)
colData(sca)$replicate <- replicate
# create a group per condition-celltype combination
colData(sca)$group <- paste0(colData(adata_)$label, ".", colData(adata_)$cell_type)
colData(sca)$group <- factor(colData(sca)$group)
# define and fit the model
zlmCond <- zlm(formula = ~ngeneson + group + (1 | replicate),
sca=sca,
method='glmer',
ebayes=F,
strictConvergence=F,
fitArgsD=list(nAGQ = 0)) # to speed up calculations
# perform likelihood-ratio test for the condition that we are interested in
summaryCond <- summary(zlmCond, doLRT='groupstim.CD14_Monocytes')
# get the table with log-fold changes and p-values
summaryDt <- summaryCond$datatable
result <- merge(summaryDt[contrast=='groupstim.CD14_Monocytes' & component=='H',.(primerid, `Pr(>Chisq)`)], # p-values
summaryDt[contrast=='groupstim.CD14_Monocytes' & component=='logFC', .(primerid, coef)],
by='primerid') # logFC coefficients
# MAST uses natural logarithm so we convert the coefficients to log2 base to be comparable to edgeR
result[,coef:=result[,coef]/log(2)]
# do multiple testing correction
result[,FDR:=p.adjust(`Pr(>Chisq)`, 'fdr')]
result = result[result$FDR<0.01,, drop=F]
result <- stats::na.omit(as.data.frame(result))
return(result)
}
We run the pipeline for monocytes.
%%time
%%R -i adata_mono -o res
res <-find_de_MAST_RE(adata_mono)
[1] "Dimensions before subsetting:" [1] 12268 5696 [1] "" [1] "Dimensions after subsetting:" [1] 1676 5696 [1] "" CPU times: user 9min 35s, sys: 2.96 s, total: 9min 38s Wall time: 9min 43s
Let's take a look at the results.
res[:5]
| primerid | Pr(>Chisq) | coef | FDR | |
|---|---|---|---|---|
| 1 | AAED1 | 8.410341e-19 | 0.709836 | 1.597106e-18 |
| 2 | ABI1 | 1.401688e-05 | 0.312797 | 1.824921e-05 |
| 3 | ABRACL | 3.920183e-06 | 0.418028 | 5.201004e-06 |
| 4 | ACADVL | 2.121110e-26 | -0.751305 | 4.656979e-26 |
| 5 | ACOT9 | 3.424174e-151 | 2.921133 | 2.689503e-150 |
We store the result in .uns as before. Note that we won't need the score column so we just pass logFC there.
res["gene_symbol"] = res["primerid"]
res["cell_type"] = "CD14_Monocytes"
sc_toolbox.tools.de_res_to_anndata(
adata,
res,
groupby="cell_type",
score_col="coef",
pval_col="Pr(>Chisq)",
pval_adj_col="FDR",
lfc_col="coef",
key_added="MAST_CD14_Monocytes",
)
adata_copy = adata.copy()
Visualization¶
我们可以使用热图和火山图来可视化这些结果。热图的每一行对应一个基因,每一列对应一个单细胞。颜色越亮,表示该基因在特定细胞中的表达水平越高。由于我们只绘制差异表达基因,因此我们希望看到两种条件之间表达的明显差异。
火山图常用于可视化统计测试的结果,X 轴表示表达变化(log fold change),Y 轴表示统计显著性(FDR 矫正后的 p 值)。我们将 FDR 矫正后的 p 值小于 0.01 且 log fold change 大于 1.5 的基因标记为特定颜色。
为了在热图中更好地观察两种条件之间表达的差异,我们会在绘制热图之前对数据进行归一化处理。
adata.X = adata.layers["counts"].copy()
sc.pp.normalize_total(adata, target_sum=1e6)
sc.pp.log1p(adata)
Next, we define a helper plotting function for the heatmaps.
FDR = 0.01
LOG_FOLD_CHANGE = 1.5
def plot_heatmap(adata, group_key, group_name="cell_type", groupby="label"):
cell_type = "_".join(group_key.split("_")[1:])
res = sc.get.rank_genes_groups_df(adata, group=cell_type, key=group_key)
res.index = res["names"].values
res = res[
(res["pvals_adj"] < FDR) & (abs(res["logfoldchanges"]) > LOG_FOLD_CHANGE)
].sort_values(by=["logfoldchanges"])
print(f"Plotting {len(res)} genes...")
markers = list(res.index)
sc.pl.heatmap(
adata[adata.obs[group_name] == cell_type].copy(),
markers,
groupby=groupby,
swap_axes=True,
)
And finally we can plot the heatmaps with DEG for CD14+ Monocytes for both edgeR and MAST with RE.
plot_heatmap(adata, "edgeR_CD14_Monocytes")
Plotting 303 genes...

plot_heatmap(adata, "MAST_CD14_Monocytes")
Plotting 436 genes...

经过分析,我们发现使用给定的调整后 p 值和 log fold change 阈值,MAST 识别出 436 个差异表达基因 (DEG),而 edgeR 识别出 303 个基因。
接下来,我们将定义用于绘制火山图的辅助函数。
FDR = 0.01
LOG_FOLD_CHANGE = 1.5
def volcano_plot(adata, group_key, group_name="cell_type", groupby="label", title=None):
cell_type = "_".join(group_key.split("_")[1:])
result = sc.get.rank_genes_groups_df(adata, group=cell_type, key=group_key).copy()
result["-logQ"] = -np.log(result["pvals"].astype("float"))
lowqval_de = result.loc[abs(result["logfoldchanges"]) > LOG_FOLD_CHANGE]
other_de = result.loc[abs(result["logfoldchanges"]) <= LOG_FOLD_CHANGE]
fig, ax = plt.subplots()
sns.regplot(
x=other_de["logfoldchanges"],
y=other_de["-logQ"],
fit_reg=False,
scatter_kws={"s": 6},
)
sns.regplot(
x=lowqval_de["logfoldchanges"],
y=lowqval_de["-logQ"],
fit_reg=False,
scatter_kws={"s": 6},
)
ax.set_xlabel("log2 FC")
ax.set_ylabel("-log Q-value")
if title is None:
title = group_key.replace("_", " ")
plt.title(title)
plt.show()
volcano_plot(adata, "MAST_CD14_Monocytes")

volcano_plot(adata, "edgeR_CD14_Monocytes")

通过热图,尤其是火山图,我们可以观察到与 MAST 识别到的数量相似的上调和下调基因相比,edgeR 在刺激组与对照组之间识别出更多的上调基因。
Key Takeaways¶
- Repeated measurements (cells) from the same experimental subject in scRNA-seq introduce correlations between measurements, which need to be adjusted by modeling the experimental units as Random Effect to mitigate the pseudoreplication issue. Account for them through a sum or mean aggregation (pseudobulk methods - by fixed- or random effect terms) or by accounting for individual as a random effect (single-cell methods) to mitigate the pseudoreplication issue.
- Determine the major axis of variation with exploratory analysis to construct an appropriate design matrix to better model the variations in the data.
- Statistical power is best increased with more samples in the experimental design.