4.3 Lineage tracing¶
这篇综述概述了用于测量细胞状态和谱系历史的检测方法,并通过小鼠肺癌模型追踪肿瘤发展这一典型案例,介绍了可用的计算管道。
动机¶
细胞谱系在生物学中无处不在。也许最著名的例子是胚胎形成:一个受精卵细胞分裂发育成为像人类这样的生物的过程。在这个过程中,细胞不断分裂产生子细胞,随着时间的推移,整个“谱系”承担起发育胚胎内不同的特化角色。这个过程的惊人复杂性几个世纪以来一直吸引着科学家们的想象力,过去一个半世纪里,我们对这一过程的理解得益于高通量测序方法和新的“谱系追踪”技术的开发,这些技术可以用于可视化和表征这一过程 cite。其中一些令人兴奋的方法允许研究人员将细胞状态的测量与它们的演化史模型联系起来,从而提供了一个窗口,让我们得以一窥分化轨迹的展开方式。
单细胞分析方法和谱系追踪方法的结合使数据集的复杂性呈指数级增长,需要开发新的计算方法进行分析。因此,迫切需要开发新的计算方法来处理这些数据集 cite。过去五年,大量借鉴群体遗传学文献,传统进化生物学概念与尖端基因工程技术的融合令人兴奋。
本章将简要概述这些新技术,并重点介绍用于分析其输出和提取生物学信息的可用计算管道。值得注意的是,我们特别关注基于 CRISPR/Cas9 的“进化”谱系追踪设置作为示例。对于其他有用的实验替代方案的更完整视图,我们推荐感兴趣的读者参考 McKenna & Gagnon cite、Wagner & Klein cite 和 VanHorn & Morris cite 的出色综述。
谱系追踪技术¶
谱系追踪技术的目标是推断观察到的细胞之间的谱系或祖先关系。 इसमें需要考虑两个主要变量:规模和分辨率。经典方法主要依靠视觉观察:例如,在 1970 年代,Sulston 及其同事通过在显微镜下仔细观察细胞分裂,推导出了线虫 C. elegans 的第一个发育谱系 cite。虽然这些方法在该领域的进展中发挥了重要作用,但对于具有更多随机发育谱系的复杂生物,这些方法无法扩展应用。
过去二十年中,革命性测序方法和微流体装置的开发促进了新的和多样化的谱系追踪方法的发展 cite。为了理解众多技术,将方法分类为“前瞻性”或“回顾性”很有帮助:
- 前瞻性谱系追踪 方法使研究人员能够追踪单个细胞 (即“克隆”或“克隆群”) 的后代。通常,这是通过向细胞(称为“克隆祖细胞”)引入可遗传标记来完成的,该标记会代代相传。
- 回顾性谱系追踪 方法利用细胞中观察到的变异性 - 例如自然发生的基因突变 - 来推断它们的谱系 (或“系统发育” ) 模型,总结克隆群中细胞分裂的历史。
已经开发了几种用于前瞻性追踪克隆群的方法:例如,组织特异性启动子下的重组酶可用于激活荧光标记,这些标记充当特定组织谱系的可遗传标记 cite, cite, cite, cite, cite。或者,可以使用慢病毒转导将随机 DNA 条形码整合到细胞基因组中,以提供可用于通过测序读数解卷积克隆身份的可遗传标记 cite, cite, cite, cite。尽管这些方法具有高度可扩展性并且通常不需要大量基因工程,但它们只能报告克隆水平的特性,例如克隆大小和组成。
回顾性谱系追踪器克服了这些限制,并且通过报告有关克隆的 亚 克隆动态特性,比前瞻性追踪器提供了额外的优势。传统上,这是通过利用细胞之间自然变异来重建细胞分裂历史来完成的,例如单核苷酸变异 cite, cite, cite, cite, cite, cite, cite] 或拷贝数变异 cite, cite]. 虽然这种方法仍然被广泛成功地用于研究人类肿瘤或组织发育史,但实验人员对突变发生的位置和频率几乎没有控制权。在实验模型中,可以通过设计可进化的谱系追踪器来利用回顾性追踪器的优势,同时改进其局限性。这种可进化的追踪器通常由设计带有可获取突变的“暂存区” (或同义词“靶位点”) 的细胞组成 cite, cite]. 例如,本章关注的一种流行方法使用 Cas9 在靶位点引入插入和缺失 (即“插入缺失”) cite, cite, cite, cite, cite, cite]. 这样,细胞谱系随着时间的推移会获得可遗传的突变,这些突变随后可以通过高通量测序平台读取并用于推断代表细胞谱系模型的系统发育树。在此基础上,出现了其他可以改善数据可解释性的技术:例如,peCHYRON cite 和 DNA 打字机 cite 等方法引入了有序的序列编辑,以便更自信地评估谱系历史。
这两类谱系追踪方法都可以利用单细胞多组学分析的最新进展。例如,研究人员经常使用单细胞 RNA 测序 (scRNA-seq) 同时读取单个细胞的功能状态及其谱系关系 cite, cite, cite, cite, cite。这种多模式读数为新的计算方法创造了机会,我们将在下面详细介绍。
正如上文所述,本章将详细介绍基于可进化 CRISPR/Cas9 的谱系追踪器数据的分析。
注意术语¶
为了方便理解,这里列出了一些章节中使用到的重要术语解释:
- 克隆群 (或 克隆):特定祖细胞所有后代的集合。
- 祖细胞 (clonal progenitor):单个细胞,它分裂增殖形成克隆群。
- 亚克隆分辨力 (subclonal resolution):对克隆群内细胞亚群之间关系的认识。
- 系统发育树 (phylogeny):克隆群细胞分裂历史的模型,通常以树状结构表示。
- 暂存区 (或 靶位点) (scratchpad/target site):人工合成的外源区域,在可进化谱系追踪技术中可以积累定向的突变。
可进化谱系追踪数据分析管道概述¶
在深入研究示例数据集的分析之前,我们将概述用于分析可进化追踪器生成的数据的计算管道(基于 cite)。通常,使用这些系统,分析将从靶位点的扩增子文库的原始测序数据开始(通常来自类似于 10X Chromium 的常规 scRNA-seq 平台)。根据所使用的技术,每个测序的扩增子长度将为 150-300bp;对于基于 CRISPR/Cas9 的可进化追踪器,每个读段将包含一个额外的 Cas9 切割位点。在此数据预处理过程中,分析人员的任务是将读取序列与参考序列比对并识别任何突变(例如插入/缺失 (indel))。
虽然这些数据集的预处理是一个关键步骤,但为了节省篇幅,我们重点关注接收预处理测序数据作为输入的分析管道,并推荐读者查阅外部预处理教程,网址为 here.
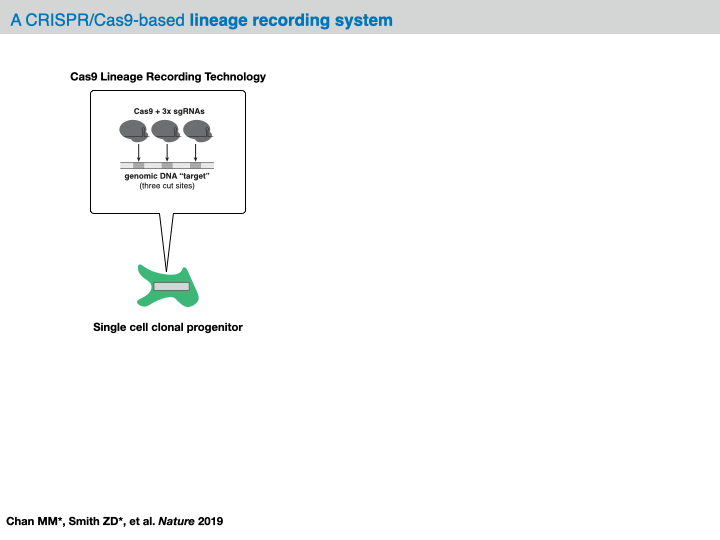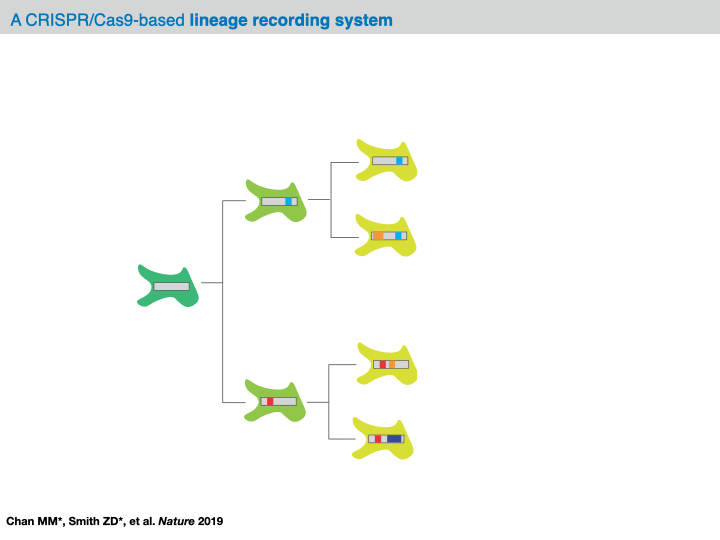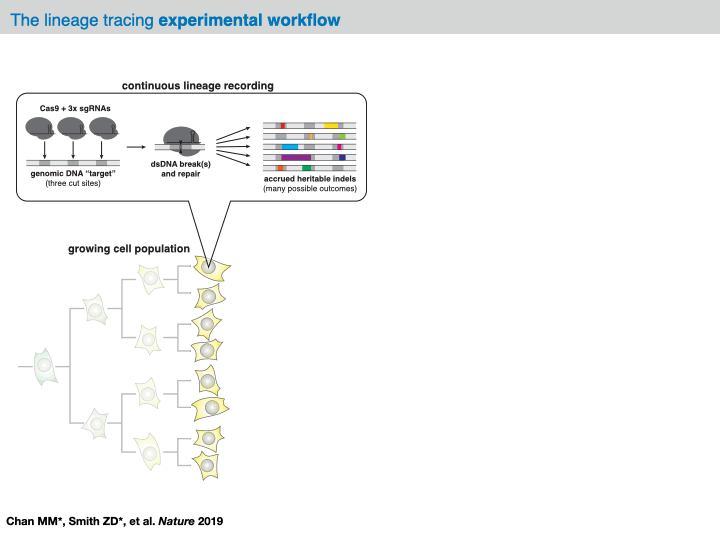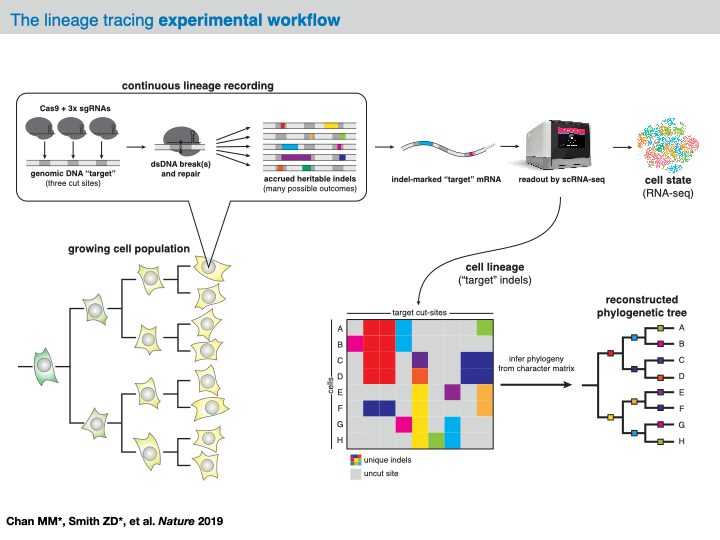
在大多数分析框架中,原始测序 reads 的预处理会产生一个称为字符矩阵的数据结构,该矩阵总结了每个细胞中所有靶位点的观察到的突变。在这个数据结构中,每一行代表一个细胞(或“样本”),每一列代表一个靶位点(或“字符”),(行,列) 的值是分类变量,表示在该特定切割位点观察到的那个细胞中的插入/缺失的标识(或“字符状态”)。根据所使用的技术,这些字符矩阵可以报告来自多达 100 个字符的 100 到 10,000 个样本的任何数据。
此时,这种数据结构概括了可进化谱系追踪测定的技术细节,并开辟了计算推断细胞上系统发育树的可能性。具体来说,目标是学习我们字符矩阵中每个细胞的层次树结构。在这棵树中,每个节点代表一个样本,每条边代表一个谱系关系。重要的是,我们通常只观察到树的叶节点,并且我们将任何未观察到的内部节点集称为祖先节点。(需要注意的是,我们倾向于宽泛地使用术语“系统发育树”,因为它具有精确的定义。实际上,我们经常推断出一个谱系图,总结细胞之间的关系。)
有许多用于从字符矩阵推断系统发育树的算法选择,这些算法可以大致分为“基于字符”和“基于距离”两种方法:
- 基于字符: 在所有可能的树拓扑中进行组合搜索,同时寻求优化字符上的函数(例如,给定字符中观察到的突变的进化历史的可能性)。
- 最大简约性 (Maximum Parsimony) {cite}
cavalli1963:找到具有最少突变的树。 - 最大似然 (Maximum Likelihood) {cite}felsenstein1981:找到具有最大可能突变历史的树 (有关谱系追踪特定算法,我们推荐读者参阅 GAPML {cite}gapml}。 - 贝叶斯系统发育推断 (Bayesian Phylogenetic Inference) {cite}huelsenbeck2001: 找到使进化历史的后验概率给定观察到的突变最大化的树。 - 基于距离: 使用细胞间距离的概念(例如它们共享的编辑次数,记为 $\delta$)来推断系统发育树,并且通常在多项式时间内运行。虽然基于距离的方法可以执行得更快,但它们需要迭代地找到最佳的细胞间差异相似性函数,这同样耗时。
- 邻接加入法 (Neighbor-Joining) {cite}
neighbor-joining:通过根据特定标准迭代地寻找使分支长度最小的配对,从给定的细胞间差异相似性矩阵生成树。 - UPGMA {cite}upgma: 使用比邻接加入法更快的算法从给定的细胞间差异相似性矩阵生成树。然而,它对生成准确的树有更严格的要求。
传统上,从这类数据推断系统发育树极具挑战性,通常需要使用可扩展性较差的组合算法。对于之前描述的单细胞可进化谱系追踪器,当采样细胞数量级大于传统系统发育研究评估的物种数量级时,这些问题会更加恶化。由于非随机缺失数据和非均匀编辑分布,这些挑战进一步加剧。幸运的是,最近出现了用于解决可进化谱系追踪器建模挑战的算法发展 cite, cite, cite,以及用于使用深度分布式计算对超大型树进行推断的扩展 cite。在实践中,大多数应用程序使用最大简约性方法或基于距离的方法(例如邻接加入法)的变体进行树推断。然而,这目前是一个活跃的研发领域,有关更深入的讨论,我们建议读者参阅总结部分。
系统发育树重建之后,下游分析有多种选择。例如,我们可以了解细胞状态在发育过程中的变化率或细胞分裂的相对倾向性。下面,我们将通过代码示例展示这些不同组件如何结合在一起,以获得对细胞谱系基础动态过程的根本见解。
使用 Cassiopeia 进行谱系追踪分析¶
本教程中,我们将主要使用用于谱系追踪分析的少数几个软件包之一 Cassiopeia cite。
通常,Cassiopeia 是一个用于处理和分析可进化谱系追踪测序数据的软件套件。该软件包含四个主要模块:
- 将来自可进化谱系追踪数据集(例如基于 CRISPR/Cas9 的谱系追踪器)的测序 reads 预处理成字符矩阵。
- 使用代码库中可用的一种算法从这些字符矩阵重建系统发育树
- 提供分析工具,从重建的系统发育树中获取见解。
- 模拟用于基准测试的真实系统发育树和谱系追踪数据
尽管这些模块在 Cassiopeia 管道中相互配合,但它们彼此独立。例如,用户可以使用不同的软件套件进行谱系推断,但可以使用 Cassiopeia 代码库中的工具进行重建后分析。
有关更多安装指南和附加文档,我们推荐读者参阅 here.
在小鼠肺癌模型中追踪肿瘤发展¶
在这个案例研究中,我们将利用 {cite}yang2022 提出的最新研究。简而言之,在这项研究中,作者将一个演变中的基于 CRISPR/Cas9 的谱系追踪器整合到了 KP 小鼠非小细胞肺癌模型中 {cite}dupage2009。具体来说,这种小鼠模型携带致癌的 Kras 和 Tp53 突变,在自然条件下不表达。然而,通过腺病毒吸入引入 Cre 重组酶后,这些致癌突变在肺气道上皮的单个细胞中被激活,从而诱导肿瘤。在这项研究中,基于 CRISPR/Cas9 的谱系追踪器在类似控制下,并在肿瘤诱导的同时变得活跃。
使用该系统,作者在大约 4.5-6 个月的时间内追踪了肿瘤从单细胞起源的发展过程,并在此时收获了侵袭性、转移性肿瘤。在肿瘤解离后,作者分析了单细胞的谱系追踪目标位点和 RNA 含量。最终获得了一个包含超过 100 个肿瘤的 70,000 多个细胞的庞大数据集,这些细胞既有谱系追踪信息,也有 scRNA-seq 信息。
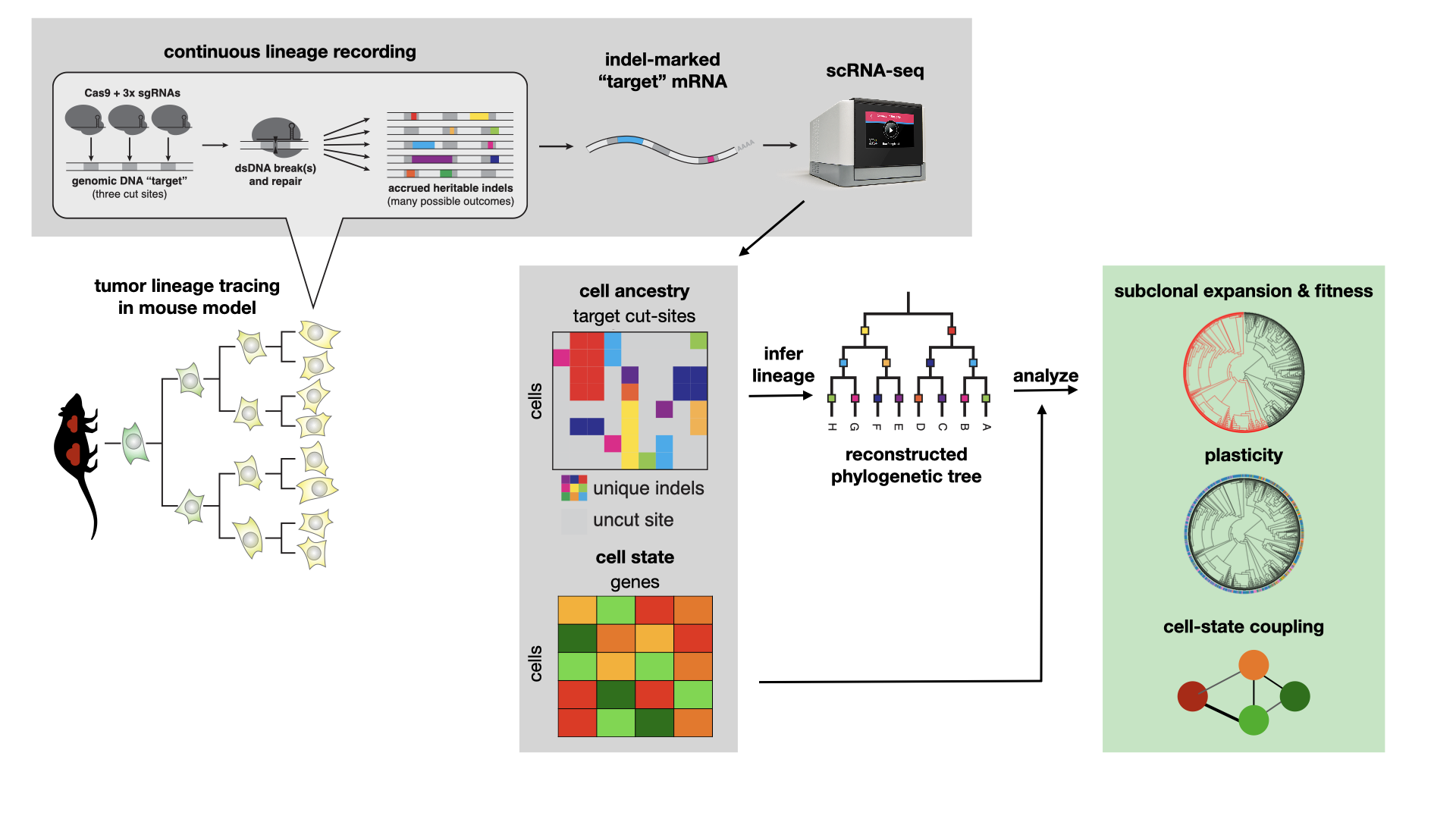
在本教程中,我们将展示用户如何使用处理过的目标位点数据来了解谱系的有趣动态特性。在整个案例研究中,每个谱系将对应于从小鼠肺部采样的单个原发性肿瘤。
在继续之前,请确保您理解我们将使用的数据类型。如上所述,我们配备了 scRNA-seq 计数矩阵和独立的谱系追踪数据(这些数据可以有多种形式;下面,我们从总结每个细胞内各个目标上观察到的插入缺失变异的等位基因表开始。具体描述见下文)。这两类数据将分别处理。谱系追踪数据将用于推断谱系,而 scRNA-seq 数据将用于解释这些谱系的有趣行为。
对于下文分析的数据集,每个细胞被设计有大约 10 个目标位点,每个位点携带串联的 3 个 Cas9 切割位点。每个目标位点都标有一个唯一的整合条形码(简称 "intBC")。总的来说,我们可以期望在每个细胞中观察到 3*(目标位点数)的字符,这些字符可以累积突变并用于谱系推断。有关测序盒结构的更多信息,请参见 Chan, Smith 等。哺乳动物胚胎发生的分子记录。自然 2019。
!wget "https://zenodo.org/record/5847462/files/KPTracer-Data.tar.gz?download=1"
--2022-06-30 10:28:22-- https://zenodo.org/record/5847462/files/KPTracer-Data.tar.gz?download=1 Resolving zenodo.org (zenodo.org)... 137.138.76.77 Connecting to zenodo.org (zenodo.org)|137.138.76.77|:443... connected. HTTP request sent, awaiting response... 200 OK Length: 1304975216 (1.2G) [application/octet-stream] Saving to: ‘KPTracer-Data.tar.gz?download=1’ KPTracer-Data.tar.g 100%[===================>] 1.21G 1.49MB/s in 7m 3s 2022-06-30 10:35:27 (2.94 MB/s) - ‘KPTracer-Data.tar.gz?download=1’ saved [1304975216/1304975216]
!tar -xvzf KPTracer-Data.tar.gz?download=1
Environment setup.¶
Before we enter this notebook's analysis, let's set up our environment.
import cassiopeia as cas
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import scanpy as sc
from cassiopeia.preprocess import lineage_utils
检查数据¶
在对目标位点测序数据进行预处理后,Cassiopeia 使用一个 allele_table 来总结在每个目标位点整合上观察到的突变。
如上所述,当前系统每个细胞大约有 10 个目标位点,每个目标位点包含三个 Cas9 切割位点。标记为 intBC 的列表示目标位点,标记为 r* 的列表示该目标位点上的 Cas9 切割位点。每个切割位点的突变存储为 CIGAR 字符串,指示观察到的插入缺失变异的大小和类型。
其他元数据也可以存储在该表中,例如与目标位点分子相关的总 UMIs 数量和读取数,分子来自哪个细胞,以及该细胞属于哪个肿瘤。
allele_table = pd.read_csv(
"KPTracer-Data/KPTracer.alleleTable.FINAL.txt", sep="\t", index_col=0
)
allele_table.head(5)
/var/folders/kt/wk3kbll507v4h18f8nmbzkl80000gq/T/ipykernel_65366/3031588044.py:1: DtypeWarning: Columns (12) have mixed types. Specify dtype option on import or set low_memory=False.
allele_table = pd.read_csv("KPTracer-Data/KPTracer.alleleTable.FINAL.txt", sep='\t', index_col = 0)
| cellBC | intBC | r1 | r2 | r3 | allele | sampleID | UMI | readCount | Tumor | MetFamily | ES_clone | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | L9.TTTGTCATCTGTCAAG-1 | TTCCCTATTTGCTA | CGCCG[111:2D]AAATG | GATAT[None]CTCTG | AATTC[220:1I]GGCGGA | CGCCG[111:2D]AAATGGATAT[None]CTCTGAATTC[220:1I... | L9 | 54 | 796 | 3724_NT_T1 | 3724_NT_T1 | 2E1 |
| 1 | L9.TTTGTCATCTGTCAAG-1 | TGTTTTTGTCTGCA | CGCCG[111:1I]AAAAAA | CATGT[151:19D]TGGTT | TTAAT[218:2D]GCGGA | CGCCG[111:1I]AAAAAACATGT[151:19D]TGGTTTTAAT[21... | L9 | 13 | 209 | 3724_NT_T1 | 3724_NT_T1 | 2E1 |
| 2 | L9.TTTGTCATCTGTCAAG-1 | TGTGAAGGTCAATA | CCGAA[113:49D]GATAT | CCGAA[113:49D]GATAT | AATTC[220:5D]GGACA | CCGAA[113:49D]GATATCCGAA[113:49D]GATATAATTC[22... | L9 | 68 | 1193 | 3724_NT_T1 | 3724_NT_T1 | 2E1 |
| 3 | L9.TTTGTCATCTGTCAAG-1 | TCAGGCGATGCGAA | CGCCG[111:1I]AAAAAA | CGATA[166:1I]TTCTCT | TAATT[219:2D]CGGAG | CGCCG[111:1I]AAAAAACGATA[166:1I]TTCTCTTAATT[21... | L9 | 43 | 745 | 3724_NT_T1 | 3724_NT_T1 | 2E1 |
| 4 | L9.TTTGTCATCTGTCAAG-1 | TATGATTAGTCGCG | CGCCG[111:1D]AAAAT | GATAT[167:54D]CGGAG | GATAT[167:54D]CGGAG | CGCCG[111:1D]AAAATGATAT[167:54D]CGGAGGATAT[167... | L9 | 27 | 530 | 3724_NT_T1 | 3724_NT_T1 | 2E1 |
We'll focus on the data from KP tumors without any additional perturbations:
all_tumors = allele_table["Tumor"].unique()
primary_nt_tumors = [
tumor
for tumor in all_tumors
if "NT" in tumor and tumor.split("_")[2].startswith("T")
]
primary_nt_allele_table = allele_table[allele_table["Tumor"].isin(primary_nt_tumors)]
如上所述,我们预计每个肿瘤群体中会有大约 10 个独特的 intBCs(即目标位点)。下面,我们总结了一些数据集的关键统计数据。
每个肿瘤的 intBC 数量¶
对于分析人员来说,一个感兴趣的统计数据是每个肿瘤的 intBC 数量,因为这提供了关于每个克隆群体信息容量的信息。
在最近的应用中({cite}yang2022),我们发现高质量克隆通常有 5 到 30 个 intBCs(对应于 15-90 个字符)。通过绘制每个肿瘤的 intBC 数量,我们可以确保在重建之前没有需要过滤掉的异常肿瘤。
primary_nt_allele_table.groupby(["Tumor"]).agg({"intBC": "nunique"}).plot(kind="bar")
plt.ylabel("Number of unique intBCs")
plt.title("Number of intBCs per Tumor")
plt.show()

正如预期的那样,我们观察到大多数肿瘤有大约 10 个 intBCs,而一个肿瘤报告的 intBC 数量较少,有几个肿瘤报告的 intBC 数量超过 25 个。接下来,我们将讨论过滤低质量肿瘤的策略。
每个肿瘤的大小¶
我们还对将要重建的肿瘤的大小感兴趣。通常可以观察到克隆的细胞数量在 100 到 10,000 之间。同样地,我们需要确保没有明显的异常值——特别是那些太小而无法重建的克隆(通常少于 100 个细胞)。
primary_nt_allele_table.groupby(["Tumor"]).agg({"cellBC": "nunique"}).sort_values(
by="cellBC", ascending=False
).plot(kind="bar")
plt.yscale("log")
plt.ylabel("Number of cells (log)")
plt.title("Size of each tumor")
plt.show()

We see that we have one very large clone (with ~30,000 cells) and the rest of the clones are in the expected range, reporting between 1,000 and 5,000 cells.
为谱系重建准备数据¶
基于 Cas9 的追踪器的一个普遍特征是,某些插入和缺失比其他插入和缺失更为常见。这可能是一个重要的建模问题,因为高概率的编辑可以独立地多次发生,这可能会导致分析人员错误地认为两个细胞彼此相关。
Cassiopeia 内置了一个工具,可以通过计算插入缺失在无关肿瘤或 intBCs 中出现的次数来估计插入和缺失的先验概率。
在观察到的插入缺失频率时,我们看到一些插入缺失(小的删除或插入)在克隆中经常出现。另一方面,我们还观察到在单个克隆中观察到的几种编辑。由于频率的差异,分析人员应尝试将这些观察到的偏差纳入后续分析中。
indel_priors = cas.pp.compute_empirical_indel_priors(
allele_table, grouping_variables=["intBC", "MetFamily"]
)
indel_priors.sort_values(by="count", ascending=False).head()
| count | freq | |
|---|---|---|
| indel | ||
| TAATT[219:2D]CGGAG | 664.0 | 0.764977 |
| CGCCG[111:1I]AAAAAA | 584.0 | 0.672811 |
| CGCCG[111:1D]AAAAT | 504.0 | 0.580645 |
| CGCCG[111:2D]AAATG | 401.0 | 0.461982 |
| AATTC[220:3D]GAGGA | 395.0 | 0.455069 |
indel_priors.sort_values(by="count").head(5)
| count | freq | |
|---|---|---|
| indel | ||
| ATATC[168:49I]GTTGTGGCCCAACATGGCAGCGTGCCGTAGCTTAGTTGTCAGGCCATTTGCTGG | 1.0 | 0.001152 |
| ACGCC[110:2D]AGAAT | 1.0 | 0.001152 |
| CATCT[101:15D]TGGCC | 1.0 | 0.001152 |
| CCCGG[111:3D]ATTGG | 1.0 | 0.001152 |
| CCGAA[113:1I]GGAATG | 1.0 | 0.001152 |
过滤低质量肿瘤¶
在肿瘤重建之前,我们将过滤掉具有以下特征的肿瘤:(i) 谱系追踪动力学较差,或 (ii) 太小而无法考虑进行分析。
在之前的出版物中({cite}quinn2021 和 {cite}yang2022),我们发现以下统计数据对于确定肿瘤质量非常有用:
- 百分比唯一性:这是肿瘤中独特谱系状态的百分比。
- 百分比切割:这是细胞群体中 Cas9 目标位点发生突变的百分比。
- 百分比耗尽:这是在细胞之间相同的目标位点的比例(即耗尽的位点)。
- 肿瘤大小:肿瘤中的细胞数量。
# utility functions for computing summary statistics
def compute_percent_indels(character_matrix):
"""Computes the percentage of sites carrying indels in a character matrix.
Args:
character_matrix: A pandas Dataframe summarizing the mutation status of each cell.
Returns:
A percentage of sites in cells that contain an edit.
"""
all_vals = character_matrix.values.ravel()
num_not_missing = len([n for n in all_vals if n != -1])
num_uncut = len([n for n in all_vals if n == 0])
return 1.0 - (num_uncut / num_not_missing)
def compute_percent_uncut(cell):
"""Computes the percentage of sites uncut in a cell.
Args:
A vector containing the edited sites for a particular cell.
Returns:
The number of sites uncut in a cell.
"""
uncut = 0
for i in cell:
if i == 0:
uncut += 1
return uncut / max(1, len([i for i in cell if i != -1]))
def summarize_tumor_quality(
allele_table,
minimum_intbc_thresh=0.2,
minimum_number_of_cells=2,
maximum_percent_uncut_in_cell=0.8,
allele_rep_thresh=0.98,
):
"""Compute QC statistics for each tumor.
Computes statistics for each clone that will be used for filtering tumors for downstream lineage reconstruction.
Args:
allele_table: A Cassipoeia allele table summarizing the indels on each molecule in each cell.
min_intbc_thresh: The minimum proportion of cells that an intBC must appear in to be considered
for downstream reconstruction.
minimum_number_of_cells: Minimum number of cells in a tumor to be processed in this QC pipeline.
maximum_percent_uncut_in_cell: The maximum percentage of sites allowed to be uncut in a cell. If
a cell exceeds this threshold, it is filtered out.
allele_rep_thresh: Maximum allele representation in a single cut site allowed. If a character has
less diversity than allowed, it is filtered out.
Returns:
A pandas Dataframe summarizing the quality-control information for each tumor.
"""
tumor_statistics = {}
NUMBER_OF_SITES_PER_INTBC = 3
# iterate through Tumors and compute summary statistics
for tumor_name, tumor_allele_table in allele_table.groupby("Tumor"):
if tumor_allele_table["cellBC"].nunique() < minimum_number_of_cells:
continue
tumor_allele_table = allele_table[allele_table["Tumor"] == tumor_name].copy()
tumor_allele_table["lineageGrp"] = tumor_allele_table["Tumor"].copy()
lineage_group = lineage_utils.filter_intbcs_final_lineages(
tumor_allele_table, min_intbc_thresh=minimum_intbc_thresh
)[0]
number_of_cutsites = (
len(lineage_group["intBC"].unique()) * NUMBER_OF_SITES_PER_INTBC
)
character_matrix, _, _ = cas.pp.convert_alleletable_to_character_matrix(
lineage_group, allele_rep_thresh=allele_rep_thresh
)
# We'll hit this if we filter out all characters with the specified allele_rep_thresh
if character_matrix.shape[1] == 0:
character_matrix, _, _ = cas.pp.convert_alleletable_to_character_matrix(
lineage_group, allele_rep_thresh=1.0
)
number_dropped_intbcs = number_of_cutsites - character_matrix.shape[1]
percent_uncut = character_matrix.apply(
lambda x: compute_percent_uncut(x.values), axis=1
)
# drop normal cells from lineage (cells without editing)
character_matrix_filtered = character_matrix[
percent_uncut < maximum_percent_uncut_in_cell
]
percent_unique = (
character_matrix_filtered.drop_duplicates().shape[0]
/ character_matrix_filtered.shape[0]
)
tumor_statistics[tumor_name] = (
percent_unique,
compute_percent_indels(character_matrix_filtered),
number_dropped_intbcs,
1.0 - (number_dropped_intbcs / number_of_cutsites),
character_matrix_filtered.shape[0],
)
tumor_clone_statistics = pd.DataFrame.from_dict(
tumor_statistics,
orient="index",
columns=[
"PercentUnique",
"CutRate",
"NumSaturatedTargets",
"PercentUnsaturatedTargets",
"NumCells",
],
)
return tumor_clone_statistics
Calculate and visualize tumors' statistics¶
tumor_clone_statistics = summarize_tumor_quality(primary_nt_allele_table)
NUM_CELLS_THRESH = 100
PERCENT_UNIQUE_THRESH = 0.05
PERCENT_UNSATURATED_TARGETS_THRESH = 0.2
low_qc = tumor_clone_statistics[
(tumor_clone_statistics["PercentUnique"] <= PERCENT_UNIQUE_THRESH)
| (
tumor_clone_statistics["PercentUnsaturatedTargets"]
<= PERCENT_UNSATURATED_TARGETS_THRESH
)
].index
small = tumor_clone_statistics[
(tumor_clone_statistics["NumCells"] < NUM_CELLS_THRESH)
].index
unfiltered = np.setdiff1d(tumor_clone_statistics.index, np.union1d(low_qc, small))
h = plt.figure(figsize=(6, 6))
plt.scatter(
tumor_clone_statistics.loc[unfiltered, "PercentUnsaturatedTargets"],
tumor_clone_statistics.loc[unfiltered, "PercentUnique"],
color="black",
)
plt.scatter(
tumor_clone_statistics.loc[low_qc, "PercentUnsaturatedTargets"],
tumor_clone_statistics.loc[low_qc, "PercentUnique"],
color="red",
label="Poor QC",
)
plt.scatter(
tumor_clone_statistics.loc[small, "PercentUnsaturatedTargets"],
tumor_clone_statistics.loc[small, "PercentUnique"],
color="orange",
label="Small lineages",
)
plt.axhline(y=PERCENT_UNIQUE_THRESH, color="red", alpha=0.5)
plt.axvline(x=PERCENT_UNSATURATED_TARGETS_THRESH, color="red", alpha=0.5)
plt.xlabel("Percent Unsaturated")
plt.ylabel("Percent Unique")
plt.title("Summary statistics for Tumor lineages")
plt.legend(loc="lower right")
plt.show()

该图总结了我们的质量控制过滤工作。每个点对应一个肿瘤,其颜色指示其过滤状态:
- 用 红色 标记的肿瘤被过滤掉,因为它们的独特状态太少或可用于重建的字符太少。
- 用 橙色 标记的肿瘤被过滤掉,因为它们太小,无法进行重建(我们使用 100 个细胞的过滤器)。
- 黑色 标记的肿瘤通过了我们的质量控制过滤,并将被考虑用于重建。
肿瘤 3726_NT_T1 似乎是一个具有满意追踪数据质量的谱系,我们将使用 Cassiopeia 重建该肿瘤谱系。
重建选定的肿瘤 (3726_NT_T1)¶
为了重建谱系,我们需要将特定肿瘤的等位基因表转换为“字符矩阵”。如上所述,这些数据结构总结了每个细胞中每个目标位点观察到的突变。默认情况下,未切割的位点用 0 表示,缺失的位点用 -1 表示。矩阵中的所有其他值对应于唯一的插入缺失变异。
肿瘤 3726_NT_T1 似乎是一个具有满意追踪数据质量的谱系,我们将使用 Cassiopeia 重建该肿瘤谱系。
tumor = "3726_NT_T1"
tumor_allele_table = primary_nt_allele_table[primary_nt_allele_table["Tumor"] == tumor]
n_cells = tumor_allele_table["cellBC"].nunique()
n_intbc = tumor_allele_table["intBC"].nunique()
print(
f"Tumor population {tumor} has {n_cells} cells and {n_intbc} intBCs ({n_intbc * 3} characters)."
)
Tumor population 3726_NT_T1 has 772 cells and 10 intBCs (30) characters.
(
character_matrix,
priors,
state_to_indel,
) = cas.pp.convert_alleletable_to_character_matrix(
tumor_allele_table, allele_rep_thresh=0.9, mutation_priors=indel_priors
)
character_matrix.head(5)
Dropping the following intBCs due to lack of diversity with threshold 0.9: ['ACTCTGCTCCAGATr2', 'ACTCTGCTCCAGATr3', 'GCCTACTTAAGTCCr1', 'GTTTATTTCCGTATr3', 'TATGATTAGTCGCGr1', 'TATGATTAGTCGCGr2', 'TGATATAAATCTTTr2', 'TTCCCTATTTGCTAr2', 'TGTTTTTGTCTGCAr1', 'ACAGGTGCTCAAATr1', 'ACAGGTGCTCAAATr2', 'ACAGGTGCTCAAATr3']
Processing characters: 0%| | 0/18 [00:00<?, ?it/s]
| r1 | r2 | r3 | r4 | r5 | r6 | r7 | r8 | r9 | r10 | r11 | r12 | r13 | r14 | r15 | r16 | r17 | r18 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L6.TTTGTCACACATCCAA-1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | -1 | -1 |
| L6.TTTGGTTTCTGAGTGT-1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| L6.TTTGGTTCATGTAAGA-1 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| L6.TTTGCGCAGCTCCTCT-1 | 1 | 2 | 3 | 3 | 3 | 3 | 1 | 3 | 3 | 3 | 1 | 1 | 3 | 3 | 3 | 3 | 3 | 0 |
| L6.TTTATGCTCGCCGTGA-1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 4 | 0 |
重建谱系¶
有多种算法可以用于推断系统发育树,其中许多可以使用 Cassiopeia 所使用的一般字符矩阵格式来实现。因此,Cassiopeia 实现了几种可用于谱系重建的算法,其他许多算法可以使用通用的 CassiopeiaSolver API 来实现。最受欢迎的算法包括:
- VanillaGreedy:一种简单且高效的启发式算法,适合进行首次谱系重建。基于 Gusfield 算法 {cite}
gusfield1991并在 {cite}jones2020中描述。 - Neighbor-Joining:一种经典的基于距离的算法,如 {cite}
neighbor-joining中所述。 - UPGMA:一种高效的基于距离的算法,对样本之间的关系有特殊假设。最初在 {cite}
upgma中描述。 - ILPSolver:一种 Steiner-Tree 优化推断算法的实现,如 {cite}
jones2020中所述。该算法较慢(通常无法扩展到超过约 1.5k 个细胞)但精确。 - HybridSolver:一种分而治之的混合算法,使用贪心自上而下的算法来分割数据,并使用精确的算法(即 ILPSolver)来解决子问题。在 {cite}
jones2020中描述。
对于本教程,我们将使用 VanillaGreedySolver 进行操作。其他 Cassiopeia 代码库 中的求解器可以作为替代方案。
为了进行推断,我们将实例化一个 CassiopeiaTree,这是一个 Cassiopeia 特定的数据结构,用于存储字符矩阵和谱系元数据,并提供用于树操作的特定工具。我们还将实例化一个 VanillaGreedySolver,它将填充 CassiopeiaTree 对象中的 tree 字段。
拥有这么多算法可供选择,可能会让人觉得难以选择“正确”的算法。因此,我们通常建议用户使用几种算法进行推断以进行比较。除此之外,用户还需要考虑数据集的大小和之前的基准研究作为选择算法的标准。根据经验,HybridSolver 在可扩展性和精度之间取得了良好的平衡,而 Neighbor-Joining 是一个方便的比较替代方案(Neighbor-Joining 还有一个优势,即它属于不同类别的算法(基于距离与基于字符),因此它可能会突出谱系的不同部分)。在本教程中,我们将使用 GreedySolver,因为它非常高效,通常可以作为数据的首次快速处理,然后再应用更精确(但较慢)的算法。
tree = cas.data.CassiopeiaTree(character_matrix=character_matrix, priors=priors)
greedy_solver = cas.solver.VanillaGreedySolver()
运行 greedy_solver.solve 将执行树推断并填充 CassiopeiaTree 对象中的 tree 字段。
推断完成后,我们可以利用 Cassiopeia 的可视化库来研究所得的树结构。我们会注意到推断中存在一些错误,但总体上,大群细胞似乎被正确地放置。如上所述,这可以作为在应用更耗时和复杂的算法之前对数据的初步处理。
greedy_solver.solve(tree)
cas.pl.plot_matplotlib(tree, orient="right", allele_table=tumor_allele_table)
filling in multiindex table: 0%| | 0/6432 [00:00<?, ?it/s]
100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 30/30 [00:11<00:00, 2.71it/s]
(<Figure size 504x504 with 1 Axes>, <AxesSubplot:>)

这种重建的表示对于评估算法性能非常有用。评估这些重建的性能可能相当困难,但随着实践会变得容易。一条好的评估性能的建议是选择一列,看看一个给定的插入缺失是否在多个不相关的细胞组中多次出现。这可能表明存在算法错误。
如您所见,大多数细胞(热图中的行)都位于具有相似编辑状态的细胞邻域中。这意味着切割位点中的特定插入缺失被干净地分组在一起(例如,左边第四列中的粉红色等位基因)。如果几个等位基因将细胞分组在一起,我们也可以对重建结果充满信心——例如,第四列中的粉红色等位基因,第六列中的淡紫色等位基因,等等。
然而,从这次重建中可以看出一些错误。例如,在第四列中,共享紫色等位基因的两个细胞组被分开了。热图中的其他等位基因表明这些细胞应该被分组在一起,这表明这是一个算法错误。
正如我们上面讨论的那样,由于分析人员有多种算法选项可用,应用几种算法进行比较通常是有用的。
Quantifying dynamic properties from the tree¶
From the tree structure learning properties about the population would be of biological interest. Some common properties would be:
- The timing and location of "expansion" events (i.e., the emergence of a population that can outgrow their neighbors)### 量化树的动态特性
通过树结构学习群体的特性在生物学上具有重要意义。一些常见的特性包括:
- “扩展”事件的时间和位置(即能够超越邻居的群体的出现)
- 个体细胞的适应性(即相对生长速率)
- 群体中细胞状态变化的次数(即可塑性)
- 群体中常见的分化程序
还有许多其他下游分析任务,这是一个不断发展的研究领域。
下面我们提供了一些示例,说明如何使用 Cassiopeia 来推断其中一些参数。
推断扩展事件¶
我们将使用 compute_expansion_pvalues 计算树中某个内部节点产生更快生长群体的概率。这个程序在 {cite}yang2022 中介绍,它对树进行深度优先搜索,并为每个内部节点注释一个概率,即后代数量在中性进化模型下生成的概率。该程序涉及一些感兴趣的超参数:
min_clade_size:考虑的最小分支大小(即内部节点下的叶子数量)。通常,太小的分支是没有信息价值的。min_depth:扩展开始的最小深度。min_depth为 0 表示根节点可以被认为是扩展的起点;一个更合理的深度是 1(根节点下的一级)。
通过这个程序,我们可以筛选节点并突出显示扩展发生的位置。例如,在 {cite}yang2022 中,作者提出了一种策略来突出显示最重要的扩展事件。下面,我们以红色突出显示了一个有趣的扩展事件。
cas.tl.compute_expansion_pvalues(tree, min_clade_size=(0.15 * tree.n_cell), min_depth=1)
# this specifies a p-value for identifying expansions unlikely to have occurred
# in a neutral model of evolution
probability_threshold = 0.01
expanding_nodes = []
for node in tree.depth_first_traverse_nodes():
if tree.get_attribute(node, "expansion_pvalue") < probability_threshold:
expanding_nodes.append(node)
cas.pl.plot_matplotlib(tree, clade_colors={expanding_nodes[6]: "red"})
(<Figure size 504x504 with 1 Axes>, <AxesSubplot:>)

在突出显示扩展发生的位置时,我们可以将树划分为更具侵略性和较不具侵略性的子群体。我们以红色突出显示了一个可能更具侵略性的群体,因为它是一个检测到的扩展。
在原始研究中,作者发现 不同的转录模式 与 扩展区域 相关,这些区域可能赋予了适应性优势。
推断树的可塑性¶
来自该模型的肿瘤由处于不同转录状态的细胞组成。可以在此数据集的低维投影(如使用 UMAP 计算的投影)上观察到该数据集的异质性:
kptracer_adata = sc.read_h5ad("KPTracer-Data/expression/adata_processed.nt.h5ad")
sc.pl.umap(
kptracer_adata,
color="Cluster-Name",
show=False,
title="Cluster Annotations, full dataset",
)
plt.show()
# plot only tumor of interest
fig = plt.figure(figsize=(10, 6))
ax = plt.gca()
sc.pl.umap(kptracer_adata[tree.leaves, :], color="Cluster-Name", show=False, ax=ax)
sc.pl.umap(
kptracer_adata[np.setdiff1d(kptracer_adata.obs_names, tree.leaves), :],
show=False,
ax=ax,
title=f"Cluster Annotations, {tumor}",
)
plt.show()


UMAP 单细胞转录组聚类的可视化揭示了肿瘤中细胞状态的连续性。正如作者所预期的,这些状态在之前的研究中已被描述,表明基于 CRISPR/Cas9 的记录并未明显扰动肿瘤的发展。
我们还可以只选择感兴趣的肿瘤中的细胞,并观察到即使在这个单一肿瘤中,细胞也占据不同的转录状态。
此外,我们可以将细胞状态分配覆盖到树的层次结构上,并评估细胞状态的遗传性。在可视化中,我们可以看到谱系的某些部分比其他部分更混合。
tree.cell_meta = pd.DataFrame(
kptracer_adata.obs.loc[tree.leaves, "Cluster-Name"].astype(str)
)
cas.pl.plot_matplotlib(tree, meta_data=["Cluster-Name"])
(<Figure size 504x504 with 1 Axes>, <AxesSubplot:>)

在上面的可视化中,树外的颜色条代表每个细胞的聚类注释。如果状态在各代之间非常稳定,我们预计相邻细胞之间的状态分配会有很强的一致性。然而,在上面的图中,我们看到细胞常常处于与其最近邻居不同的状态(特别是在系统发育树的右侧)。
这些细胞状态的不稳定性被称为“有效可塑性”,可以使用几种算法进行量化。一种方法是使用 Fitch-Hartigan 最大简约算法来推断细胞状态必须改变的最小次数,以产生观察到的模式。该功能已在 Cassiopeia 中实现,可以如下使用:
parsimony = cas.tl.score_small_parsimony(tree, meta_item="Cluster-Name")
plasticity = parsimony / len(tree.nodes)
print(f"Observed effective plasticity score of {plasticity}.")
Observed effective plasticity score of 0.2356902356902357.
这种有效可塑性评分介于 0 和 1 之间,表示谱系的整体混合程度。得分为 0 表示完全没有混合,得分为 1 表示每个细胞与其姐妹细胞的状态不同。
通常,我们更关心单细胞的有效可塑性测量。我们可以如下计算单细胞的可塑性评分:
# compute plasticities for each node in the tree
for node in tree.depth_first_traverse_nodes():
effective_plasticity = cas.tl.score_small_parsimony(
tree, meta_item="Cluster-Name", root=node
)
size_of_subtree = len(tree.leaves_in_subtree(node))
tree.set_attribute(
node, "effective_plasticity", effective_plasticity / size_of_subtree
)
tree.cell_meta["scPlasticity"] = 0
for leaf in tree.leaves:
plasticities = []
parent = tree.parent(leaf)
while True:
plasticities.append(tree.get_attribute(parent, "effective_plasticity"))
if parent == tree.root:
break
parent = tree.parent(parent)
tree.cell_meta.loc[leaf, "scPlasticity"] = np.mean(plasticities)
cas.pl.plot_matplotlib(tree, meta_data=["scPlasticity"])
kptracer_adata.obs["scPlasticity"] = np.nan
kptracer_adata.obs.loc[tree.leaves, "scPlasticity"] = tree.cell_meta["scPlasticity"]
# plot only tumor of interest
fig = plt.figure(figsize=(10, 6))
ax = plt.gca()
sc.pl.umap(kptracer_adata[tree.leaves, :], color="scPlasticity", show=False, ax=ax)
sc.pl.umap(
kptracer_adata[np.setdiff1d(kptracer_adata.obs_names, tree.leaves), :],
show=False,
ax=ax,
)
plt.title(f"Single-cell Effective Plasticity, {tumor}")
plt.show()


在比较树上的推断有效可塑性模式和低维可视化时,我们可以观察到,正如预期的那样,转录状态混合更多的区域具有更高的有效可塑性。我们还可以观察到,有效可塑性似乎在“中期”聚类中更为丰富,如“AT1-like”和“高可塑性”状态。
有关这些模式的更多讨论,请参阅原始研究 {cite}yang2022。
结论¶
在本章中,我们概述了谱系追踪技术,并展示了一个使用最新数据集进行基于 CRISPR/Cas9 的谱系追踪分析的案例研究。在结束时,我们指出了谱系追踪分析和工具开发的其他资源,并强调了新用户的关键要点。
由于这是一个新兴领域,我们预计将看到大规模时间序列谱系追踪数据集数量的增加,以及专门用于分析这些数据的新计算工具的开发 {cite}rodriguez2022 和 {cite}mukhopadhyay2022。我们预计,主要重点将放在将基因表达与谱系信息整合的方法上,从而提供更完整的个体生物学图景。此外,遵循以下详细说明的新方向,我们预计方法将重点关注从时间序列数据中推断轨迹。
New directions¶
新的系统发育推断算法¶
在新的谱系推断算法方面,有许多有前途的方向:
可扩展的贝叶斯推断:与更传统的系统发育算法的趋势类似,一个潜在的方向是将贝叶斯方法扩展到更大的输入规模。虽然大多数贝叶斯算法利用马尔可夫链蒙特卡洛(MCMC)来估计后验分布 {cite}
huelsenbeck2001,但变分推断的进展将极大地提高贝叶斯算法的可扩展性 {cite}zhang2018。这种进展的概率性质将支持对树的不确定性进行高通量估计,并且与其他单细胞转录组贝叶斯方法(如 scVI {cite}lt:Lopez2018)自然契合。改进的基于距离的算法:基于距离的算法的一个基本方面是根据样本的突变数据和已知的数据显示估计样本之间的相异性。考虑到这一点,一个有前途的方向是开发更具统计稳健性和一致性的相异性函数,以适应不断发展的谱系追踪器,通过考虑突变的产生方式和特定突变的可能性先验。这在 DCLEAR {cite}
gong2021和 {cite}fang2022中已经证明是成功的。继续在这一领域的进展将大大推动基于距离的算法,因为这些算法可以在多项式时间内运行,并在具有适当相异性函数的情况下生成非常准确的树。
解释时间序列谱系追踪数据的计算工具¶
包含时间序列信息的谱系追踪研究的日益复杂性必须伴随着计算方法的发展,这些方法不仅限于构建系统发育树。即,整合多组学测量与谱系追踪和时间信息的方法,以恢复控制细胞状态、分化和行为的程序 {cite}mukhopadhyay2022。
由于这一领域刚刚起步,目前可用的工具数量仍然有限,但值得强调一些领先的方法:
LineageOT针对不断发展的 CRISPR/Cas9 设置 {cite}forrow2021:一种通用方法,用于从带有每个时间点谱系信息的 scRNA-seq 时间序列中推断发育轨迹,适用于不断发展的 CRISPR/Cas9 设置。该方法作为 Waddington OT 算法 {cite}schiebinger2019的扩展,考虑了谱系关系在将细胞从早期映射到后期时间点时的重要性。在计算时间点对之间的转移矩阵时,LineageOT 基于其谱系相似性校正后期时间点的表达谱。该方法已成功应用于重建 C. elegans 发育的时间序列。此外,通过模拟证明LineageOT可以准确恢复复杂的轨迹结构,而这些结构无法仅通过细胞状态的测量来恢复。更多详细信息和教程请参阅 https://lineageot.readthedocs.io。CoSpar针对静态条形码谱系追踪数据 {cite}wang2022:一种从单细胞转录组学中推断细胞动态并与静态条形码谱系追踪数据结合的计算方法。该方法基于生物动态的两个基本假设:(i) 处于相似状态的细胞行为相似,(ii) 细胞限制其可能的动态以给出稀疏转变。CoSpar的应用展示在造血、重编程和定向分化数据集上。这些例子表明,CoSpar可以识别先前未检测到的早期命运偏差,预测涉及命运选择的转录因子和受体。文档和详细示例可在 https://cospar.readthedocs.io/ 找到。
开发新计算方法的资源¶
尽管现有的分析工具库非常强大,但仍有很大的发展空间。为了促进未来的努力,我们重点介绍一些用于算法基准测试的工具:
- 最近的 Allen Institute 系谱重建 DREAM 挑战 {cite}
gong2021生成了三个基准数据集,用于评估新算法。这些数据集中有两个是合成的(即模拟的),而另一个是使用 intMEMOIR 技术 {cite}chow2021生成的。最近发布的结果提供了成功算法的行为见解,并提供了这些基准数据集的访问权限 {cite}gong2021。 Cassiopeia-benchmark是 Cassiopeia 中的一个模块,允许用户高效生成模拟的谱系数据,以对新的谱系重建算法进行基准测试。尽管该模块主要关注基于 CRISPR/Cas9 的进化追踪器模拟,但一般模拟器 API 可以扩展以适应其他技术考虑。作者在他们的网站上提供了使用该基准测试套件的详细教程。TedSim是一个模拟框架,不仅可以模拟条形码数据,还可以在谱系上模拟转录组数据 {cite}pan2022。这个模拟框架对于测试下游分析工具非常有用,例如那些设计用来从谱系追踪数据中推断发育轨迹的工具。
关键要点¶
- 请记住,实验数据的分辨率仍然有限。应进行统计分析和谱系信息质量评估,以识别可应用谱系重建的个体。如上所示,有专门的工具可以轻松提供相关统计数据。
- 通常很难预先确定哪个算法在重建中表现最佳,即使表现良好的算法也可能突出谱系的不同部分。建议您应用几种算法进行重建以便进行比较。
- 像任何没有确凿真相的分析一样,任何结论都必须谨慎得出。首先,始终是一个好习惯,将谱系与插入缺失热图一起可视化,以确保没有明显的重建错误(有关如何通过目测评估重建准确性的更多提示,请参阅“重建谱系”部分)。此外,得出更强结论的有效策略是比较用不同算法推断的同一谱系的下游分析结果。当然,正如大多数涉及计算结论的问题一样,我们通常建议在实验系统中验证结论。