5.4 Perturbation modeling¶
Motivation¶
单细胞实验方案的进步允许进行大规模多重实验,以在数千种独特条件下测量数十万细胞。这些通常被称为“扰动”,即由外部影响引起的临时或永久性变化 {cite}srivatsan2020。最近,这些技术已被改编用于多模态读数的CRISPR-Cas9分析 {cite}Papalexi2021,frangieh2021、全基因组扰动 {cite}replogle2021 和组合扰动 {cite}wessels2022。尽管实验有所进展,但探索组合基因敲除或药物组合的大规模扰动空间仍然具有挑战性。广泛的探索空间促使开发用于建模单细胞扰动响应的计算方法 {cite}ji2021。
扰动建模涉及以下领域 {cite}ji2021:
- 扰动响应:在给定对照和处理条件信息的情况下,预测扰动后的组学特征。预测可以通过预测特征与真实值的相关性来评估。还可以预测表型测量值,如IC50值、剂量反应曲线下面积、毒性和生存率。
- 目标和机制:使用组学测量预测扰动的目标和机制。即使对于未表征的化合物,也可以通过扰动建模识别药物作用方式。
- 扰动相互作用:预测扰动的组合效应,以了解遗传和药物或药物组合之间的相互影响。
- 化学性质:使用组学测量预测扰动的化学性质,如分子指纹、R基团、药效团甚至完整化合物。
所有这些步骤的强大且可访问的工具仍在开发中。因此,我们将在以下部分中仅介绍三种可以使用单细胞扰动数据处理的任务方法:
Identifying the cell types most affected by perturbations¶
Motivation¶
扰动很少对所有细胞产生相同的影响。特别是,不同的细胞类型或处于细胞周期不同状态的细胞可能受到不同程度的影响。为此,我们将利用Skinnidier等人开发的 Augur {cite}Skinnider2021,Squair2021Augur,这是一种量化响应程度的方法。
Augur 模型
Augur 旨在根据单细胞基因表达数据对实验扰动的响应来对细胞类型进行排序或优先级排列。基本思想是在分子测量空间中,对诱导扰动反应强烈的细胞比对扰动反应较小或没有反应的细胞类型更容易被分为扰动组和未扰动组。通过测量实验标签(例如,处理和对照)在每种细胞类型内的预测能力来量化这种可分离性。Augur 训练一个机器学习模型,在多次交叉验证运行中预测每种细胞类型的实验标签,然后根据度量模型准确性的指标得分来优先排序细胞类型响应。对于分类数据,曲线下面积是默认指标;对于数值数据,一致性相关系数被用作模型准确性的代理,从而近似扰动响应。
Limitations of Augur¶
由于 Augur 需要不同的细胞类型来确定扰动响应的程度,如果由于基因表达的持续、平滑过程或轨迹(例如细胞分化)而导致细胞类型标记具有挑战性,Augur 可能无法进行足够细致的排名。构建描述不同簇之间关系的聚类树(用于细胞类型注释),并将 Augur 应用于所有可能的聚类分辨率,可以确定最适合的分辨率,从而注释感兴趣的扰动 {cite}Squair2021Augur。
此外,介导组织或生物体对特定扰动的响应的细胞类型本身可能包含响应者和非响应者的亚群。将细胞分配为响应者和非响应者本身可能不准确,因为给定细胞类型的细胞可能沿着连续的扰动响应强度轨迹分布。然而,Augur 并不解析个别细胞的扰动响应,而是简单地将它们聚合为细胞类型的平均值 {cite}pemo:Squair2021。
某些扰动效应可能主要来源于特定细胞类型的相对丰度变化。然而,Augur 中的子抽样过程会丢弃任何关于相对丰度的信息。实际上,Augur 在交叉验证之前从每种条件中抽取相等数量的细胞。特定细胞类型丰度的任何变化都可能伴随着该细胞类型内在转录谱的变化。强烈的扰动甚至可能导致特定细胞类型完全耗尽或仅在受到扰动时突然出现。因此,建议对每种细胞类型执行差异丰度测试,以帮助解释 Augur 分析结果 {cite}Squair2021Augur。
Predicting cell type prioritization for IFN-β stimulation¶
为了演示 Augur,我们将使用 Kang 数据集,这是一个基于 10x 水滴法的 scRNA-seq 外周血单核细胞(PBMC)数据集,包含来自 8 位狼疮患者在INF-β 处理前后6小时的样本(共16个样本){cite}pemo:kang2018。
我们的目标是解读哪些细胞类型受INF-β处理的影响最大。
First, we import pertpy and scanpy.
import warnings
warnings.filterwarnings("ignore")
warnings.simplefilter("ignore")
# This is required to catch warnings when the multiprocessing module is used
import os
os.environ["PYTHONWARNINGS"] = "ignore"
import pertpy as pt
import scanpy as sc
Installed version 0.2.0 of pertpy is newer than the latest release 0.1.0! You are running a nightly version and features may break!
pertpy 提供了一个方便的数据加载器来访问 Kang 数据集。
adata = pt.dt.kang_2018()
我们将label重命名为condition,并对条件本身进行替换以提高可读性。
adata.obs.rename({"label": "condition"}, axis=1, inplace=True)
adata.obs["condition"].replace({"ctrl": "control", "stim": "stimulated"}, inplace=True)
This dataset contains PBMCs {cite}pemo:kang2018 across seven different cell-types.
adata.obs.cell_type.value_counts()
CD4 T cells 11238 CD14+ Monocytes 5697 B cells 2651 NK cells 1716 CD8 T cells 1621 FCGR3A+ Monocytes 1089 Dendritic cells 529 Megakaryocytes 132 Name: cell_type, dtype: int64
我们现在使用 pertpy 创建一个 Augur 对象,根据我们感兴趣的估计器来测量数据集中每种细胞类型的扰动标签的可预测性。估计器的选项包括 random_forest_classifier 或 logistic_regression_classifier(用于分类数据)和 random_forest_regressor(用于数值数据)。所有估计器都使用一个 Params 类来定义进一步的参数。这里我们将使用 random_forest_classifier,这通常是一个可靠且快速的选择,适合我们的分类数据。
ag_rfc = pt.tl.Augur("random_forest_classifier")
接下来,我们需要将 AnnData 对象加载到 Augur 可以处理的格式。这可以通过 Augur 对象的 load 函数轻松完成。
loaded_data = ag_rfc.load(adata, label_col="condition", cell_type_col="cell_type")
loaded_data
AnnData object with n_obs × n_vars = 24673 × 15706
obs: 'nCount_RNA', 'nFeature_RNA', 'tsne1', 'tsne2', 'label', 'cluster', 'cell_type', 'replicate', 'nCount_SCT', 'nFeature_SCT', 'integrated_snn_res.0.4', 'seurat_clusters', 'y_'
var: 'name'
obsm: 'X_pca', 'X_umap'
这使我们可以使用 predict 函数运行 Augur。通常,Augur 可以在两种模式下运行:
- 基于原始 Augur 实现的特征选择(
select_variance_feature=True),也是默认选项。该方法去除了在该细胞类型中细胞间变异较小的特征。有关更多细节,请参阅 Augur Nature Protocols 论文{cite}Squair2021Augur。选择此特征选择时,结果与 R 中的 Augur 实现非常相似。 - 基于
scanpy.pp.highly_variable_genes的特征选择。这种特征选择减少了用于模型训练的基因数量,可能导致 Augur 分数膨胀,因为高变异基因对区分细胞类型非常有用。然而,这种模式更快,并且能够很好地恢复扰动对细胞类型的影响。我们推荐它用于预期对特定细胞类型有强烈影响的扰动的大型数据集。
由于我们不期望INF-β对特定细胞类型有特殊的影响,因此我们使用原始的 Augur 特征选择来运行 Augur。为了进一步提高分辨率,我们将 subsample_size 设置为20(默认值为50),这对应于每种细胞类型随机抽取的细胞数量。
v_adata, v_results = ag_rfc.predict(
loaded_data, subsample_size=20, n_threads=4, select_variance_features=True, span=1
)
v_results["summary_metrics"]
Set smaller span value in the case of a `segmentation fault` error.
Set larger span in case of svddc or other near singularities error.
Output()
Installed version 0.2.0 of pertpy is newer than the latest release 0.1.0! You are running a nightly version and features may break! Installed version 0.2.0 of pertpy is newer than the latest release 0.1.0! You are running a nightly version and features may break! Installed version 0.2.0 of pertpy is newer than the latest release 0.1.0! You are running a nightly version and features may break! Installed version 0.2.0 of pertpy is newer than the latest release 0.1.0! You are running a nightly version and features may break!
| CD14+ Monocytes | CD4 T cells | Dendritic cells | NK cells | CD8 T cells | B cells | FCGR3A+ Monocytes | Megakaryocytes | |
|---|---|---|---|---|---|---|---|---|
| mean_augur_score | 0.920476 | 0.669376 | 0.847007 | 0.673299 | 0.626247 | 0.783628 | 0.888934 | 0.512619 |
| mean_auc | 0.920476 | 0.669376 | 0.847007 | 0.673299 | 0.626247 | 0.783628 | 0.888934 | 0.512619 |
| mean_accuracy | 0.817033 | 0.601172 | 0.744249 | 0.610330 | 0.575714 | 0.674505 | 0.780330 | 0.510513 |
| mean_precision | 0.835072 | 0.630593 | 0.779699 | 0.642700 | 0.587059 | 0.742801 | 0.792996 | 0.526175 |
| mean_f1 | 0.806636 | 0.564695 | 0.735157 | 0.591230 | 0.552114 | 0.633906 | 0.780933 | 0.406510 |
| mean_recall | 0.826349 | 0.575714 | 0.756032 | 0.616984 | 0.580000 | 0.622540 | 0.816508 | 0.388413 |
结果表包含拟合模型的多个评估指标。对于解释IFN-β响应的细胞类型优先级,只有 mean_augur_score 是相关的,它对应于 mean_auc。值越高,拟合模型就越容易区分对照和受扰动的细胞状态。因此,该细胞类型的扰动效应更强。让我们来可视化这个效应。
lollipop = pt.pl.ag.lollipop(v_results)

正如观察到的那样,CD14+单核细胞受IFN-β的影响最大,而巨核细胞受影响最小。这大致对应于原始出版物中各细胞类型的差异表达基因数量 {cite}pemo:kang2018。
相应的 mean_augur_score 也保存在 v_adata.obs 中,并且可以在 UMAP 中绘制。
sc.pp.neighbors(v_adata)
sc.tl.umap(v_adata)
sc.pl.umap(adata=v_adata, color=["augur_score", "cell_type", "label"])

Determining the most important genes for the prioritization¶
对优先级贡献最大的基因,如 Augur 分数所反映的,对应于我们模型的特征重要性。这些特征重要性保存在结果对象中,并且可以轻松绘制。
important_features = pt.pl.ag.important_features(v_results)

这些基因现在可以进一步探讨,例如它们在通路或其他基因集中的作用。然而,由于 Augur 在细胞类型层面进行推断,它并不直接确定参与扰动响应的个别基因。Augur 的作者们建议使用差异基因表达测试,在概念上和实际操作上都是对个别基因层面的推断更为合适的方法 {cite}Squair2021Augur。
Differential prioritization¶
Augur 还可以通过执行置换测试来进行差异优先排序,以识别在两次不同的细胞类型优先排序之间曲线下面积(AUC)的统计显著性差异(例如药物A和B的响应与未经处理的对照相比)。
Bhattacherjee 等人给小鼠提供了可卡因,并在撤药后48小时和15天时从前额叶皮质取样进行scRNA-seq {cite}Bhattacherjee2019。
我们现在将评估 withdraw_15d_Cocaine 和 withdraw_48h_Cocaine 条件相对于 Maintenance_Cocaine 的效果。基本上,差异优先排序是通过对两组细胞类型优先排序之间AUC差异的置换测试获得的,并与随机置换样本标签后相同两次优先排序之间的预期AUC差异进行比较 {cite}Squair2021Augur。在此过程中,用户首先分别对药物A(withdraw_15d_Cocaine)和药物B(withdraw_48h_Cocaine)进行细胞类型优先排序,然后计算药物A和药物B之间的AUC差异。为了计算AUC差异的统计显著性,通过置换样本标签然后在置换的数据中重复细胞类型优先排序,为每种细胞类型计算AUC差异的经验零分布。然后计算置换P值。该过程能够识别条件之间细胞类型优先排序的统计显著性差异,以及细胞类型在何种条件下在转录上更具可分离性。
首先,我们使用 pertpy 获取 bhattacherjee 数据集,并创建一个使用随机森林分类器的 Augur 对象。
bhattacherjee_adata = pt.dt.bhattacherjee()
ag_rfc = pt.tl.Augur("random_forest_classifier")
Output()
IOPub message rate exceeded. The Jupyter server will temporarily stop sending output to the client in order to avoid crashing it. To change this limit, set the config variable `--ServerApp.iopub_msg_rate_limit`. Current values: ServerApp.iopub_msg_rate_limit=1000.0 (msgs/sec) ServerApp.rate_limit_window=3.0 (secs)
接下来,我们使用 augur_mode=default 和 augur_mode=permute 运行 Augur,比较 Maintenance_Cocaine 和 withdraw_15d_Cocaine。注意,我们对数据集进行对数归一化以进行简单的归一化。
sc.pp.log1p(bhattacherjee_adata)
# Default mode
bhattacherjee_15 = ag_rfc.load(
bhattacherjee_adata,
condition_label="Maintenance_Cocaine",
treatment_label="withdraw_15d_Cocaine",
)
bhattacherjee_adata_15, bhattacherjee_results_15 = ag_rfc.predict(
bhattacherjee_15, random_state=None, n_threads=4
)
bhattacherjee_results_15["summary_metrics"].loc["mean_augur_score"].sort_values(
ascending=False
)
Filtering samples with Maintenance_Cocaine and withdraw_15d_Cocaine labels.
Output()
Oligo 0.916088 Astro 0.912823 Microglia 0.907143 OPC 0.900397 Endo 0.780590 Excitatory 0.688617 NF Oligo 0.678730 Inhibitory 0.660023 Name: mean_augur_score, dtype: float64
# Permute mode
bhattacherjee_adata_15_permute, bhattacherjee_results_15_permute = ag_rfc.predict(
bhattacherjee_15,
augur_mode="permute",
n_subsamples=100,
random_state=None,
n_threads=4,
)
Output()
Now let's do the same for Maintenance_Cocaine and withdraw_48h_Cocaine.
# Default mode
bhattacherjee_48 = ag_rfc.load(
bhattacherjee_adata,
condition_label="Maintenance_Cocaine",
treatment_label="withdraw_48h_Cocaine",
)
bhattacherjee_adata_48, bhattacherjee_results_48 = ag_rfc.predict(
bhattacherjee_48, random_state=None, n_threads=4
)
bhattacherjee_results_48["summary_metrics"].loc["mean_augur_score"].sort_values(
ascending=False
)
Filtering samples with Maintenance_Cocaine and withdraw_48h_Cocaine labels.
Output()
Inhibitory 0.675272 Astro 0.664728 OPC 0.628141 Microglia 0.614116 Endo 0.603107 Oligo 0.589728 NF Oligo 0.587596 Excitatory 0.559229 Name: mean_augur_score, dtype: float64
# Permute mode
bhattacherjee_adata_48_permute, bhattacherjee_results_48_permute = ag_rfc.predict(
bhattacherjee_48,
augur_mode="permute",
n_subsamples=100,
random_state=None,
n_threads=4,
)
Output()
Skipping NF Oligo cell type - 79 samples is less than min_cells 100.
这使我们可以在散点图中查看两次运行的 Augur 分数。对角线是恒等函数。如果值相同,它们将位于该线上。
scatter = pt.pl.ag.scatterplot(bhattacherjee_results_15, bhattacherjee_results_48)

要确定在比较 withdraw_48h_Cocaine 和 withdraw_15d_Cocaine 时哪个细胞类型受影响最大,我们可以运行差异优先级分析。
pvals = ag_rfc.predict_differential_prioritization(
augur_results1=bhattacherjee_results_15,
augur_results2=bhattacherjee_results_48,
permuted_results1=bhattacherjee_results_15_permute,
permuted_results2=bhattacherjee_results_48_permute,
)
pvals
| cell_type | mean_augur_score1 | mean_augur_score2 | delta_augur | b | m | z | pval | padj | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | Endo | 0.780590 | 0.603107 | -0.177483 | 1000 | 1000 | -12.768461 | 0.001998 | 0.002331 |
| 1 | OPC | 0.900397 | 0.628141 | -0.272256 | 1000 | 1000 | -14.543180 | 0.001998 | 0.002331 |
| 2 | Excitatory | 0.688617 | 0.559229 | -0.129388 | 1000 | 1000 | -8.456439 | 0.001998 | 0.002331 |
| 3 | Microglia | 0.907143 | 0.614116 | -0.293027 | 788 | 1000 | -19.857981 | 0.425574 | 0.425574 |
| 4 | Inhibitory | 0.660023 | 0.675272 | 0.015249 | 1000 | 1000 | 3.218067 | 0.001998 | 0.002331 |
| 5 | Astro | 0.912823 | 0.664728 | -0.248095 | 1000 | 1000 | -17.167407 | 0.001998 | 0.002331 |
| 6 | Oligo | 0.916088 | 0.589728 | -0.326361 | 1000 | 1000 | -19.445162 | 0.001998 | 0.002331 |
根据 R Augur 的实现,p 值的计算使用了 b 和 m。其中,b 是置换值大于原始值的次数,m 是运行的置换次数。由于 b 对所有细胞类型(除了小胶质细胞)都是相同的,因此这些细胞的 p 值也是相同的。
diff = pt.pl.ag.dp_scatter(pvals)

在这种情况下,"Inhibitory" 细胞类型在两个比较的细胞类型之间没有太大差异,这可能表明已经造成了永久性损伤。
Predicting IFN-β response for CD4-T cells¶
许多扰动反应建模方法旨在预测未观测人群对刺激(如药物、基因敲除或疾病)的转录组学反应,以帮助实验设计和假设生成。当由于实验或样本失败(例如,细胞分选失败)、高昂的实验成本阻碍了所有可能性的探索以及某些细胞类型的发现频率较低等原因而无法捕捉到接受扰动处理的细胞时,就可能发生这种情况。在上述所有情况下,针对缺失人群的计算机预测可以为是否进行新实验提供有依据的决策。
许多扰动反应建模方法旨在预测未观测人群对刺激(如药物、基因敲除或疾病)的转录组学反应,以帮助实验设计和假设生成。当由于实验或样本失败(例如,细胞分选失败)、高昂的实验成本阻碍了所有可能性的探索以及某些细胞类型的发现频率较低等原因而无法捕捉到接受扰动处理的细胞时,就可能发生这种情况。在上述所有情况下,针对缺失人群的计算机预测可以为是否进行新实验提供有依据的决策。
变分自编码器
自编码器的基本原理是由两部分组成,即编码器和解码器。编码器尝试学习输入数据(通常是基因表达)的潜在空间,解码器可以以最小的重建误差对其进行解码。潜在空间的维度通常比输入空间的维度低。
自编码器的一种扩展是变分自编码器(VAE),它通过为整个空间提供生成能力来解决自编码器潜在空间不规则的问题。不规则的潜在空间只对形成聚类的不同类别提供强大的采样能力。著名的手写数字MNIST数据集只允许在确定的10个聚类中的0-9数字进行采样。然而,如果在聚类之外尝试采样,则会产生无效输入。与自编码器的编码器输出潜在向量不同,VAE的编码器为每个输入输出潜在空间中预定义分布的参数。通过VAE强制执行的正态分布潜在分布,潜在空间得到了正则化。
在这里,我们演示了scGen {cite}lotfollahi2019 的应用,这是一种结合了向量算术的变分自编码器模型。该模型学习数据的潜在表示,并在其中估计对照(未处理)细胞和扰动(处理)细胞之间的差异向量。然后,将估计的差异向量添加到感兴趣的细胞类型或群体的对照细胞中,以预测每个单细胞的基因表达反应。在这里,我们应用scGen来预测CD4-T细胞群体对IFN-β的反应,这些细胞在训练期间被人为排除(未见)以模拟上述现实场景之一。我们再次利用包含来自8位接受IFN-β治疗或未接受治疗的狼疮患者的外周血单核细胞(PBMCs)的数据集 {cite}pemo:kang2018 ,跨越七种不同的细胞类型。
首先,我们导入scanpy 和 scgen 以便处理AnnData对象并使用scGen。
import scanpy as sc
import pertpy as pt
import scgen
Global seed set to 0
Setting up the Kang dataset for scGen¶
We will again use pertpy to get the Kang dataset.
adata = pt.dt.kang_2018()
scGen在对数转换的数据上效果最佳。选择高变异基因可以通过减少特征空间来加速计算。
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata)
我们将 label 重命名为 condition,并对条件本身进行重命名以提高可读性。
adata.obs.rename({"label": "condition"}, axis=1, inplace=True)
adata.obs["condition"].replace({"ctrl": "control", "stim": "stimulated"}, inplace=True)
该数据集包含七种不同细胞类型的外周血单核细胞(PBMCs) {cite}pemo:kang2018。
adata.obs.cell_type.value_counts()
CD4 T cells 11238 CD14+ Monocytes 5697 B cells 2651 NK cells 1716 CD8 T cells 1621 FCGR3A+ Monocytes 1089 Dendritic cells 529 Megakaryocytes 132 Name: cell_type, dtype: int64
我们从训练数据 (adata_t) 中移除所有 CD4T 细胞,以模拟在实验中未能捕捉到特定群体的现实场景。
adata_t = adata[
~(
(adata.obs["cell_type"] == "CD4 T cells")
& (adata.obs["condition"] == "stimulated")
)
].copy()
cd4t_stim = adata[
(
(adata.obs["cell_type"] == "CD4 T cells")
& (adata.obs["condition"] == "stimulated")
)
].copy()
scGen 需要数据采用特定格式,这可以通过 AnnData 和 setup_anndata 函数来实现。它需要样本的键,即 batch_key(在我们的例子中为 "condition")以及细胞类型标签的键,即 labels_key(为 "cell_type")。
scgen.SCGEN.setup_anndata(adata_t, batch_key="condition", labels_key="cell_type")
Model construction and training¶
scGen 需要修改后的 AnnData 对象 (adata_t) 来构建模型对象,该对象可用于训练模型。此函数接收多个用户输入,包括模型在瓶颈层(网络的中间层)之前的每个隐藏层的节点数 (n_hidden) 以及此类层的数量 (n_layers)。此外,用户可以调整瓶颈层的维度,该维度用于计算扰动细胞和对照细胞之间的差异向量。这里使用的默认参数取自原始发表的文章。实际上,更宽的隐藏层可提高重建精度,这对于我们预测多个基因的扰动反应至关重要。
model = scgen.SCGEN(adata_t, n_hidden=800, n_latent=100, n_layers=2)
scGen 是一个神经网络,有成千上万个参数来学习低维数据表示。在这里,我们使用 train 方法来估计这些参数,利用训练数据进行训练。这里有多个参数,max_epochs 是模型允许更新其参数的最大迭代次数,这里设置为100。更高的训练轮数将需要更多的计算时间,但可能会带来更好的结果。batch_size 是模型在更新其参数时所看到的样本数量(单个细胞)。在 scGen 的情况下,较低的数量通常会带来更好的结果。最后,有 early_stopping,如果在 early_stopping_patience 训练轮后模型的结果没有改进,该机制将使模型停止训练。早停机制可以防止训练数据的潜在过拟合,从而导致对未见人群的泛化能力差。
model.train(
max_epochs=100, batch_size=32, early_stopping=True, early_stopping_patience=25
)
GPU available: False, used: False TPU available: False, using: 0 TPU cores IPU available: False, using: 0 IPUs
Epoch 27/100: 27%|██▋ | 27/100 [1:01:41<2:46:48, 137.11s/it, loss=140, v_num=1] Monitored metric elbo_validation did not improve in the last 25 records. Best score: 665.237. Signaling Trainer to stop.
为了可视化模型学习的数据表示,我们使用UMAP算法绘制模型的潜在表示。get_latent_representation()函数为每个细胞返回一个100维的向量。我们将这些潜在表示存储在AnnData对象的.obsm槽中。
adata_t.obsm["scgen"] = model.get_latent_representation()
接下来,我们使用计算得到的潜在表示重新计算邻居图和UMAP嵌入,最终在UMAP图中可视化新的嵌入。
sc.pp.neighbors(adata_t, use_rep="scgen")
sc.tl.umap(adata_t)
sc.pl.umap(adata_t, color=["condition", "cell_type"], wspace=0.4, frameon=False)

如上所述,IFN-β刺激引起了所有细胞类型的强烈转录变化。
Predicting CD4T responses to IFN-β stimulation¶
在模型训练完成后,我们可以要求模型模拟训练数据中每个对照CD4T细胞对IFN-β反应的效果。通过predict方法可以实现这一预测,该方法接收AnnData对象condition列中相应的标签(如下所示的ctrl_key和stim_key),以估算“对照”细胞和“刺激”细胞在潜在空间中的全局差异向量。然后将该向量添加到指定的每个单细胞中(此处为CD4T)。
pred, delta = model.predict(
ctrl_key="control", stim_key="stimulated", celltype_to_predict="CD4 T cells"
)
# we annotate the predicted cells to distinguish them later from ground truth cells.
pred.obs["condition"] = "predicted stimulated"
INFO Received view of anndata, making copy. INFO Input AnnData not setup with scvi-tools. attempting to transfer AnnData setup INFO Received view of anndata, making copy. INFO Input AnnData not setup with scvi-tools. attempting to transfer AnnData setup INFO Received view of anndata, making copy. INFO Input AnnData not setup with scvi-tools. attempting to transfer AnnData setup
Evaluating the predicted IFN-β response¶
在前面的章节中,我们预测了对照群体中每个CD4T细胞对IFN-β的反应。由于单细胞测序是破坏性的,这意味着细胞不能在特定扰动前后进行测量,因此无法直接评估同一细胞在IFN-β刺激后的预测结果。然而,我们在数据中有一组经过IFN-β处理的细胞,可以用来衡量预测的细胞群体与真实细胞的对齐程度。为此,我们通过观察对照、预测和实际的CD4T IFN-β细胞在主成分分析(PCA)空间中的嵌入来定性评估预测结果。此外,我们还通过定量测量预测细胞和IFN-β在所有基因以及IFN-β刺激后差异表达基因中的平均基因表达之间的相关性来评估预测结果。
首先,我们构建一个包含对照细胞、预测刺激细胞和实际刺激细胞的AnnData对象。
ctrl_adata = adata[
((adata.obs["cell_type"] == "CD4 T cells") & (adata.obs["condition"] == "control"))
]
# concatenate pred, control and real CD4 T cells in to one object
eval_adata = ctrl_adata.concatenate(cd4t_stim, pred)
eval_adata.obs.condition.value_counts()
stimulated 5678 control 5560 predicted stimulated 5560 Name: condition, dtype: int64
We first look at the PCA co-embedding of control, IFN-β stimulated and predicted CD4T cells.
sc.tl.pca(eval_adata)
sc.pl.pca(eval_adata, color="condition", frameon=False)

如上所述,预测的刺激细胞向IFN-β刺激的CD4T细胞移动。然而,我们还应查看差异表达基因(DEGs),以验证预测的刺激细胞中是否也存在最显著的DE基因。下面,我们查看预测细胞和实际细胞之间的总体平均相关性。在此之前,我们提取对照细胞和刺激细胞之间的DEGs:
cd4t_adata = adata[adata.obs["cell_type"] == "CD4 T cells"]
We estimate DEGs using scanpy's implementation of the Wilcoxon test.
sc.tl.rank_genes_groups(cd4t_adata, groupby="condition", method="wilcoxon")
diff_genes = cd4t_adata.uns["rank_genes_groups"]["names"]["stimulated"]
diff_genes
array(['ISG15', 'IFI6', 'ISG20', ..., 'EEF1A1', 'FTH1', 'RGCC'],
dtype=object)
scGen具有一个reg_mean_plot功能,它计算预测的IFN-β细胞和实际IFN-β细胞的平均基因表达之间的R²相关性。R²值越高(最大为1),预测结果与真实情况越接近。图中用红色高亮显示的基因是IFN-β刺激后上调的前10个差异表达基因(DEGs),这些基因对于成功的预测至关重要。可以看到,模型在预测高平均值基因方面表现良好,但在一些平均表达值在0-1之间的基因方面表现较差。我们还通过非DEGs测量准确性,因为模型在改变DEGs表达的同时,不应改变未受扰动影响的基因表达。
r2_value = model.reg_mean_plot(
eval_adata,
axis_keys={"x": "predicted stimulated", "y": "stimulated"},
gene_list=diff_genes[:10],
top_100_genes=diff_genes,
labels={"x": "predicted", "y": "ground truth"},
show=True,
legend=False,
)

我们还可以查看IFN-β上调的主要基因的分布。例如,我们绘制了ISG15的表达分布,这是一个在IFN刺激后诱导的知名基因。可以看到,模型识别出这个基因在IFN-β刺激后应该上调,并且其表达值确实在真实(被刺激的)细胞中转移到了一个类似的范围。
sc.pl.violin(eval_adata, keys="ISG15", groupby="condition")

总体而言,我们展示了scGen作为扰动响应模型的应用示例,用于预测在期望扰动下未见过的细胞群体的基因表达。虽然扰动响应模型可以提供计算机内预测,但它们不能替代实际实验的进行。同样,不清楚预测的响应有多少是由细胞类型特异性响应或跨细胞类型的响应引起的。然而,正如在scGen案例中所观察到的那样,它可以预测高表达基因的总体响应,但对于低表达基因的预测较差,这需要进一步优化,并激励开发更复杂和更健壮的方法。
Analysing single-pooled CRISPR screens¶
扩展的CRISPR兼容CITE-seq (ECCITE-seq)能够捕获单导向RNA (sgRNA)序列,同时测量转录组和表面蛋白,以抗体衍生标签 (ADT) 的形式呈现。这使得可以在进行多种基因扰动的同时,结合转录组学读数来探索和验证扰动对蛋白质表达的影响,从而使这种检测方法非常强大。然而,这种强大不仅带来了巨大的责任,也带来了复杂性。
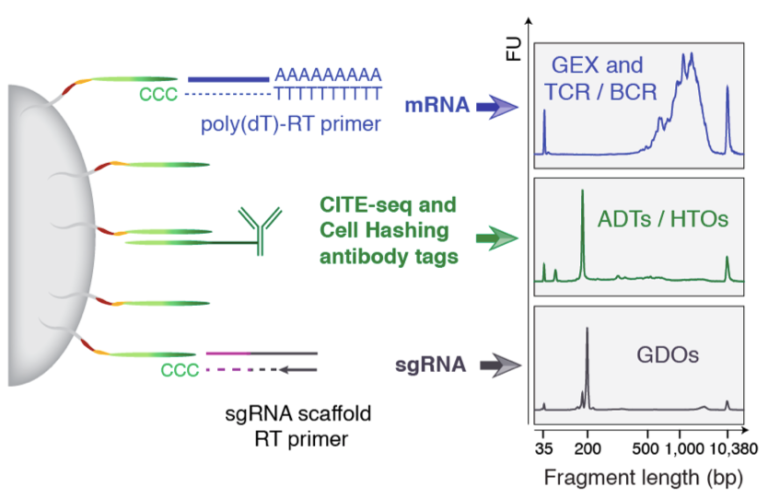
ECCITE-seq概述。使用抗体衍生标签测量mRNA和表面蛋白表达。通过hashtag衍生的寡核苷酸解决生物学重复。导向RNA的分配使用导向衍生的寡核苷酸完成。图片来自(https://cite-seq.com/eccite-seq)。
为了进行此次分析,我们将使用Papalexi 2021数据集{cite}Papalexi2021。该数据集包含约20000个使用了111个gRNA的刺激THP-1细胞。使用IFN-γ、去甲基化药物(DAC)和转化生长因子(TGF)-β1的组合刺激THP-1细胞,结果诱导了三个免疫检查点的表达:程序性死亡配体1(PD-L1)、PD-L2和CD86。ECCITE-seq实验的目的是研究调节PD-L1表达的分子网络,因为PD-L1在人体癌症中经常出现,并且可以抑制T细胞介导的免疫反应{cite}Papalexi2021。
对于此次具体分析,我们的目标是:
- 去除混杂的变异来源,如细胞周期效应或批次效应。
- 确定哪些细胞受到了期望扰动的影响,哪些细胞逃逸了。
- 可视化扰动响应。
为了进行此次分析,我们将再次使用pertpy,它实现了Mixscape管道,用于scverse生态系统,速度约为Seurat生态系统的5-10倍。该管道最初是为Seurat生态系统开发的,我们基本遵循了Seurat Vignette的说明(https://satijalab.org/seurat/articles/mixscape_vignette.html)。
首先,我们导入pertpy、scanpy和muon,这些工具将用于蛋白质数据的预处理。
import pertpy as pt
import muon as mu
import scanpy as sc
第一步,我们使用pertpy获取数据集。数据加载器返回一个MuData对象,其中包含转录组测量、ADT测量和导向RNA计数
mdata = pt.dt.papalexi_2021()
mdata
Output()
MuData object with n_obs × n_vars = 20729 × 18778
4 modalities
rna: 20729 x 18649
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_HTO', 'nFeature_HTO', 'nCount_GDO', 'nCount_ADT', 'nFeature_ADT', 'percent.mito', 'MULTI_ID', 'HTO_classification', 'guide_ID', 'gene_target', 'NT', 'perturbation', 'replicate', 'S.Score', 'G2M.Score', 'Phase'
var: 'name'
adt: 20729 x 4
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_HTO', 'nFeature_HTO', 'nCount_GDO', 'nCount_ADT', 'nFeature_ADT', 'percent.mito', 'MULTI_ID', 'HTO_classification', 'guide_ID', 'gene_target', 'NT', 'perturbation', 'replicate', 'S.Score', 'G2M.Score', 'Phase'
var: 'name'
hto: 20729 x 12
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_HTO', 'nFeature_HTO', 'nCount_GDO', 'nCount_ADT', 'nFeature_ADT', 'percent.mito', 'MULTI_ID', 'HTO_classification', 'guide_ID', 'gene_target', 'NT', 'perturbation', 'replicate', 'S.Score', 'G2M.Score', 'Phase'
var: 'name'
gdo: 20729 x 111
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_HTO', 'nFeature_HTO', 'nCount_GDO', 'nCount_ADT', 'nFeature_ADT', 'percent.mito', 'MULTI_ID', 'HTO_classification', 'guide_ID', 'gene_target', 'NT', 'perturbation', 'replicate', 'S.Score', 'G2M.Score', 'Phase'
var: 'name'
原始的Seurat对象包含四个分析,已被转换为单个AnnData对象,从而创建了下载的MuData对象。各个模式如下:
adt:四个捕获的抗体衍生标签(CD86、PDL1、PDL2、CD366)的计数矩阵,在此设置中通常简称为“蛋白质”。gdo:111个使用的导向RNA(gRNA)。为了给每个细胞分配gRNA身份,检查了导向衍生寡核苷酸(GDO)计数。如果一个细胞对所有gRNA序列的计数少于五个,则其被分类为阴性。对于所有其他细胞,计数最高的gRNA被分配给该细胞。计数高于一个gRNA的细胞被分类为双重体。hto:为了跟踪每个生物学重复的身份,按照细胞标记协议使用标签衍生寡核苷酸(HTO)对样本进行了标记{cite}Stoeckius2018。rna:这对应于所有细胞的转录组测量,是通常的细胞基因计数矩阵。
Preprocessing¶
我们保持RNA和ADT的预处理过程简单。首先,使用scanpy的normalize_total对RNA进行归一化,然后进行对数转换和高变异基因选择。对于ADT的归一化,我们应用中心化对数比率(centered log-ratio, CLR)转换{cite}Stoeckius2017。
sc.pp.normalize_total(mdata["rna"])
sc.pp.log1p(mdata["rna"])
sc.pp.highly_variable_genes(mdata["rna"], subset=True)
mu.prot.pp.clr(mdata["adt"])
Data exploration¶
To get a feeling for our dataset we visualize the replicates, cell-cycle phases and perturbations in a UMAP embedding.
sc.pp.pca(mdata["rna"])
# We calculate neighbors with the cosine distance similarly to the original Seurat implementation
sc.pp.neighbors(mdata["rna"], metric="cosine")
sc.tl.umap(mdata["rna"])
sc.pl.umap(mdata["rna"], color=["replicate", "Phase", "perturbation"])

查看UMAP时,我们发现两个明显的问题:
- 许多细胞按重复ID分开。这是批次效应的常见标志。
- 细胞周期阶段在嵌入中是一个混杂因素。
因此,我们现在尝试通过计算局部扰动特征,将细胞投射到扰动空间,以缓解这些问题。
Calculating local perturbation signatures¶
为了缓解上述问题,我们将计算局部扰动特征。核心思想是,通过从每个细胞中减去对照池(=NT)中最近的k个细胞的平均表达,提取每个细胞中仅反映基因扰动的部分。最近邻的k个细胞必须处于与目标细胞匹配的生物学状态,但不能被任何gRNA靶向。得到的成分被称为局部扰动特征。默认情况下,邻居数k设置为20。根据Papalexi等人的建议,我们推荐将k设置在20到30之间。过小或过大的k都不太可能去除数据集中的技术变异{cite}Papalexi2021。
现在,我们使用pertpy创建一个Mixscape对象并计算扰动特征。
ms = pt.tl.Mixscape()
ms.perturbation_signature(
mdata["rna"],
pert_key="perturbation",
control="NT",
split_by="replicate",
n_neighbors=20,
)
# We create a copy of the object to recalculate the PCA.
# Alternatively we could replace the X of the RNA part of our MuData object with the `X_pert` layer.
adata_pert = mdata["rna"].copy()
adata_pert.X = adata_pert.layers["X_pert"]
sc.pp.pca(adata_pert)
sc.pp.neighbors(adata_pert, metric="cosine")
sc.tl.umap(adata_pert)
sc.pl.umap(adata_pert, color=["replicate", "Phase", "perturbation"])

使用扰动特征来计算邻居图和最终的嵌入,可以消除技术变异,并揭示一个额外的扰动特定簇。现在我们的数据大部分已摆脱混杂效应,我们需要确定针对哪些目标细胞(即扰动的细胞),其扰动是成功的(=KO)还是不成功的(=NP)。
Identifying cells with no detectable perturbation¶
我们主要的假设是每个目标基因类别是两个高斯分布的混合体。其中一个代表成功敲除(KO)的细胞,另一个代表未扰动(NP)的细胞。NP细胞的分布应与表达非靶向gRNA(NT)的细胞相同。在估计出KO细胞的分布后,Mixscape计算一个细胞属于KO分布的后验概率,并将概率超过0.5的细胞分类为KOs。将这一方法应用于所有11个目标基因类别,可以识别出所有KO细胞,同时评估不同gRNA的靶向效果。
ms.mixscape(adata=mdata["rna"], control="NT", labels="gene_target", layer="X_pert")
We can now plot the class distributions for all 111 gRNAs.
pt.pl.ms.barplot(mdata["rna"], guide_rna_column="guide_ID")

<ggplot: (8793801394204)>
我们检测到每个类别中gRNA靶向效率的变化。例如,针对STAT1的gRNA 4似乎效果不佳,而gRNA 1-3则效果显著。
让我们检查一个示例目标基因(IFNGR2)的扰动评分。
pt.pl.ms.perturbscore(
adata=mdata["rna"], labels="gene_target", target_gene="IFNGR2", color="orange"
)

<ggplot: (8793804547229)>
正如预期的那样,NT类和IFNGR2 NP类的分布非常匹配,而IFNGR2 KO类的分布则明显偏移。这也应该在后验概率中有所反映。
sc.settings.set_figure_params(figsize=(10, 10))
pt.pl.ms.violin(
adata=mdata["rna"],
target_gene_idents=["NT", "IFNGR2 NP", "IFNGR2 KO"],
groupby="mixscape_class",
)

后验概率明显将两类区分开来,几乎没有不清楚的情况。这一点可以通过在Mixscape类之间运行一个简单的差异表达(DE)测试并将结果可视化在热图上,通过排序后验概率进一步突出显示。
pt.pl.ms.heatmap(
adata=mdata["rna"],
labels="gene_target",
target_gene="IFNGR2",
layer="X_pert",
control="NT",
)
WARNING: dendrogram data not found (using key=dendrogram_mixscape_class). Running `sc.tl.dendrogram` with default parameters. For fine tuning it is recommended to run `sc.tl.dendrogram` independently. WARNING: Groups are not reordered because the `groupby` categories and the `var_group_labels` are different. categories: IFNGR2 KO, IFNGR2 NP, NT var_group_labels: NT

到目前为止,我们仅使用了转录组数据,但现在我们可以利用测量的蛋白质数据来证明只有IFGN通路敲除(KO)细胞才会出现PDL1表达的减少。
mdata["adt"].obs["mixscape_class_global"] = mdata["rna"].obs["mixscape_class_global"]
pt.pl.ms.violin(
adata=mdata["adt"],
target_gene_idents=["NT", "JAK2", "STAT1", "IFNGR1", "IFNGR2", "IRF1"],
keys="PDL1",
groupby="gene_target",
hue="mixscape_class_global",
)

Visualizing perturbation responses with Linear Discriminant Analysis¶
Mixscape流程的最后一步是计算和可视化扰动特定簇。具体方法是应用线性判别分析(LDA)并使用确定的结果重新计算UMAP。LDA尝试通过使用基因表达和标签来最大化已知标签的可分离性,在我们的例子中,这些标签是Mixscape类。
ms.lda(adata=mdata["rna"], labels="gene_target", layer="X_pert")
pt.pl.ms.lda(adata=mdata["rna"])

LDA突出显示了至少两种主要的扰动类型,这可以通过两个岛状分布看出。然而,我们要强调的是,只有更严格的分析,例如通路分析和随后的生物学验证,才能确定多个敲除具有相似的效果。
Key Takeaways¶
- 在应用Augur时,确保能够自信地标记细胞类型。使用差异丰度测试找到混杂效应,并使用差异基因表达找到基因水平的扰动效应来源。
- 使用scGen等工具预测扰动响应对于高表达基因效果良好,但对于低表达基因则具有挑战性。
- Mixscape流程的线性判别分析步骤主要用于可视化。UMAP中的距离应谨慎解释,因此敲除之间的相似性和差异性并不总能一目了然地推断出来。
Quiz¶
- Why is it required to have confident cell type labels when applying Augur?
- Why should scGen primarily only be applied to highly expressed genes?
- The mixscape pipeline results in clusters of genetic perturbations. Why must the distances in the UMAPs be interpreted with caution? What does it mean when some genetic perturbations are close or distant in the UMAP embedding?