scGPT toward building a foundation model for single cell multi omics using generative AI
摘要¶
生成预训练模型在诸如语言和计算机视觉等多个领域取得了卓越的成功。特别是,结合大规模多样性数据集和预训练 transformer 被视为开发基础模型的一种有前景的方法。通过将语言与细胞生物学 (在细胞生物学中,文本由单词组成; 类似地,细胞由基因定义) 作比较,我们的研究探讨了基础模型在推进细胞生物学和遗传学研究方面的适用性。利用新兴的单细胞测序数据,我们构建了一个基于 3300 多万个细胞库的单细胞生物学基础模型 scGPT,这是一种生成式预训练 transformer。我们的研究结果说明,scGPT 有效地提取了关于基因和细胞的关键生物学见解。通过进一步适应迁移学习,scGPT 可以优化以在多种下游应用中实现卓越性能。这包括细胞类型注释、多批次整合、多组学整合、干扰响应预测和基因网络推断等任务。
单细胞 RNA 测序 (scRNA-seq) 通过对不同细胞类型进行细致表征并推进我们对疾病发生机制的理解,为细胞异质性探索、系谱追踪、病理机制阐明,以及最终实现个性化治疗策略铺平了道路。scRNA-seq 的广泛应用导致了全面的数据图谱,如人类细胞图谱,现已包含数千万个细胞。测序技术的最新进展促进了数据模态的多样性,并将我们的理解从基因组扩展到表观基因组、转录组和蛋白组,从而提供了多模态见解。这些突破也引发了新的研究问题,如参考映射、干扰预测和多组学整合。与此同时,并行开发能够有效利用、增强和适应不断扩大的测序数据的方法至关重要。
生成预训练基础模型是解决这一挑战的一种有前景的方法。基础模型通常建立在自注意力 transformer 架构之上,因为它在学习表达性数据表示方面效果良好,是一类在大规模多样数据集上进行预训练的深度学习模型,可方便地适用于各种下游任务。这种模型最近在计算机视觉和自然语言生成 (NLG) 等各个领域取得了前所未有的成功,例如 DALL-E 2、GPT-4 和最近用于生物学应用的 Enformer。更有趣的是,这些生成式预训练模型始终优于从头训练的特定任务模型。这表明这些领域存在与任务无关的知识理解,启发我们探索将其应用于单细胞组学研究。然而,目前单细胞研究中基于机器学习的方法相当零散,专门用于不同分析任务的特定模型。因此,每项研究所使用的数据集通常在广度和规模上都有限。为了应对这一限制,我们需要一种经过大规模数据预训练,能够理解跨不同组织间基因之间复杂相互作用的基础模型。
为了增强对大规模单细胞测序数据的建模能力,我们借鉴了 NLG 中自监督预训练工作流程的 inspiration,其中自注意力 transformer 已经证明了对输入词元建模的强大能力。虽然文本由词语组成,但细胞可以通过它们编码的基因和蛋白产物来描述。通过同时学习基因和细胞嵌入,我们可以更好地理解细胞特征。此外,transformer 输入词元的灵活性可以轻松纳入其他特征和元信息。最近在 Geneformer 中也探索了这一方向,其中基于 transformer 的编码器使用按表达水平排序的基因进行训练,并展现出对细胞类型和基因功能预测的能力。在此基础上,我们认为有必要量身打造一个预训练工作流程,直接对非序列组学数据的复杂性进行建模,并将其适用性扩展到更广泛的任务。
在这项工作中,我们在超过 3300 万个细胞上进行预训练,提出了单细胞基础模型 scGPT。我们为非序列组学数据建立了统一的生成式预训练工作流程,并调整了 transformer 架构以同时学习细胞和基因表示。此外,我们提供了具有特定任务目标的微调流水线,旨在促进预训练模型应用于各种不同任务。
scGPT 通过三个关键方面展示了单细胞基础模型的变革潜力。首先,scGPT 代表了一种大规模生成基础模型,可实现跨不同下游任务的迁移学习。通过在细胞类型注释、基因干扰预测、批次校正和多组学整合方面实现最先进的性能,我们展示了 " 通用预训练,按需微调 " 这一普遍方法作为单细胞组学计算应用的一站式解决方案的有效性。其次,通过比较微调后和原始预训练模型之间的基因嵌入和注意力权重,scGPT 发现了有关各种情况下 (如细胞类型和干扰状态) 特定基因 - 基因相互作用的宝贵生物学见解。第三,我们的观察揭示了一种缩放效应: 更大的预训练数据量产生更优质的预训练嵌入,进而导致在下游任务上的性能提高。这一发现凸显了令人兴奋的前景: 随着研究界可用测序数据的不断扩大,基础模型也将持续改进。基于这些发现,我们预见,采用预训练基础模型将极大扩展我们对细胞生物学的理解,并为未来的发现奠定坚实基础。发布 scGPT 模型和工作流程旨在赋能并加速这些领域及更多领域的研究。
结果¶
单细胞 transformer 基础模型概述¶
单细胞测序可以在单个细胞水平上检测分子特征。例如,scRNA-seq 测量 RNA 转录物的丰度,提供了有关细胞身份、发育阶段和功能的见解。我们引入了 scGPT,一种采用生成式预训练方法的单细胞领域基础模型。该核心模型包含堆叠的带有多头注意力的 transformer 层,能同时生成细胞和基因嵌入。
scGPT 包括两个训练阶段: 首先在大规模细胞图谱上进行通用预训练,然后在更小的数据集上针对特定应用进行微调。在预训练阶段,我们引入了特别设计的注意力掩码和生成式训练流水线,以自监督的方式训练 scGPT,共同优化细胞和基因表示。这一技术解决了基因表达的非序列性质,以适应 NLG 的序列预测框架。在训练过程中,模型逐渐学会根据细胞状态或基因表达线索生成细胞的基因表达。在微调阶段,预训练模型可针对新数据集和特定任务进行调整。我们提供了灵活的微调流水线,适用于单细胞研究中各种基本任务,包括 scRNA-seq 整合与批次校正、细胞类型注释、多组学整合、干扰预测和基因调控网络推断。
为了收集丰富广泛的测序数据,用于 scGPT 的自监督预训练,我们从 CELLxGENE 集合中汇集了来自 3300 万个正常 (非疾病) 人类细胞的 scRNA-seq 数据。这个综合数据集涵盖来自 51 个器官或组织、441 项研究的各种细胞类型,为人体内细胞异质性提供了丰富的表示。预训练后,我们使用均一流形近似和投影 (UMAP) 可视化对 3300 万细胞中 10% 的人体细胞进行了 scGPT 细胞嵌入可视化。得到的 UMAP 图清晰有趣,不同的细胞类型由不同颜色在局部区域和簇中准确表示。考虑到数据集包含 400 多项研究,这展示了预训练提取生物学变化的卓越能力。
scGPT 提高了细胞类型注释的精度¶
为了将预训练的 scGPT 用于细胞类型注释,一个神经网络分类器将 scGPT transformer 输出的细胞嵌入作为输入,输出细胞类型的分类预测。整个模型在带有专家注释的参考数据集上通过交叉熵进行训练,然后用于对留出的查询数据分区进行细胞类型预测。我们在不同数据集上进行了大量实验,以评估 scGPT 在细胞类型注释方面的表现。首先,我们将 scGPT 用于预测人胰腺数据集中的细胞类型。我们在图 2a 中可视化了预测结果。值得注意的是,除了在参考分区中细胞数量极少的罕见细胞类型外,scGPT 在大多数细胞类型上实现了较高精度 (>0.8),如混淆矩阵所示 (图 2b)。例如,在 10600 个细胞的参考集中,属于肥大细胞和主要组织相容性复合体 (MHC)II 类细胞类型的细胞不到 50 个。图 2c 可视化了微调后 scGPT 中的细胞嵌入,展现了较高的细胞类型内相似性。
接下来,我们在一个多发性硬化症 (MS) 疾病数据集上测试了该模型。该模型在健康人类免疫细胞的参考分区上进行微调,并评估了对 MS 状态细胞的预测。微调后的模型与原始研究提供的细胞类型注释高度一致,达到约 0.85 的高准确率 (图 2f,g)。此外,我们将该模型应用于一个更具挑战性的情况,即跨疾病类型进行泛化,使用肿瘤浸润性髓细胞数据集。该模型在参考数据分区的六种癌症类型上进行微调 (方法),并在三种未见过的癌症类型的查询分区上进行评估 (图 2d)。结果显示,在区分免疫细胞亚型方面精度很高 (图 2e,h),细胞嵌入在不同细胞类型之间也表现出明显的可分离性 (图 2i)。
最后,我们在这三个数据集上将微调后的 scGPT 与另外两种最新的基于 transformer 的方法 TOSICA 和 scBERT 进行了基准测试 (方法)。scGPT 在所有分类指标上,包括准确率、精确率、召回率和宏 F1 值,均持续优于其他方法 (图 2j)。
除细胞类型分类外,我们进一步探索了 scGPT 将未见过的查询细胞投影到参考数据集的能力,即参考映射 (补充说明 1 和补充图 11)。我们发现,scGPT 仅使用预训练权重就能达到与现有方法相当的性能。通过在参考数据集上进行微调,性能可以进一步提高。
scGPT 预测未知的基因干扰响应¶
测序和基因编辑技术的最新进展极大地促进了大规模干扰实验,使人们能够描述细胞对各种基因干扰的响应。这一方法为揭示新的基因相互作用和推进再生医学带来了巨大希望。然而,潜在基因干扰的庞大组合空间很快就会超出实验可行性的实际限制。为了克服这一限制,scGPT 可用于利用已知实验中细胞响应获得的知识,并将其外推以预测未知响应。在基因维度上利用自注意力机制,可以编码受干扰基因与其他基因响应之间的错综复杂相互作用。通过利用这一能力,scGPT 可以有效地从现有实验数据中学习,并准确预测未知干扰的基因表达响应。
预测未知基因干扰。对于干扰预测任务,我们使用三个白血病细胞系的 Perturb-seq 数据集来评估模型:Adamson 数据集包含 87 个单基因干扰; 整理后的 Replogle 数据集包含 1823 个单基因干扰;Norman 数据集包含 131 个双基因干扰和 105 个单基因干扰。为评估 scGPT 的干扰预测能力,我们在一个子集上对模型进行微调,给定输入的对照细胞状态和干预基因,以预测受干扰的表达谱。接下来,模型在涉及未见过基因的干扰上进行测试 (方法)。我们计算了 Pearson△指标,用于测量预测值与观测到的干扰后表达变化之间的相关性。此外,我们在每个干扰的前 20 个最显著变化基因上报告了这一指标,记为在差异表达基因上的 Pearson△值。有关指标计算的详情,请参阅补充说明 12。我们进行了 scGPT 与另外两种方法 GEARS 和线性回归基线的性能比较 (方法)。我们的结果表明,scGPT 在所有三个数据集上均取得了最高分 (图 3a 和补充表 6)。特别是在预测干扰后的变化方面,scGPT 表现出众,始终以 5-20% 的优势领先于其他方法。此外,我们在图 3b 中可视化了 Adamson 数据集中两个示例干扰的预测,scGPT 准确预测了所有前 20 个差异表达基因的表达变化趋势。
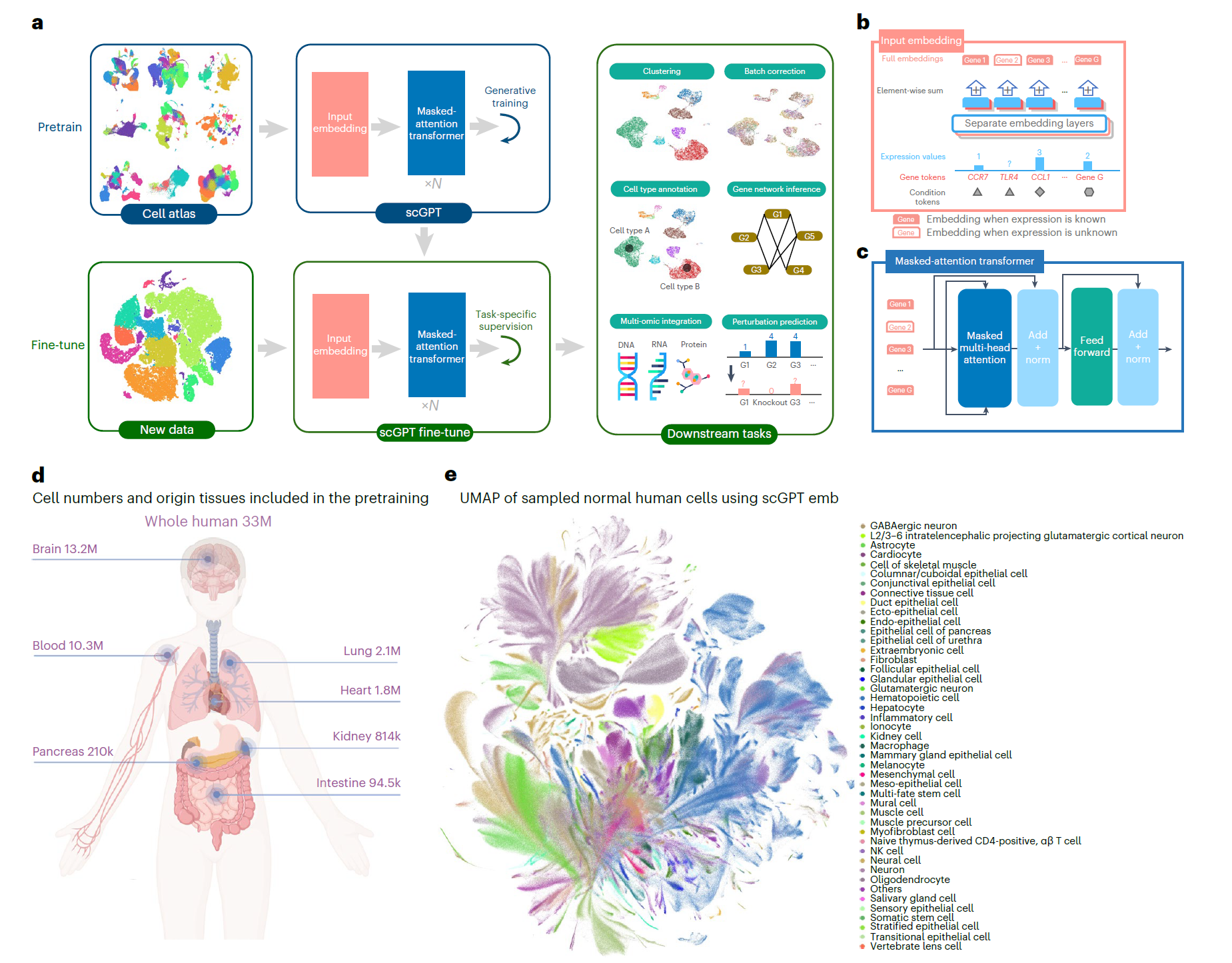
图 1 | 模型示意图。a,scGPT 的工作流程。该模型在来自细胞图谱的大规模 scRNA-seq 数据上进行生成式预训练。scGPT 的核心组件包含堆叠的 transformer 块,具有专门为生成式训练设计的注意力掩码。对于下游应用,预训练模型参数可以在新数据上进行微调。我们将 scGPT 应用于多种任务,包括细胞类型注释、批次校正、多组学整合、基因干扰预测和基因网络推断。b,输入数据嵌入的详细视图。输入包含三层信息: 基因符号、表达值和条件符号 (模态、批次、干扰条件等)。c,scGPT transformer 层的详细视图。我们在 masked multi-head attention 块中引入了专门设计的注意力掩码,以对单细胞测序数据进行生成式预训练。Norm 表示层归一化操作。d,描绘训练数据规模和器官来源的图示。scGPT 全人体模型是在 3300 万个正常人体细胞的 scRNA-seq 数据上进行预训练的。k 代表千。e,预训练 scGPT 细胞嵌入 (emb; 随机 10% 子集) 的 UMAP 可视化,按主要细胞类型着色。GABA 代表γ- 氨基丁酸。
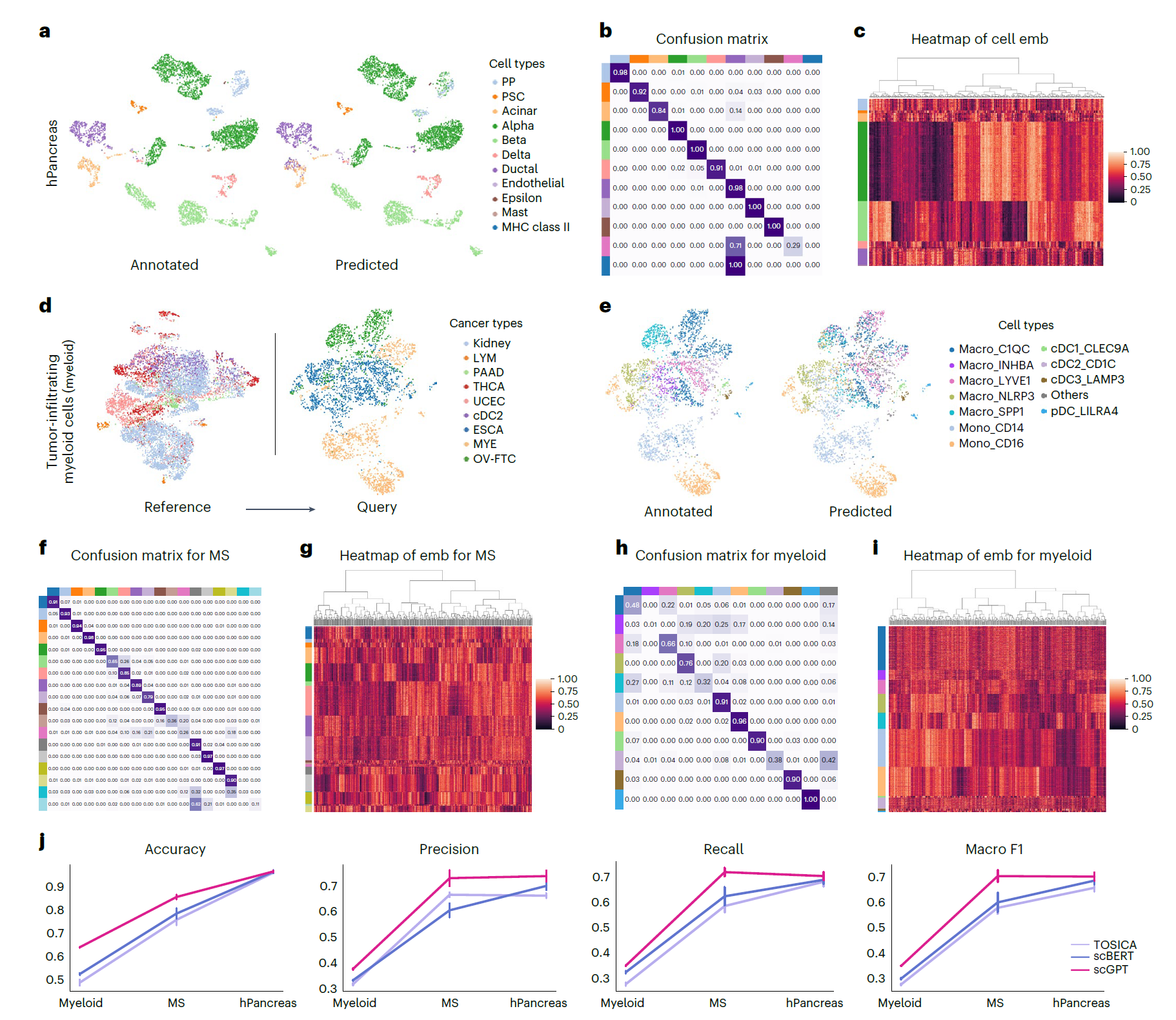
图 2 | 使用 scGPT 进行细胞类型注释的结果。a,人胰腺数据集细胞的基因表达 UMAP,按原始研究中的细胞类型注释 (左侧) 和由微调后的 scGPT 预测的细胞类型 (右侧) 着色。PP,胰岛素促肽细胞;PSC,胰腺星状细胞。b,人胰腺数据集中预测的细胞类型与注释的细胞类型之间的混淆矩阵。c,scGPT 在人胰腺数据集中 512 维细胞嵌入的热图。d,髓细胞数据集的 UMAP 可视化,按癌症类型着色。scGPT 在参考分区 (左侧) 上进行微调,并在查询分区 (右侧) 上进行评估。这两个数据分区包含不同的癌症类型。cDC2,2 型 (CD1A+CD172A+) 常规树突状细胞;ESCA,食管癌;LYM,淋巴瘤;MYE,骨髓瘤;OV-FTC,卵巢或滤泡状甲状腺癌;PAAD,胰腺腺癌;THCA,甲状腺癌;UCEC,子宫内膜癌。e,在查询分区上,UMAP 按原始研究中的细胞类型注释 (左侧) 和 scGPT 预测的细胞类型着色。f,h,分别为 MS 和髓细胞数据集中预测的细胞类型与实际注释之间的混淆矩阵。g,i,分别显示了 scGPT 中 MS 和髓细胞数据集细胞 512 维嵌入的热图。j,通过对髓细胞、MS 和人胰腺数据集进行 n=5 次随机训练 - 验证集划分,评估 scGPT 的细胞注释性能。测试集的性能指标呈现为均值±标准误。
预测未知的干扰响应的能力可以扩大干扰实验的范围,如图 3c 所示。为探索预测干扰响应的扩展空间,我们使用 Norman 数据集进行了聚类分析,以验证与生物学相关的功能信号。原始 Perturb-seq 研究覆盖了针对 105 个基因的 236 个干扰。然而,考虑到这些目标基因的所有可能组合,总共有 5,565 种潜在干扰,这表明实验 Perturb-seq 数据仅代表了整个干扰空间的 5%。因此,我们应用微调后的 scGPT 扩展原位干扰,并使用 UMAP 在图 3d 中可视化每种干扰的预测平均响应。使用原始研究中的注释,我们发现相同功能组的干扰条件聚集在相邻区域 (补充图 4)。接下来,我们使用 Leiden 对预测的表达进行了聚类,并观察到这些簇与干扰组合中的 " 主导基因 " 高度相关。例如,与 KLF1 基因相关的圆圈簇表明,该簇中的数据点经历了涉及 KLF1 和另一个基因 (即 KLF1 + X) 的组合干扰。以 KLF1 和 CNN1 簇为例,我们进一步验证了相应的预测表达仅在这些区域高度表达 (图 3e),这与 Norman 数据集中 CRISPRa(CRISPR 介导的转录激活)Perturb-seq 实验的预期结果一致。主导基因簇体现了 scGPT 揭示干扰组合与特定响应模式之间关联的能力。
原位反向干扰预测。scGPT 还能够给定最终细胞状态预测基因干扰的来源,我们称之为原位反向干扰预测。理想情况下,执行此类反向预测的模型可用于推断影响细胞系发育的重要驱动基因,或促进发现潜在治疗基因靶点。这种能力的一个假设应用示例可能是预测影响细胞从疾病状态恢复的 CRISPR 靶向基因。为展示反向干扰预测的有效性,我们使用了 Norman 数据集中涉及 20 个基因的一个子集 (图 3f)。这个组合空间共包括 210 种单基因或双基因干扰组合。我们使用已知的 39 种 (18%) 干扰 (图 3f 中的训练组) 对 scGPT 进行微调。然后,我们在未见过的受干扰细胞状态查询上测试模型,scGPT 成功预测了可产生观测结果的干扰源 (在最高排名的预测中)。例如,scGPT 将正确的 CNN1 + MAPK1 基因干扰排名为一个测试样例的第一预测,而将正确的 FOSB + UBASH3B 基因干扰排名为另一个案例的第二预测 (图 3f)。总的来说,scGPT 在 Top 1 预测中平均识别出 91.4% 相关干扰 (7 个中的 6.4 个)(图 3g 中的蓝条),在 Top 8 预测中识别出 65.7% 正确干扰 (7 个测试案例中的 4.6 个)(图 3g 中的粉红条),远远优于 GEARS 和差异基因基线。我们预计这些预测可用于规划干扰实验,最大限度提高获得目标细胞状态的可能性。与随机尝试相比,后者平均需要在该子集中 210 种可能干扰中尝试 105.5 次,以更少的尝试次数找到遗传变化的正确来源,可为加速发现重要的遗传驱动因子和优化干扰实验提供一种有价值的工具。
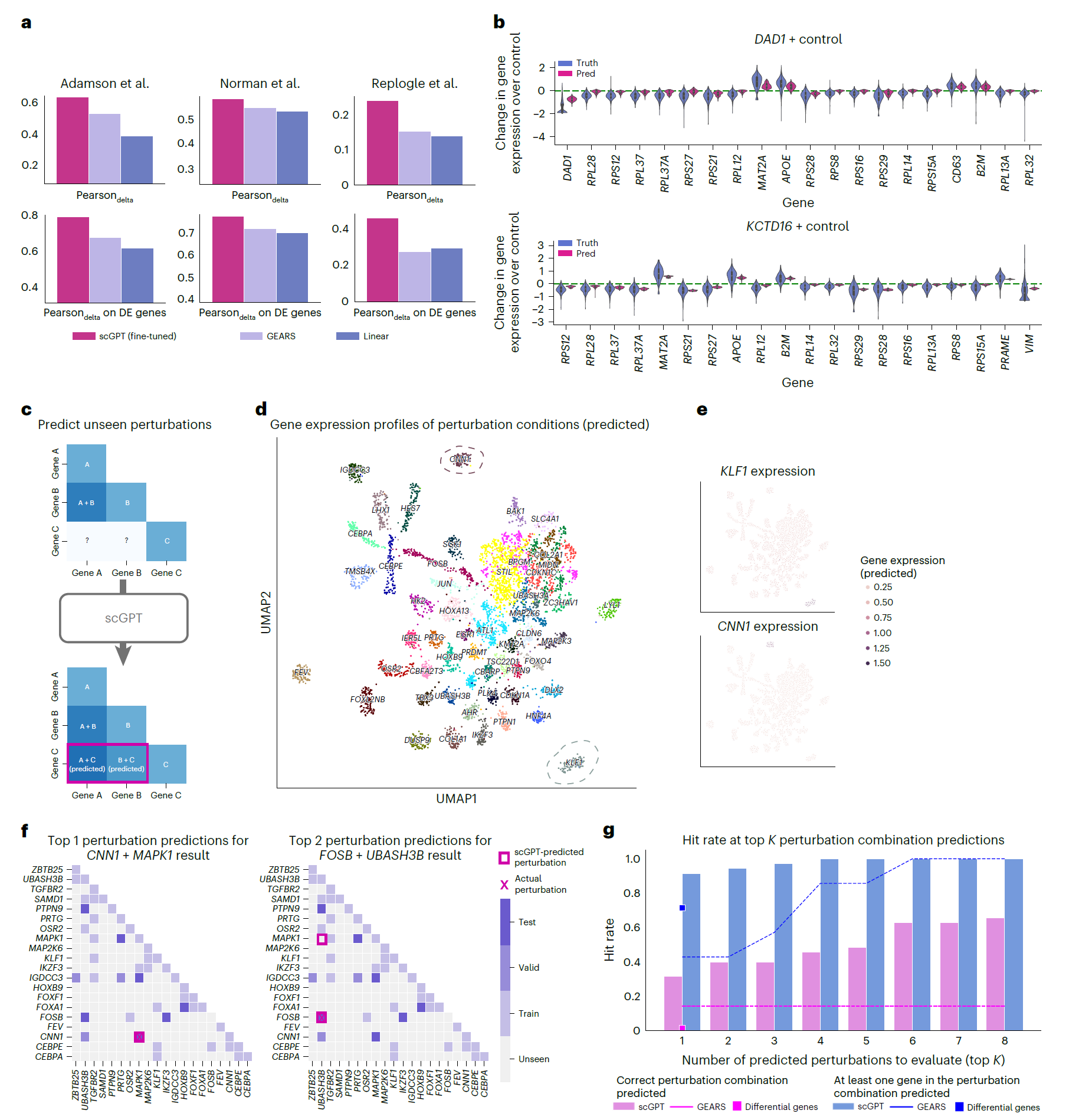
图 3 | 干扰响应和反向干扰预测结果。 a, scGPT 与其他干扰预测方法的比较。报告了预测值与实际基因表达变化之间的 Pearson 相关性。该指标分别计算了全部基因和前 20 个差异表达基因。b,Adamson 测试数据集中两个示例干扰,前 20 个差异表达基因的预测 (预测值;n=300 个细胞) 和实际基因表达变化 (KCTD16 基因干扰为 n=405 个细胞,DAD1 基因干扰为 n=618 个细胞) 的分布。箱体表示表达变化的四分位数范围。中位数由每个箱体内的中心线标记。须状线延伸至 1.5 倍的四分位数范围。水平虚线代表基因表达变化的空值基线。c,使用 scGPT 预测未知干扰响应的示意图。d,干扰条件的预测基因表达谱的 UMAP。UMAP 图按 Leiden 簇着色,并标记了每个簇的主导基因。e,两个选定的受干扰基因 (KLF1 和 CNN1) 在干扰条件 UMAP 上的表达模式。f,在 20 个基因的干扰组合空间中可能的干扰组合的可视化。网格按实验类型 (训练、验证、测试、未见) 着色。所有预测的干扰均用方框高亮显示,实际源干扰用十字标记。g,scGPT 在七个测试案例中对正确和相关预测的 Top 1-8 准确率,与 GEARS 和 Top 2 差异基因的天真基线进行了基准测试。相关预测 (蓝色) 表示至少有一个干扰基因在干扰组合中被发现了。scGPT 的命中率由条形图表示,GEARS 的命中率由线条显示,而差异基因 (仅针对 Top 1 预测) 由方形标记表示。
scGPT 实现了多批次和多组学整合
多批次 scRNA-seq 整合。整合来自不同批次的多个 scRNA-seq 数据集面临独特的挑战,即同时保留综合数据的生物学变异并去除技术批次效应。为整合测序样本,我们以自监督的方式对 scGPT 进行微调,学习统一的细胞表示以恢复被掩蔽的基因表达 (方法)。在我们的基准测试中,我们将 scGPT 与三种流行的整合方法 scVI、Seurat 和 Harmony 进行了比较。评估是在三个整合数据集上进行的,即 COVID-19(18 个批次)、外周血单核细胞 (PBMC)10k(两个批次) 和扁桃体皮质 (两个批次) 数据集。在 PBMC 10k 数据集中,scGPT 成功分离了所有细胞类型 (图 4a)。scGPT 优异的整合性能进一步得到了其高生物学保真分数的支持,AvgBIO 得分为 0.821,比对比方法高 5-10%。AvgBIO 分数是归一化互信息 (NMIcell)、调整后的兰德指数 (ARIcell) 和平均轮廓宽度 (ASWcell) 三个细胞类型聚类指标的综合,详见补充说明 12。值得注意的是,scGPT 在没有微调的情况下也展现出了很好的 PBMC 10k 数据集整合性能 (补充图 5),突出了预训练的广泛适用性。在扁桃体皮质数据集的情况下,scGPT 的性能与所有其他方法都相当 (补充图 6c)。这一发现凸显了从全人体数据集学习到的特征在应用于特定器官或组织 (如大脑) 时的可转移性和稳健性。此外,scGPT 在所有整合指标上都保持竞争力分数,并很好地保留了生物学信号 (补充表 3 和补充图 6 和 7)。另外,我们还开发了加速整合任务微调过程的策略,包括冻结特定模型层和排除不表达的基因,同时保持与原始方法相当的结果 (补充说明 3)。
单细胞多组学整合。单细胞多组学 (scMultiomic) 数据结合了多种遗传调控视角,如表观遗传、转录和翻译活动,在聚合细胞表示的同时保留生物学信号方面面临独特挑战。scGPT 通过有效提取不同组学数据集的综合细胞嵌入来应对这一挑战。对于包括基因表达和染色质可及性联合测量的 10x Multiome PBMC 数据集,我们将 scGPT 与两种最先进的方法 scGLUE 和 Seurat(v.4) 进行了比较。如图 4b 所示,scGPT 是唯一能够成功生成 CD8+naive 细胞的独特簇的方法。接下来,我们在图 4c 所示的骨髓单核细胞 (BMMCs) 配对的基因表达和蛋白丰度数据集上测试了 scGPT。该数据集包含额外的复杂性,如大量数据 (90,000 个细胞)、多个批次 (12 个供体) 和细粒度亚组注释 (48 种细胞类型)。与 Seurat(v.4) 相比,scGPT 呈现出更明确的聚类结构,AvgBIO 得分提高了 9%。值得注意的是,scGPT 能够将 CD4+naive T 细胞和 CD4+ 活化 T 细胞分离为两个独立的簇。它还将整合素β7+ 活化 CD4+ T 细胞与其他 CD4+ T 细胞区分开来,进一步证实了该模型捕获免疫细胞亚组之间细微差异的能力。在镶嵌式数据集整合设置中,测序样本共享一些但不是全部数据模态,给整合方法带来了挑战。为展示 scGPT 在这种情况下的能力,我们以 ASAP 人 PBMC 数据集为例,该数据集包括四个测序批次的三种数据模态。在与 scMoMat 的基准实验中,如图 4d 所示,scGPT 在 B 细胞、髓细胞和自然杀伤细胞 (NK 细胞) 组中表现出优异的批次校正能力。总体而言,scGPT 展现出卓越的细胞类型聚类性能,并在多种基准生物学保真指标上表现出稳健性 (补充表 4)。
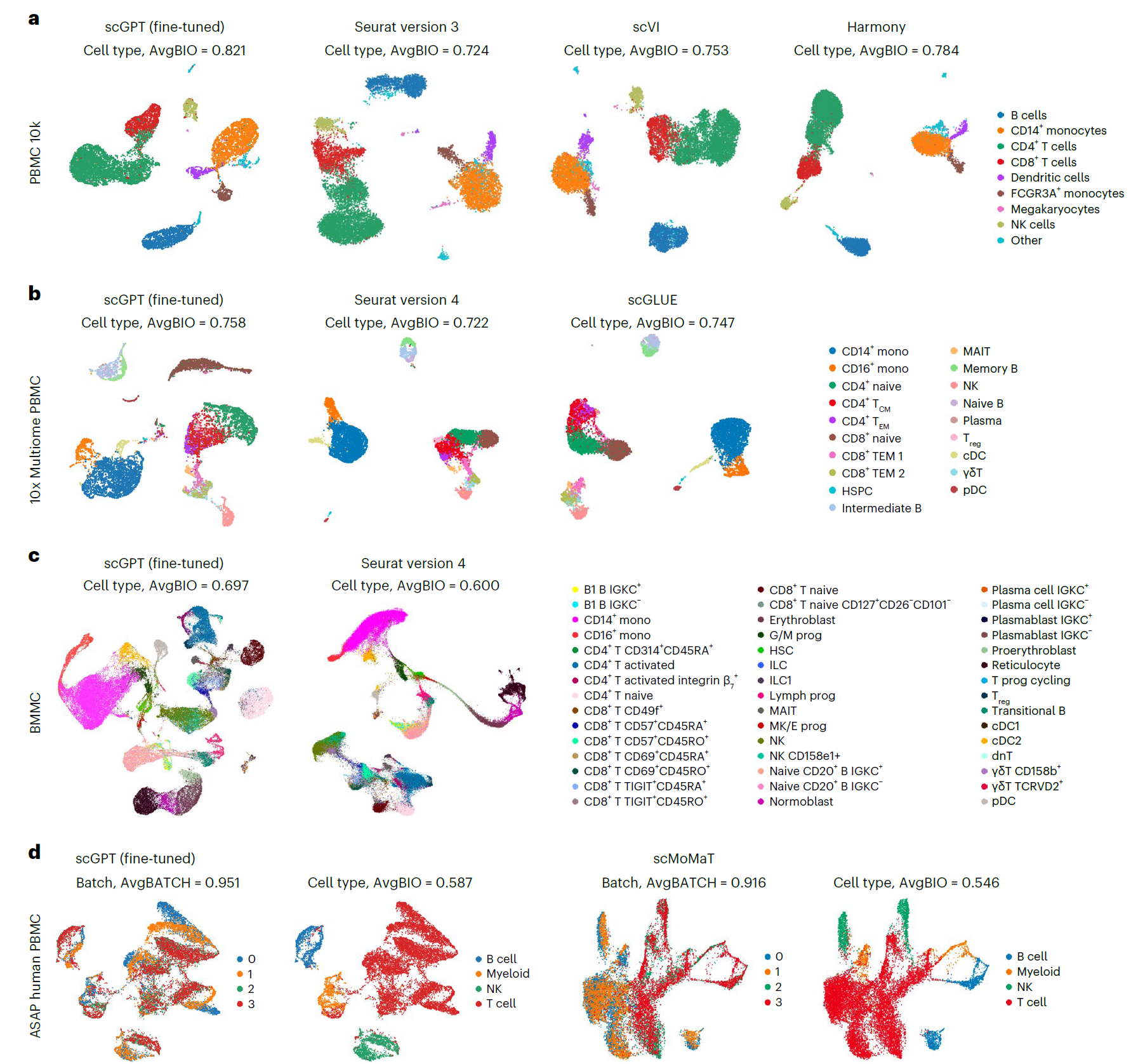
图 4 | 多批次和多组学整合结果。a,在 PBMC 10k 数据集上对微调后的 scGPT 进行细胞类型聚类任务的基准测试。学习到的细胞嵌入的 UMAP 图按细胞类型着色。b,在 10x Multiome PBMC 数据集 (配对 RNA 和转座酶可及染色质测定 (ATAC) 数据) 上,微调后的 scGPT 模型与 scGLUE 和 Seurat (v.4) 进行细胞类型聚类任务的基准测试。γδT,γδT 细胞;HSC,造血干细胞;HSPC,造血干细胞和祖细胞;ILC,先天性淋巴细胞;MAIT,黏膜相关不变 T 细胞;mono,单核细胞;pDC,浆细胞样树突状细胞;TCM,中央记忆 T 细胞;TEM,效应记忆 T 细胞;Treg,调节性 T 细胞。c,在 BMMC 数据集 (配对 RNA 和蛋白数据) 上,微调后的 scGPT 模型与 Seurat (v.4) 进行细胞类型聚类任务的基准测试。dnT,双阴性 T 细胞;G/M,粒细胞/巨噬细胞;MK/E,巨核细胞/红细胞;Prog,祖细胞。d,在 ASAP PBMC 数据集 (镶嵌 RNA、ATAC 和蛋白数据) 上,scGPT 与 scMoMat 进行批次校正和细胞类型聚类任务的基准测试。学习到的基因嵌入的 UMAP 图按测序批次 (左侧) 和细胞类型 (右侧) 着色。
通过学习到的基因符号嵌入,scGPT 展示了将功能相关基因分组以及区分功能不同基因的能力。在图 5a 中,我们通过可视化预训练 scGPT 模型的基因嵌入获得的人类白细胞抗原 (HLA) 蛋白相似性网络,进行了一次健全性检查。在这种零次设置下,scGPT 模型成功突出了与两个已充分描述的 HLA 类别对应的两个集群:HLA I 类和 HLA II 类基因。这些类别编码抗原呈递蛋白,在免疫环境中扮演不同角色。例如,HLA I 类蛋白 (由 HLA-A、HLA-C 和 HLA-E 等基因编码) 被 CD8+ T 细胞识别并介导细胞毒性效应,而 HLA II 类蛋白 (由 HLA-DRB1、HLA-DRA 和 HLA-DPA1 编码) 被 CD4+ T 细胞识别并触发更广泛的辅助功能。
另外,我们在 " 免疫人体 " 数据集上对 scGPT 模型进行了微调,并探索了该数据集中存在的免疫细胞类型特异的 CD 基因网络。我们使用与整合任务相同的微调策略 (方法) 来进行基因调控网络分析。预训练的 scGPT 模型成功识别了编码 T 细胞激活所需的 T3 复合物的一组基因 (CD3E、CD3D 和 CD3G),以及 B 细胞信号传导所需的 CD79A 和 CD79B,以及作为 HLA I 类分子的辅助受体的 CD8A 和 CD8B。此外,微调后的 scGPT 模型还突出了 CD36 与 CD14 之间的联系。
scGPT 能够发现表现出细胞类型特异性激活的有意义的基因程序。基因程序随后使用来自 scGPT 的基因嵌入进行选择和聚类 (方法)。在图 5c 中,我们可视化了在 " 免疫人体 " 数据集中由微调后的 scGPT 模型从高变异基因 (HVG) 中提取的基因程序及其在不同细胞类型中的表达。我们观察到,一组 HLA II 类基因被识别为第 2 组。同样,参与 T3 复合物的 CD3 基因被识别为第 3 组,在 T 细胞中表达最高。为系统地验证提取的基因程序,我们对 Reactome 数据库进行了通路富集分析,并使用严格的多重测试校正 (https://mathworld.wolfram.com/BonferroniCorrection.html) 识别了高置信度的 " 通路命中 "(方法)。在图 5d 中,我们将来自 scGPT 的结果与来自共表达网络的结果进行了比较。值得注意的是,scGPT 在所有聚类分辨率下都显示出大幅更高的富集通路数量。此外,我们如图 5e 所示,检查了 scGPT 和共表达网络之间识别的通路的相似和差异之处。两种方法都识别出 15 条共同通路,包括与细胞周期和免疫系统相关的通路。scGPT 另外独特地识别出 22 条额外通路,其中 14 条与免疫相关。值得注意的是,scGPT 特别突出了与适应性免疫系统、T 细胞受体信号传导、PD-1 信号传导和 MHC II 类呈递相关的通路。这与微调数据集中存在适应性免疫群体的事实是一致的。这些发现展示了 scGPT 捕获错综复杂的基因 - 基因连接并在更广泛的生物学背景中阐明具体机制的卓越能力。富集通路的详细列表见补充表 5。
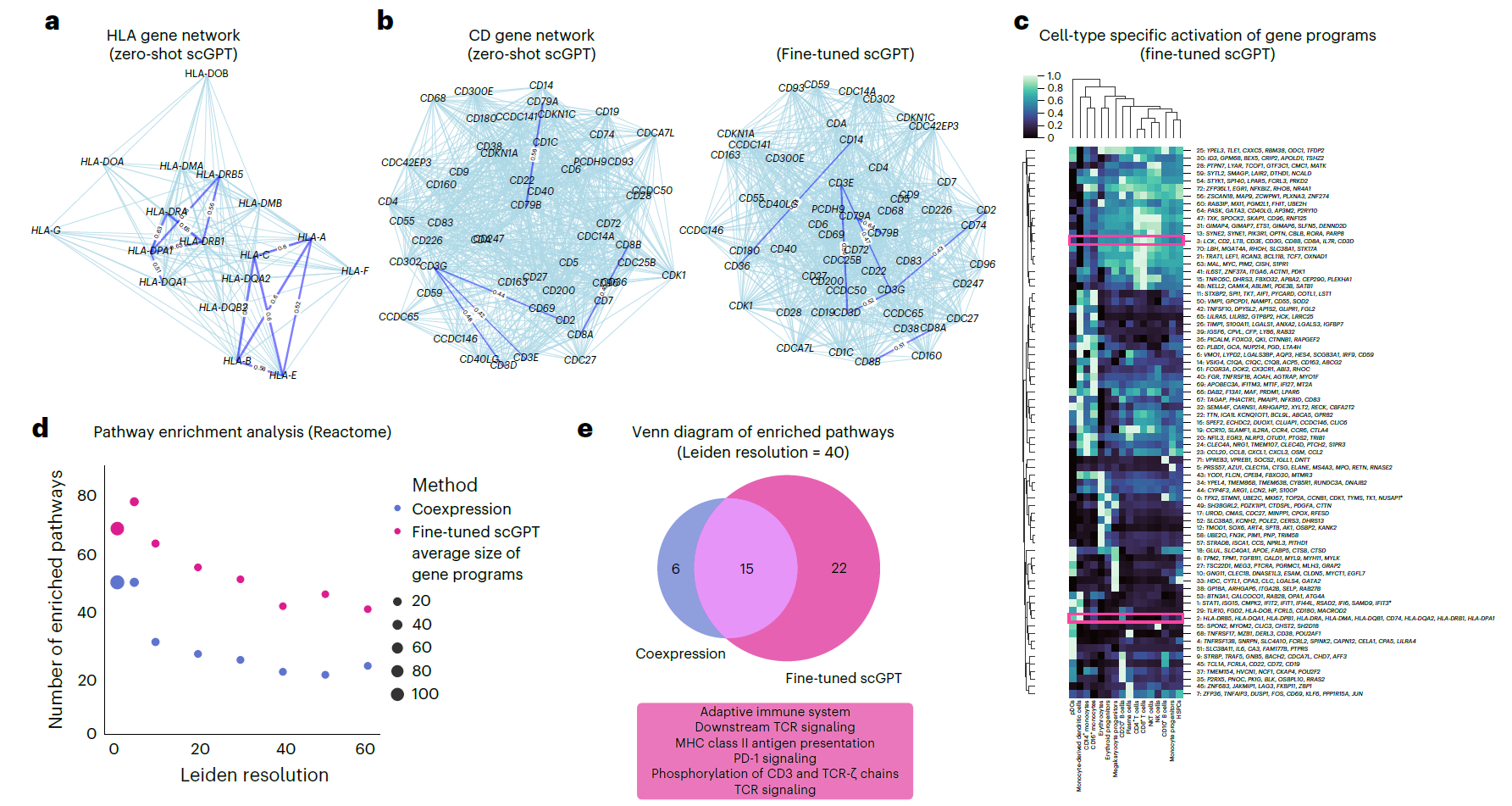
图 5 | 基因符号嵌入分析。a,零次 scGPT 的 HLA 基因网络。b,在 " 免疫人体 " 数据集上,零次 (即预训练) 和微调后 scGPT 的 CD 基因网络。c," 免疫人体 " 数据集中 scGPT 提取的基因程序在不同细胞类型中的特异性激活情况。颜色表示平均基因表达量。对于包含超过 10 个基因的基因程序 (用星号表示),为简单起见,仅显示前 10 个基因。d,scGPT 和共表达网络在 " 免疫人体 " 数据集中提取的基因程序的通路富集分析。将 scGPT 基因程序的富集通路数量与共表达方法在不同 Leiden 分辨率下的结果进行了比较。e,用文氏图比较了共表达和 scGPT 识别的富集通路之间的重叠和差异。一些 scGPT 独有的、特异于适应性免疫功能的示例通路在文本框中高亮显示。TCR 代表 T 细胞受体。
除了使用基因嵌入进行数据集级基因网络推断外,scGPT 的注意力机制使其能够在单个细胞水平捕获基因 - 基因相互作用。scGPT 通过汇总注意力图中的单个细胞信号提取细胞状态特定的网络激活数据。这为研究提供了洞察力,可了解个体细胞内特定背景下的基因调控相互作用,在不同细胞状态和条件下可能存在差异。例如,在干扰实验中,scGPT 检查干扰前后基因网络激活的变化,以推断哪些基因受到每个受扰基因的最大影响(见图 6a 和方法)。在 Adamson CRISPR 干扰数据集中,scGPT 确定了受 DDIT3(编码转录因子)抑制影响最大的前 20 个基因,这些基因在 ChIP-Atlas 数据库中均被发现是 DDIT3 的信号靶点(见图 6b)。此外,scGPT 捕获了在控制组与 DDIT3 敲除组中受 DDIT3 影响最大的前 100 个基因之间的不同通路激活模式。值得注意的是,在 DDIT3 敲除组中确定的 ATF6 转录因子通路已知介导未折叠蛋白反应并调节细胞凋亡。同样,在 BHLHE40 抑制的情况下,发现受其影响最大的前 20 个基因中有 19 个是由染色质免疫沉淀测序(ChIP-seq)预测的该转录因子的靶点(见图 6c)。突出显示 DNA 合成和有丝分裂的通路激活模式反映了转录因子 BHLHE40 在细胞周期调节中的作用。这些基于注意力的发现进一步验证了 scGPT 在细胞状态水平上学习的基因网络,为模型学到的生物学提供了额外的可解释性。
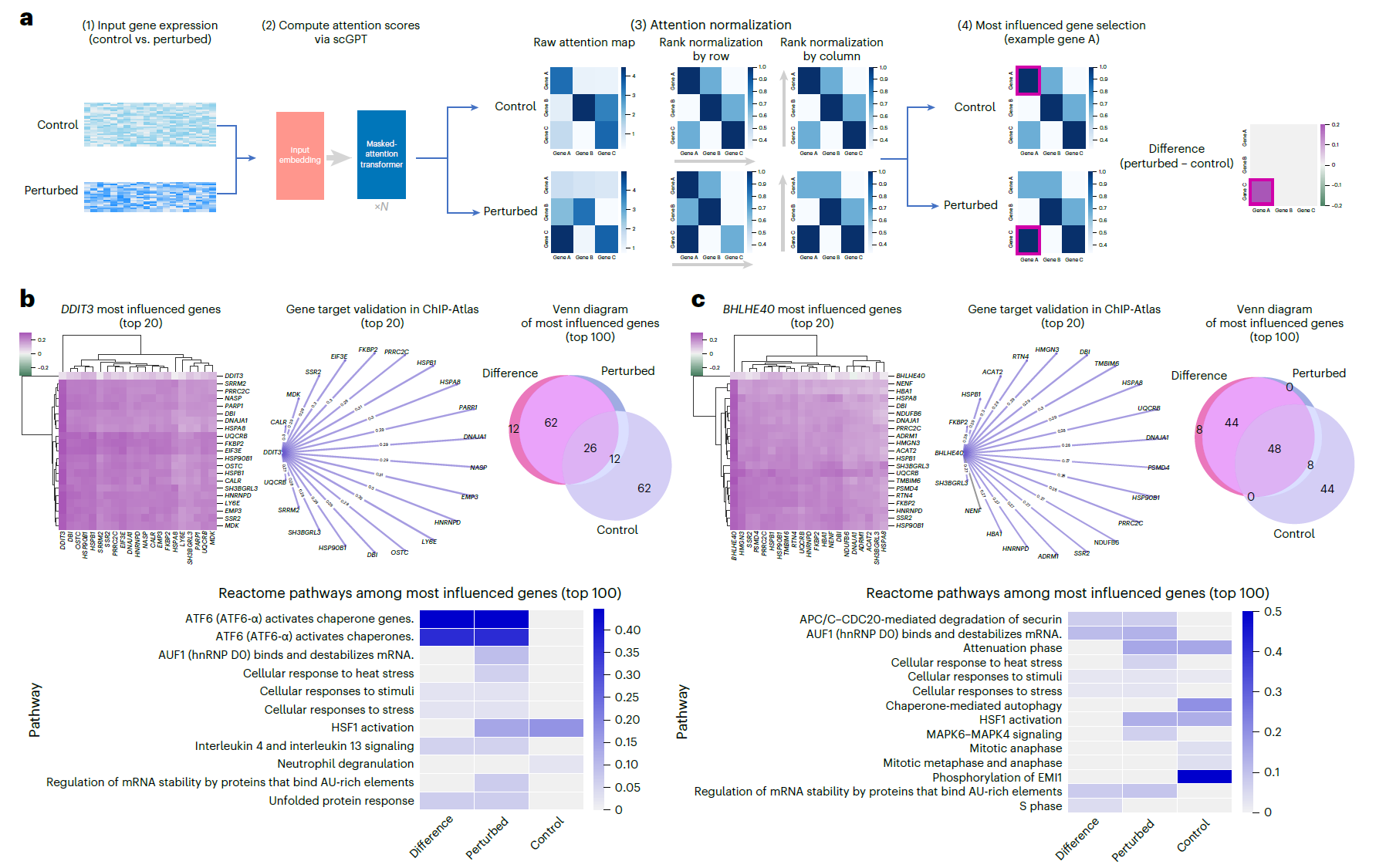
图 6 | 基于注意力的基因相互作用分析。a、干扰数据的基于注意力的 GRN 发现工作流程。连续按行和列获取和排名控制和干扰细胞状态中的注意力分数。相应地选择控制、干扰和差异设置中受影响最大的基因。b、DDIT3 抑制的 GRN 分析。基因连接性热图呈现 DDIT3 抑制对最受 DDIT3 影响的前 20 个基因网络的干扰后变化。基因靶标网络图展示了在 ChIP-Atlas 数据库中验证的前 20 个基因,ChIP–seq 预测的靶标以紫色突出显示。Venn 图比较了在三个选择设置(即,控制、干扰和干扰后差异)中识别的前 100 个最受影响基因集之间的重叠和差异。通路热图展示了在这三个选择设置中从前 100 个受影响最大的基因中识别出的 Reactome 通路的差异。颜色表示每个通路的代表性程度,以基因重叠的百分比表示。c、BHLHE40 抑制的 GRN 分析,以类似的方式可视化。
在转移学习中的规模化和上下文效应
在前面的章节中,scGPT 已经通过微调展示了以转移学习方式的巨大潜力。我们通过将其与从头开始为每个下游任务训练的类似变压器模型进行比较,进一步确认了使用基础模型的好处(标记为 scGPT(从头开始))。结果见附表 2–4,微调后的 scGPT 始终在集成和细胞类型注释等任务中展示出性能增益。鉴于基础模型对下游任务的贡献,我们进一步对影响转移学习过程的因素感兴趣。
首先,我们深入探讨了预训练数据量与微调模型性能之间的关系:对于某个分析任务,通过将更多测序数据加入预训练图谱,能获得多少改进?我们预训练了一系列参数相同但使用不同数量数据的 scGPT 模型,从 30,000 到 3300 万个测序的正常人细胞。附图 13a 展示了使用这些不同预训练模型进行微调的结果性能。我们观察到,随着预训练数据量的增加,微调模型的性能提高了(附注 4)。这些结果表明了一种规模效应,即更大的预训练数据量导致更好的预训练嵌入和在下游任务上的性能改进。值得注意的是,我们的发现也与自然语言模型中报告的规模定律一致,突显了数据量在模型性能中的重要作用。预训练数据量在微调结果中的关键作用,预示了预训练模型在单细胞领域有着光明的未来。随着更大更多样化的数据集的出现,我们可以预期模型性能的进一步提高,推进我们对细胞过程的理解。
我们探讨的第二个因素是上下文特定预训练的影响。在这里,上下文使用指的是一个在特定细胞类型上进行预训练,然后在类似细胞类型上进行下游任务微调的 scGPT 模型。为了探索这个因素的影响,我们在来自各个主要器官的正常人细胞(图 1d)上预训练了七个器官特异性模型,以及另一个用于泛癌细胞的模型。我们通过可视化预训练数据的细胞嵌入来验证预训练的有效性:泛癌模型的细胞嵌入能准确区分不同的癌症类型。器官特异性模型能够揭示相应器官的细胞异质性。接下来,我们对 COVID-19 数据集上的各个模型进行微调,以检查预训练上下文的影响。我们的分析显示了模型的上下文在预训练中的相关性与其后续数据整合任务的性能之间存在明显的相关性。在数据整合任务中表现最好的是预训练于整体人体、血液和肺部数据集的模型,这与 COVID-19 数据集中存在的细胞类型密切相关。值得注意的是,即使是大脑预训练模型,尽管经过了 1300 万个细胞的大量数据训练,与类似数据量的血液预训练模型相比,性能也降低了 8%。这强调了在预训练中将细胞上下文与目标数据集对齐对于下游任务的优越结果的重要性。虽然考虑细胞上下文是必要的,但整体人体预训练模型是一个多才多艺且可靠的选择,适用于各种应用场景。
讨论¶
我们引入了 scGPT,这是一个利用大量单细胞数据的预训练变压器的基础模型。借鉴了语言模型中自监督预训练的成功,我们在单细胞领域采用了类似的方法来揭示复杂的生物相互作用。在 scGPT 中使用变压器使得同时学习基因和细胞嵌入成为可能,这有助于对细胞过程的各个方面进行建模。通过利用变压器的注意力机制,scGPT 捕获了单细胞水平的基因 - 基因相互作用,提供了额外的可解释性层次。
我们通过广泛的实验,在零样本和微调设置下展示了预训练的好处。预训练模型展示了强大的能力,能够推广到未见过的数据集,并在零样本实验中呈现出与细胞类型相符的有意义的聚类模式。此外,scGPT 中学到的基因网络与已知的功能组具有强烈的一致性。此外,预训练模型的知识可以通过微调转移到多个下游任务中。在诸如细胞类型注释、干扰预测以及多批次和多组学数据集成等各种任务中,微调后的 scGPT 模型始终优于从头开始训练的模型。这显示了预训练模型对下游任务的价值,使得分析更准确和具有生物学意义。值得注意的是,目前的预训练并不能本质上减轻批次效应,因此在存在大量技术变化的数据集上,模型的零样本性能可能受到限制。由于常常缺乏确定的生物学基本事实,并且数据质量存在变化(详见附注 10),评估模型也是复杂的。
对于未来的方向,我们计划在更大规模的数据集上进行预训练,包括多组学数据、空间组学和各种疾病情况。将干扰和时间数据纳入预训练阶段也很有趣,使模型能够学习因果关系,并推断基因和细胞如何随着时间变化而做出响应。我们还计划探索单细胞数据的上下文指导学习。这涉及开发技术,使预训练模型能够在零样本设置中理解和适应不同的任务和上下文,而无需微调。通过使 scGPT 掌握不同分析的细微差别和特定要求,我们可以增强其在各种研究场景中的可用性和适用性。我们预见,预训练范式将被轻松地整合到单细胞研究中,并作为一个基础,利用呈指数增长的细胞图谱中的现有知识进行新的发现。