scRank infers drug responsive cell types from untreated scRNA seq data using a target perturbed gene regulatory network

引言¶
细胞对药物的反应表现出巨大的异质性,这主要归因于细胞群体的多样性。在不同细胞的特定生物网络中,药物与其靶点(如受体或酶)相互作用会产生不同的效果。例如,激酶抑制剂可能会阻止肿瘤细胞增殖,而对正常细胞影响最小,因为不同的途径激活有所不同。因此,理解异质的药物反应需要对复杂组织中的细胞亚群进行表征。传统上,组织转录组分析仅提供聚合的表达数据,掩盖了不同细胞群体的独特分子特征。这些独特的分子特征在介导细胞对治疗的不同反应中至关重要。最近的单细胞 RNA 测序(scRNA-seq)技术进展提供了在单细胞水平上解析细胞异质性的机会。这一方法揭示了大量信息,包括细胞类型的识别、其空间分布、细胞间通信模式以及理解疾病进展和细胞对药物反应的基因调控机制。因此,药物开发策略已从针对特定因素转向针对关键细胞类型,从而为精准医学开辟了新途径。最近的研究,包括使用治疗集群识别可药物作用的细胞群体和深度学习方法在疾病背景下优先考虑细胞类型,进一步加深了我们对药物开发中细胞靶标的理解。此外,将整体表达数据与 scRNA-seq 分析相结合的工具为确定表型相关的细胞群体提供了补充见解。尽管 scRNA-seq 取得了进展,但识别对药物治疗有反应的关键细胞类型仍是一个重要挑战,因为这需要理解药效动力学,包括药物与其靶点的直接相互作用及其对细胞途径的后续级联效应。这些知识对于理解细胞水平的药物作用机制和开发副作用更少的精确治疗策略至关重要。虽然之前大多数分解药物反应的努力都集中在识别基因表达或蛋白质活性显著变化上,但哪个细胞类型对药物治疗表现出最高反应性的问题尚未得到充分解决。
在本研究中,细胞类型的药物反应被定义为不同细胞类型在暴露于药物后表现出细胞状态变化的程度。分析细胞类型药物反应的常用方法是基于差异表达基因(DEGs)的数量。最近,一种称为 Augur 的新方法使用高维空间内的可分离性来确定细胞类型的反应优先级。尽管这些策略允许量化药物反应,但它们不可避免地忽略了药物靶标的先验知识。此外,由于这些分析需要疾病 - 治疗配对数据集,它们不适用于从仅包含疾病状态而缺乏药物干预的数据集中推断药物反应细胞类型。由于充足的疾病数据集通常可用但缺乏药物治疗数据集,现有方法在疾病样本中的可扩展性受到限制。实际上,随着疾病状态数据的日益增加,仅从未处理的数据集中推断对治疗有反应的关键细胞类型具有扩展我们对药物在细胞水平上影响的知识的潜力,因此可以指导进一步的实验设计。因此,迫切需要一种方法,能够结合药物靶标知识和疾病细胞状态的知识,从未处理的数据集中识别药物反应细胞类型。鉴于基因广泛相互作用,药物反应在很大程度上依赖于与特定药物靶标相关的转录程序,基因调控网络(GRNs)可能代表推断细胞药物反应的合理基础。
为此,我们提出了 scRank,这是一种使用靶标扰动 GRN(tpGRN)在未处理的 scRNA-seq 数据中模拟和评分药物扰动的药物反应细胞类型推断方法。我们的工作基于两个假设。首先,不同细胞类型的内在细胞状态可以通过细胞类型特异性 GRN 反映出来。其次,细胞中的药物(抑制剂)扰动可以建模为 GRN 中药物靶基因节点的删除,这会导致全局和局部效应。scRank 算法的设计遵循这些假设。首先,scRank 对细胞类型进行聚类网络推断。为了模拟 GRN 中的药物效应,scRank 删除了药物靶基因节点的所有边缘,以创建 tpGRN。为了量化每个细胞类型中 tpGRN 的药物扰动程度并优先考虑它们,我们使用流形对齐算法和网络扩散评估了全局和局部扰动效应。我们进行了广泛的模拟,以评估 scRank 在识别药物反应细胞类型方面的准确性和鲁棒性。我们进一步使用未处理的 scRNA-seq 数据集验证了 scRank 的性能,这些数据包括来自多种复杂疾病(如癌症和糖尿病)的体内和体外数据。我们显示 scRank 可以成功识别药物靶向细胞类型,并且其性能优于其他现有方法。此外,我们将 scRank 应用于我们之前在心肌梗死小鼠模型中的巨噬细胞 scRNA 数据集,识别了丹参酮 IIA 的另一个潜在靶标,并通过实验验证了这一点。简而言之,scRank 使得仅从未处理的 scRNA-seq 数据中推断药物反应细胞类型成为可能,从而揭示药物的治疗机制。
结果¶
scRank 概述¶
我们提出了 scRank(https://github.com/ZJUFanLab/scRank),这是一种基于细胞类型对指定药物潜在基因网络活动进行排名的计算工具(图 1)。药物扰动不仅对单一靶点产生影响,还会影响下游基因。这些与靶点相关的下游基因往往在共同的路径中共同表达,通常协同作用以响应药物治疗。药物通过与靶点的相互作用,不仅通过直接靶点调节发挥作用,还通过这些共同表达的下游基因启动一系列信号事件,最终导致特定的生物功能。因此,对药物影响的综合评估必须超越仅仅检查靶基因表达,涵盖药物在更广泛的生物网络中的整体影响。我们使用 scRank 的方法利用网络方法来预测药物在不同细胞类型中的影响程度。我们假设化合物扰动通过抑制靶点,应该沿分子网络传播,最终影响网络的整体结构和强度。通过扰动基于共表达关系重构的基因表达谱中的分子网络中的药物直接靶基因节点,我们可以捕捉药物的系统性影响。我们将这种扰动效应建模为计算机模拟药物扰动所产生的全局网络差异和靶相关信号的局部网络扩散的组合。通过使用 tpGRN 实施这一假设,我们使用扰动评分来评估计算机模拟药物扰动对每个细胞类型的影响并对它们进行排序。由于这种计算机模拟药物扰动,我们的方法允许仅基于未处理的数据推断药物反应中的细胞类型优先级。
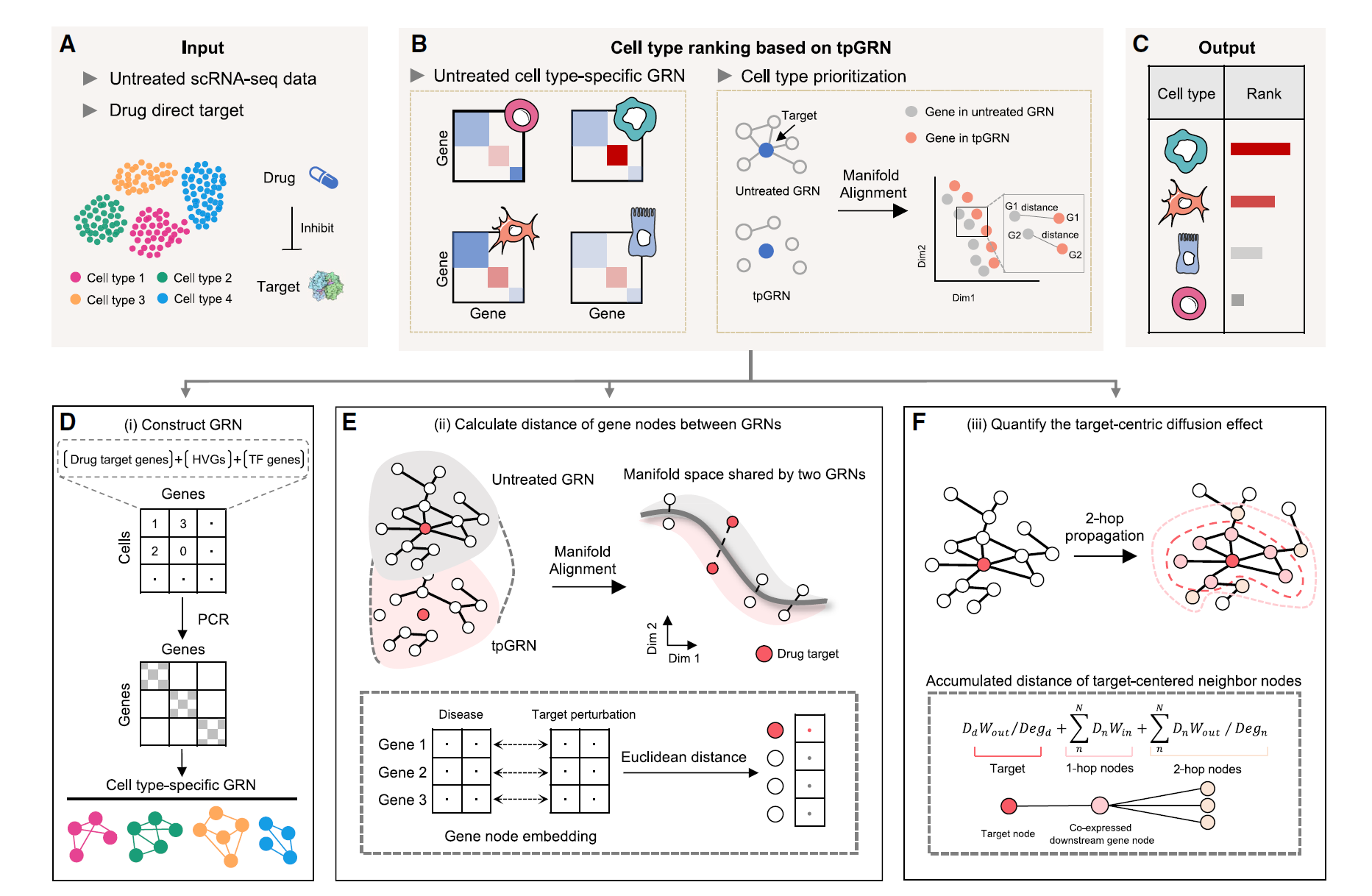
图 1:scRank 方法工作流程 > (A) 未处理的单细胞 RNA 测序数据和直接靶基因。 (B) 基于 tpGRN 对药物响应的细胞类型进行排名的概念。首先构建细胞类型特异性的 GRNs。在确定靶基因后,通过切断靶基因节点的所有边缘创建 tpGRN。通过比较未处理的 GRNs 和 tpGRNs,量化药物扰动以对细胞类型进行排名。 (C) 结合局部和全局扰动效应,以优先考虑每种细胞类型的药物反应 (D) 构建 GRN 的过程。抑制剂药物的靶基因从 DGIdb 收集,前 2,000 个高变异基因和转录因子被考虑用于构建 GRN。使用主成分回归获得共表达邻接矩阵。对于每种细胞类型,使用相同的基因节点创建特定的 GRN。 (E) 通过流形对齐计算全局扰动效应的示意图 (F) 使用网络扩散计算局部扩散扰动效应的示意图。Dd 表示药物靶点节点的距离,Dn 表示 1 跳基因节点 n 的距离,Wout 表示输出边的权重,Win 表示输入边的权重,Deg 表示基因节点的度。
scRank 方法一般以单细胞转录组数据和感兴趣的药物直接靶点作为输入数据,输出药物响应细胞类型排名(图 1A)。输入数据仅包括从疾病状态派生的未处理表达计数矩阵,以及用于执行计算机模拟药物扰动的药物直接靶点的基因名称。通过比较每种细胞类型在未处理和计算机模拟药物靶点扰动条件下的 GRN,使用流形对齐和网络扩散评估药物扰动(图 1B)。输出的扰动评分表示细胞类型对药物的反应程度(图 1C)。
对于单细胞 GRN 重建,首先根据细胞类型对未处理的表达数据进行分离,以构建一组细胞类型特异性 GRN。为了保留疾病状态的主要特征,并结合尽可能多的生物过程和药物靶基因以有效构建 GRN,我们只考虑有限的与表达相关的特征,即前 2,000 个高变异基因(HVGs)、转录因子和药物靶基因。在实践中,使用 Seurat 确定 HVGs,从 AnimalTFDB 获取转录因子,从 DGIdb 获取药物靶基因,并将其整合为输入基因特征来构建 GRN(图 1D)。这种特定组合有效地预测了药物反应(图 S1)。受 scTenifoldNet 和 scTenifoldKnk 的启发,我们采用了基于机器学习的框架来构建网络。然而,为了确保在适应特定药物反应推断问题时对生物过程的稳健表征,我们对框架进行了某些修改,包括输入特征和子样本策略。在实践中,使用主成分回归算法结合随机采样策略和张量分解,能够从表达数据中确定基因之间的共表达关系,从而产生表示细胞类型 GRN 的基因 - 基因邻接矩阵。
为了对药物反应细胞类型进行排名,首先通过将未处理 GRN 中靶基因的边权重设置为零来生成 tpGRN,以模拟抑制剂效应(图 1E)。我们还测试了通过 tpGRN 模拟激动剂效应的能力(图 S2)。然后,使用流形对齐算法比较未处理网络和靶标扰动网络。在共享的流形空间中计算不同条件下相同基因节点之间的距离,以衡量网络结构方面的差异,代表全局扰动。为了考虑局部扩散,即扰动效应将通过共表达边从靶基因传播到下游基因,我们考虑了 2 跳邻域扩散并对其进行了评分(图 1F)。然后将网络中的全局和局部效应结合为扰动评分,以量化扰动程度并对细胞类型进行排名。
使用合成真实数据集验证 scRank¶
我们首先在三种情况下对合成数据上的 scRank 进行测试,以评估其在识别药物反应细胞类型方面的性能。在我们的研究中,合成数据集设计用于模拟疾病状态下的细胞转录组。为实现这一点,我们使用 SERGIO 从预定义的 GRN 模拟单细胞基因表达数据集,其中疾病相关基因相互作用并形成疾病模块。我们假设如果药物的靶基因位于高活性疾病模块内,那么药物的扰动效应在细胞网络内将更为显著。因此,具有活性靶基因的细胞类型应对这种扰动表现出更显著的反应,从而产生细胞类型排名的真实情况(图 2A)。具体来说,我们开发了三个不同的场景,每个场景都有定义好的药物反应细胞类型的真实排名(图 2B)。这种方法使我们能够在不同情况下评估模型的预测准确性。
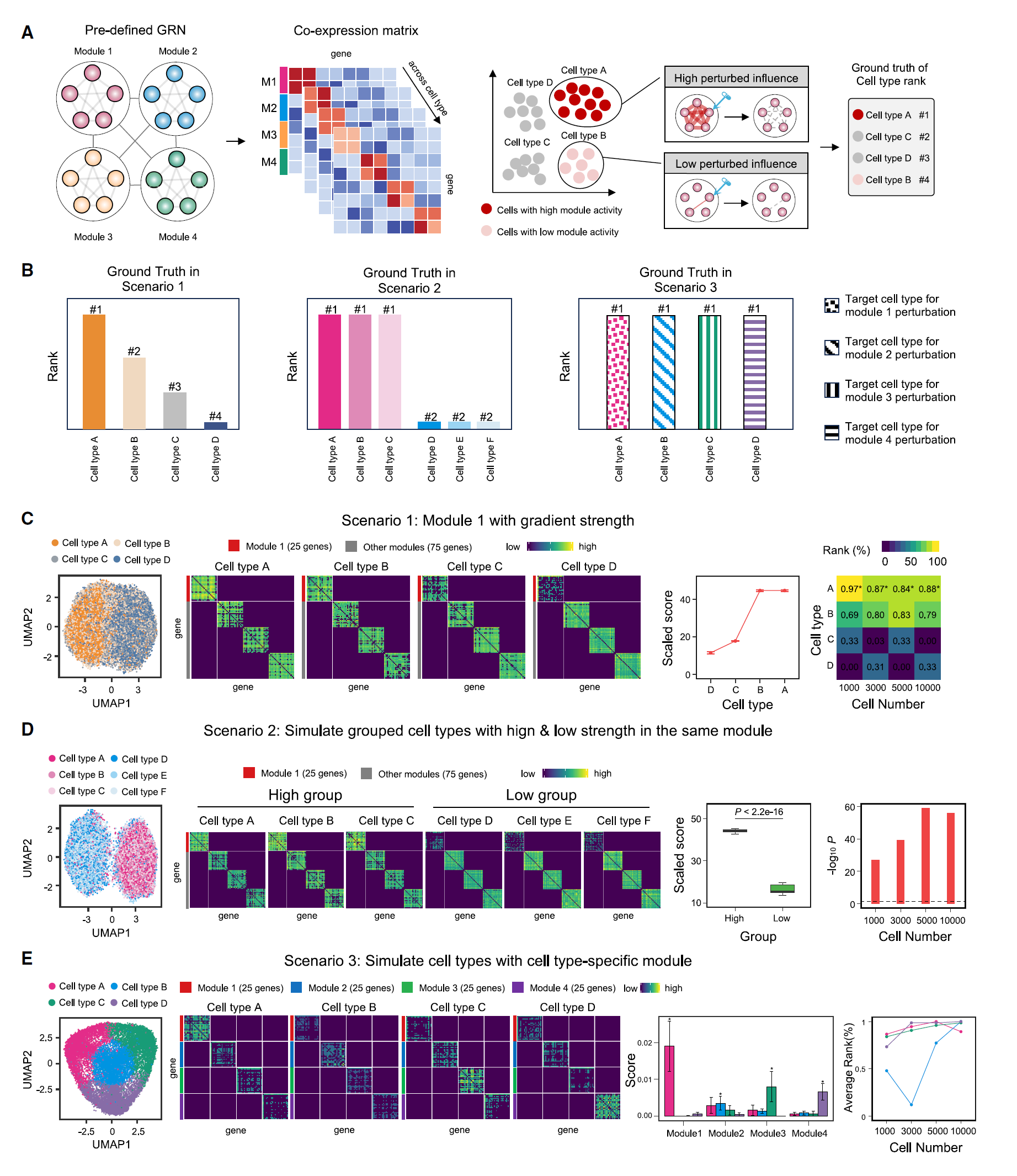
图 2:scRank 在模拟数据集中的基准测试 > (A) 生成模拟真实数据的示意图。我们使用预定义的 GRNs 通过 SERGIO 模拟三种情况下不同细胞类型中具有不同模块活动的疾病 scRNA 数据。我们假设具有高药物相关模块活性的细胞类型可能会受到较大的扰动。药物反应细胞类型排名的真实情况可以通过预定义的模块活动获得。 (B) 每种情况下的细胞类型真实排名。 (C) 四种细胞类型的 3,000 个模拟细胞,具有递增的模块 1 活性梯度。scRank 在不同细胞类型中的扰动评分按比例缩放后以点线图呈现。数据以中位数 ±SD 表示。每种细胞类型的数据点数量为 25。scRank 预测的具有递增细胞数量的细胞类型的平均排名以热图表示,其中星号表示每个数据集的排名最高的细胞类型。 (D) 六种细胞类型的 3,000 个模拟细胞,可根据它们的二进制模块 1 活性分为高低两组。scRank 计算的两组的扰动评分按比例缩放后以箱线图表示(最小值,第 25 百分位数,中位数,第 75 百分位数和最大值)。每组的数据点数量为 75。p 值通过 Wilcoxon 秩和检验计算,随细胞数量增加 (E) 四种细胞类型的 3,000 个模拟细胞,具有细胞类型特异性模块高活性。条形图为 scRank 计算的不同模块基因的扰动评分。数据以中位数 ±SD 表示。每个模块的数据点数量为 25。不同类型扰动下排名最高的细胞类型随细胞数量增加而变化的点线图。
在场景 1 中,我们模拟了具有四种细胞类型的 scRNA-seq 数据集,这些细胞类型在基因模块 1 的活性上表现出逐步增加,以说明疾病进展的常见模式(图 2C)。假设药物靶点在模块 1 中,我们测试了 scRank 是否能够根据与药物相关的基因活性正确优先排序细胞类型。结果显示,scRank 能够通过扰动模块 1 中的基因成功识别细胞类型,并在大多数情况下随着细胞数量的增加一致地将细胞类型 A 排在最高(图 2C)。
在场景 2 中,模块 1 不是线性增加,而是以二进制方式表现为两组细胞类型(图 2D),反映了不同的疾病模式。结果表明,scRank 也能够区分基因模块 1 中的“高组”和“低组”,在每种情况下都有显著差异。在场景 3 中,基因模块以细胞类型特异性的方式表现(图 2E),这对于理解各种细胞类型间的差异药物反应至关重要。每种细胞类型都有一个特定的高活性基因模块。然后我们迭代扰动模块 1 到 4 的基因。对于每个特定模块基因的扰动,目标细胞类型的扰动评分都比其他任何细胞类型更高,目标细胞类型的平均排名在大多数情况下最高。总的来说,在合成数据集中的验证证明了 scRank 能够准确排名细胞类型。
scRank 与其他方法的性能比较¶
为了进一步评估 scRank 在识别药物靶向细胞类型方面的性能,我们将其与现有方法进行了比较。这些方法基于不同条件下(治疗前和治疗后)相同细胞类型之间的相对 DEGs 数量或可分离性来优先排序对药物反应最强的细胞类型。差异表达分析方法,如 Wilcoxon 秩和检验、MAST、二模态似然比检验和 DESeq2,专注于识别统计显著的 DEGs。
需要注意的是,这些方法都需要具有治疗前和治疗后数据集,而 scRank 只需要治疗前数据集和药物信息,不需要任何治疗后数据。因此,为了将 scRank 与这些方法进行比较,我们首先将它们应用于合成的配对条件数据集(场景 4,图 3A)。数据集包含两种条件:疾病和治疗。对于疾病条件,四种细胞类型在模块 1 的活性上表现为梯度增加,代表疾病中异常激活的基因模块。考虑到引入药物抑制模块 1 中的基因,模块 1 的活性应当降至各细胞类型的基础水平。我们推测,基因模块活性较高的细胞类型将受到药物更大的影响,从而生成细胞类型的真实排名。因此,采用 Spearman 秩检验来比较预测的细胞类型排名与真实排名。通过增加数据集中的细胞数量,各种方法的细胞类型排名与真实情况一致。然而,scRank 在使用四个基准数据集进行的推断中表现出稳健性能,在推断药物反应细胞类型方面优于现有方法(图 3A)。
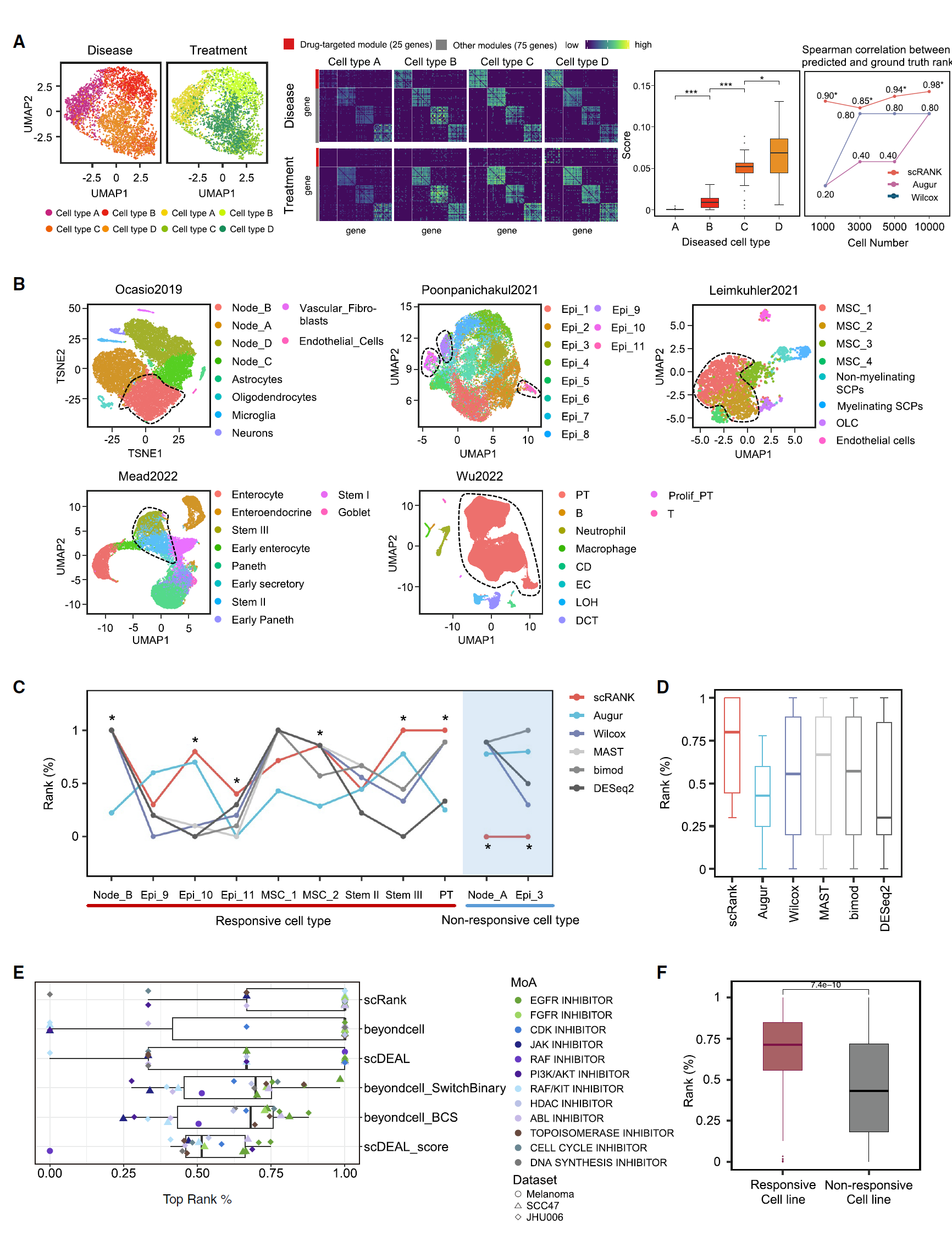
图 3:scRank 优于现有方法的性能
(A) 模拟数据中具有治疗前和治疗后条件的四种细胞类型。跨细胞类型和条件的共表达网络以热图表示。scRank 计算的疾病条件下各细胞类型的扰动评分以箱线图表示(最小值,第 25 百分位数,中位数,第 75 百分位数和最大值)。每种细胞类型的数据点数量为 25。p 值使用 Wilcoxon 检验计算。∗p ≤ 0.05 和 ∗∗∗p ≤ 0.001。scRank 与现有细胞类型优先级方法(Augur,通过 Wilcoxon 秩和检验的差异表达分析)在模拟数据中的性能比较通过点线图表示,细胞数量递增。星号表示特定数据集中所有细胞类型的最佳优先级。 (B) 具有真实靶细胞类型的数据集的低维投影。靶细胞类型用虚线圈出。 (C) scRank 与其他方法(Augur 和差异表达分析方法,包括 Wilcox, MAST, bimod 和 DESeq2)在九种响应细胞类型和两种非响应细胞类型中的性能比较。星号表示与真实情况一致的响应和非响应细胞类型的最佳优先级。 (D) 各方法的平均性能。数据以箱线图表示(最小值,第 25 百分位数,中位数,第 75 百分位数和最大值)。每种方法的数据点数量为 9 (E) scRank 与其他药物反应预测方法(beyondcell 和 scDEAL)在体外样本中的性能比较,平均准确率分别为 scRank 的 77.8%,beyondcell 的 72.2% 和 scDEAL 的 64.8%。数据以箱线图表示(最小值,第 25 百分位数,中位数,第 75 百分位数和最大值)。每种方法的数据点数量为 18。beyondcell 方法包括三种策略(beyondcell, bc_SwitchBinary, bc_BCS)来排名细胞类型。scDEAL 方法包括两种策略(scDEAL, scDEAL_binary)来排名细胞类型。 (F) 箱线图表示 scRank 预测的响应和非响应细胞系的排名,涵盖了 53 种药物中 179 个细胞系的分析。每组的数据点数量为 159。p 值通过单侧配对 Wilcoxon 检验计算。
此外,为了进一步比较 scRank 与现有方法在具有真实数据的 scRNA-seq 数据上的性能,我们收集了五个单细胞药物扰动数据集,这些数据集中包含九种已知的靶细胞类型(图 3B)。每个数据集包括配对条件,作者提供了药物靶向的细胞类型。在实践中,我们使用具有两种条件的数据集进行差异表达分析和 Augur,而 scRank 仅应用于疾病条件数据集。scRank 在五个数据集中识别了九种药物靶向细胞类型,并将其排在最高排名(图 3C 和 S3),在大多数情况下超过其他方法的排名。此外,scRank 将作为阴性对照的两种非响应细胞类型排在最低位置(图 3C),其排名低于任何其他方法的排名。平均而言,scRank 在比较其他方法时将靶细胞类型排在更高的位置(图 3D),表明它能够更准确地从仅包含未处理条件的数据集中确定药物靶向细胞类型。此外,我们还评估了 scRank 与其他扰动预测方法(如 scGen、scVIDR 和 CellOracle)的性能,scRank 在识别响应细胞类型方面优于所有其他工具(图 S4)。我们进一步使用三个额外的癌症 scRNA-seq 数据集验证了 scRank 的性能,重点关注五种经典治疗方法:抗 PD1、抗 CTLA4、抗 CD40、抗 PDL1 和多西他赛(图 S5)。通过这些数据集以及我们研究中包含的其他体内样本,我们确认了 scRank 在识别药物响应细胞类型方面的有效性。这一验证不仅限于仅使用直接靶标或下游靶标的表达,突出了 scRank 在不同治疗背景下的鲁棒性和多功能性(图 S6)。
接下来,我们结合了三个额外的癌症细胞系 scRNA-seq 数据集,以证明 scRank 在低细胞异质性情况下的性能,并使用这些数据集将 scRank 与药物反应预测工具(如 beyondcell 和 scDEAL)进行比较(图 3E 和 S7)。结果表明,即使在低变异性的体内样本中,scRank 仍保持优越的性能。此外,我们将 scRank 应用于包括 Kinker 等人研究中的 179 个不同细胞系的 scRNA-seq 数据。我们利用每个细胞系的 CTRP 数据集中的药物敏感性信息,评估 scRank 在 53 种药物中识别响应细胞系的性能。结果表明,scRank 适用于各种药物,总体准确率为 71.3%(图 3F 和 S8)。值得注意的是,响应性细胞系的排名显著高于非响应性细胞系。
识别髓母细胞瘤中对 vismodegib 敏感和耐药的肿瘤亚型¶
作为在真实 scRNA-seq 数据集上的基准扩展,我们选择了一个基准数据集,该数据集是从携带定义的药物敏感和耐药细胞类型的小鼠产生的,以展示 scRank 如何识别药物靶向细胞类型及其相关的生物机制(图 4A)。使用预处理条件下的细胞和 vismodegib 直接靶基因 Smo 作为输入数据(图 4A),scRank 成功识别了药物敏感和耐药的肿瘤亚型(Node_B 和 Node_A),分别基于其最高和最低的扰动评分,这与原始论文的发现一致(图 4B)。
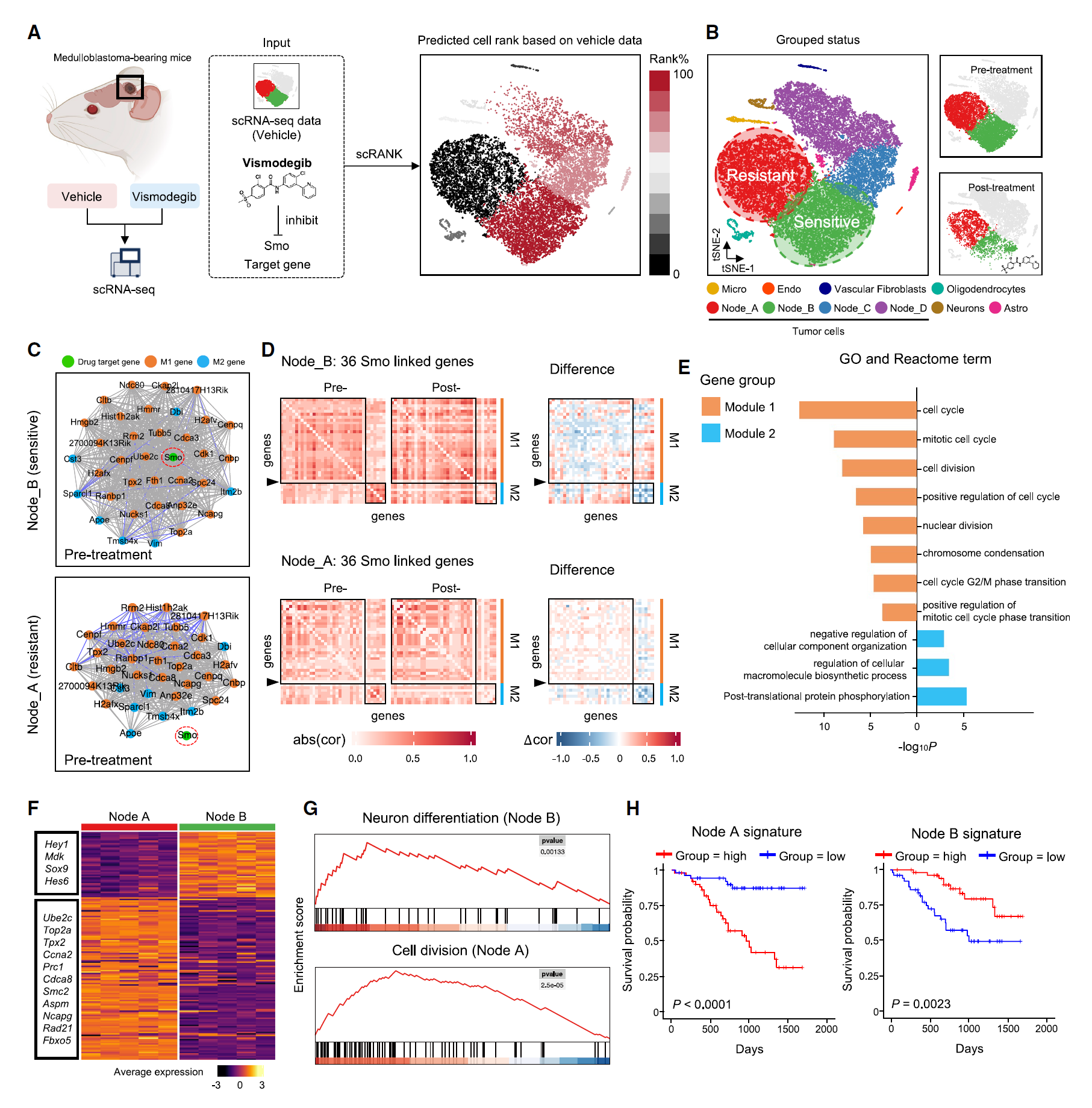
图 4:验证髓母细胞瘤中 vismodegib 药物敏感和耐药肿瘤亚型的预测
(A) 采用两种治疗条件下携带髓母细胞瘤的小鼠的 scRNA 数据。scRank 的输入为车辆 scRNA 数据和 vismodegib 的靶基因 Smo。t- 分布随机邻嵌(t-SNE)图按每种细胞类型中预测的最高排名百分比着色。 (B) 两种条件下原始样本的 t-SNE 图按细胞类型着色。Node_A 到 Node_D 代表肿瘤细胞。根据治疗前后相对差异,Node_A 和 Node_B 分别定义为耐药和敏感肿瘤亚型,如原始研究中所述。 (C) 预处理条件下 Node_A 和 Node_B 的 Smo 相关子网络。网络节点按基因模块类别着色。 (D) 两个肿瘤亚型的 GRN 热图及其差异(治疗后减去治疗前) (E) 不同模块基因的基因本体(GO)分析。 (F) Node_A 和 Node_B 中差异表达基因的热图。条形代表每个样本中的平均表达。 (G) 基因集合富集分析(GSEA)确定的 Node_B(上部)和 Node_A(下部)中显著激活的通路。 (H) Node_A 和 Node_B 标志基因的生存概率分析。在耐药肿瘤亚型(Node_A)中,标志基因的高表达表现出较低的生存概率。在药物敏感肿瘤亚型(Node_B)中,标志基因的高表达表现出较高的生存概率。p 值通过双侧 log-rank 检验计算。
为了阐明髓母细胞瘤对 Smo 抑制剂 vismodegib 的分子机制,我们应用 scRank 重建了治疗前条件下两种肿瘤亚型的细胞类型特异性基因网络(图 4C 和 4D)。我们重点研究了与 Smo 相关的基因,并可视化了 GRNs。图 4C 所示,网络中的基因被分为两个基因模块(M1 和 M2),药物靶基因(Smo)位于模块 1 中。基因模块 1 参与细胞周期进程、细胞分裂和 G2/M 期转换,而基因模块 2 主要涉及蛋白质磷酸化(图 4E)。值得注意的是,在治疗前条件下,Smo 相关的共表达模式在 Node_A 和 Node_B 的网络中显著不同,其中 Smo 在 Node_A 的网络中形成了一个孤立节点,而在 Node_B 的网络中形成了一个紧凑结构(图 4C)。这种与 Smo 的共表达关系表明,该基因在 Node_B 中的细胞增殖中高度相关,而在 Node_A 中则不然(图 4E),这与观察到的 Node_B 中 M1 和 M2 模块活性显著高于 Node_A 相一致(图 S9)。此外,两个细胞类型的治疗前后网络差异表明,Node_B 中的细胞周期相关模块 M1 的活性被抑制,而 Node_A 中的活性上调(图 4D),提供了药物效应依赖于靶基因相关子网络内基因活性的证据。有趣的是,Smo 和 Cnbp 之间的关系被发现位于 Node_B 的 M1 模块中(图 4C 和 S9),表明 Smo 信号可以通过这一非经典途径传递以促进生长。因此,抑制 Node_B 中的 Smo 信号,而不是 Node_A,可能对高活性 Shh 通路的细胞增殖抑制有效。这些结果表明,基因网络水平的差异,而不是表达水平的差异(图 S9),导致了不同细胞类型之间的药物反应差异。
为了进一步验证临床样本中对 SHH 抑制剂治疗的预测细胞类型优先级,我们采用两种肿瘤细胞标志对 178 个人类声波刺猬亚型髓母细胞瘤(SHH-MB)整体 RNA-seq 样本进行评分。我们发现,Node_A 和 Node_B 从分裂到分化的连续发展路径。Node_A 定义了细胞周期相关基因表达的增殖状态,而 Node_B 定义了高表达 HEY1、MDK、SOX9 和 HES6 的分化状态(图 4F 和 4G)。肿瘤显示出更大增殖且可能具有更高 Node_A 活性的患者预后显著较差(p < 0.0001,图 4H)。与此一致的是,肿瘤显示出更高分化的患者预后显著更好(p = 0.0023,图 4H)。因此,scRank 预测的最高排名细胞类型预计确实代表治疗靶标。
表征对 SSRIs 响应的 MDD 中兴奋性神经元亚型¶
我们随后检查了 scRank 是否可以应用于识别对选择性 5- 羟色胺再摄取抑制剂(SSRIs)响应的靶细胞类型。SSRIs 是治疗重度抑郁症(MDD)最常用的药物,通过抑制由 SLC6A4 基因编码的 5- 羟色胺转运蛋白来阻止 5- 羟色胺的再摄取,从而增加神经细胞周围的 5- 羟色胺可用性(图 5A)。然而,尽管对这种作用机制有全面的理解,但对 SSRI 响应的神经元亚群仍然未知。为此,我们收集了来自健康和未治疗 MDD 人脑的背外侧前额叶皮层区域的单核 RNA 测序(snRNA-seq)数据。在使用未治疗的 MDD 脑组织中的 snRNA-seq 数据和 SSRI 氟西汀的直接靶基因 SLC6A4 作为输入数据后,scRank 对氟西汀响应的细胞类型进行了排名(图 5B)。
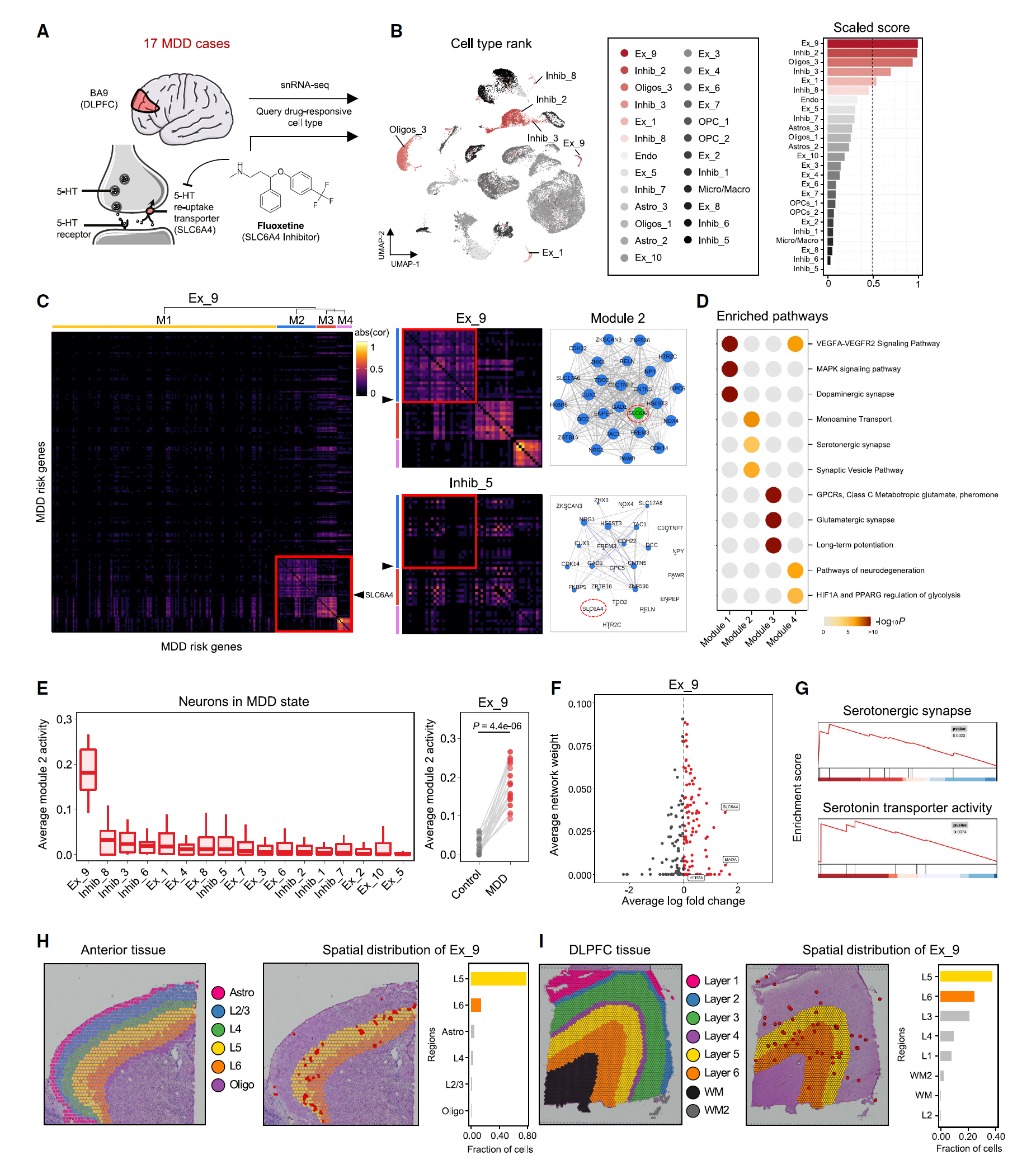
图 5:利用 scRank 识别 MDD 中由氟西汀靶向的神经元亚型
(A) 顶部图像显示了从 17 名 MDD 患者的 Brodmann 区域 9(BA9)的背外侧前额叶皮层(DLPFC)派生的 snRNA-seq 数据集。下部图像显示了氟西汀治疗 MDD 的机制示意图。MDD 状态的 scRNA-seq 数据和氟西汀靶基因是 scRank 的输入数据。 (B) 使用 UMAP 可视化的 25 种细胞类型数据,根据药物反应预测排名着色。条形图显示每种细胞类型的缩放扰动评分。Ex,兴奋性神经元;Inhib,抑制性神经元;Oligos,少突胶质细胞;Endo,内皮细胞;Astro,星形胶质细胞;OPC,少突胶质前体细胞。 (C) 兴奋性神经元簇 9(Ex_9)的 GRN 热图,其中包含 205 个 MDD 风险基因,这些基因分为四个模块。右上角的热图表示 Ex_9 模块 2-4 的子网络,而右下角的热图表示抑制性神经元簇 5(Inhib_5)的网络。两张热图均表示未处理样本中的 GRN。热图右侧的图形提供了相应细胞类型中模块 2 的网络可视化,其中基因节点根据其模块着色。 (D) 使用 Metascape 网页工具确定的模块基因显著富集的生物过程和通路 (E) MDD 中每种神经元亚型的平均模块 2 活性。数据以箱线图表示(最小值,第 25 百分位数,中位数,第 75 百分位数和最大值)。每组的数据点数量为 26。右侧显示了 Ex_9 在对照组和 MDD 组之间药物靶相关模块的比较,评估了 26 个模块 2 基因的平均边权重,通过配对双侧 Wilcoxon 检验计算。 (F) 健康状态和疾病状态之间 MDD 风险基因的平均对数折叠变化。 (G) 使用 GSEA 确定的 Ex_9 中显著激活的不同表达的 MDD 风险基因通路。 (H) 和 (I) 使用 CellTrek 的 Ex_9 空间映射。(H) 和 (I) 左侧的图像表示小鼠前脑组织 (H) 或人类背外侧前额叶皮层组织 (I) 中每个空间点的层注释。(H) 和 (I) 右侧的图像表示 Ex_9 的空间分布,其中红色像素特别标记这些神经元的位置。特别突出较深层(层 5 和 6)。随后的条形图显示每层细胞的相对比例。
我们首先使用 scRank 检查了排名最高和排名最低的细胞类型(Ex_9[兴奋性神经元簇 9] 和 Inhib_5[抑制性神经元簇 5])的 GRN。为了检查与疾病相关的生物过程,我们仅选择了 MDD 风险基因。我们发现,在 Ex_9 中,MDD 风险基因网络被模块化为四个基因模块,其中基因模块 2、3 和 4 表现出更高的活性(图 5C)。基因模块在涉及多巴胺能突触、5- 羟色胺能突触、谷氨酸能突触和神经退行性变的不同通路中富集(图 5D)。有趣的是,Ex_9 在模块 2 中表现出异常高的活性,其中包含编码药物靶基因 SLC6A4,表明该兴奋性神经元亚型可能在单胺运输中更活跃,因此可能是 SLC6A4 抑制剂的靶标。然而,在排名最低的细胞类型 Inhib_5 中,与 Ex_9 相比,基因模块 2 中的 SLC6A4 形成了一个孤立节点,整个网络非常松散,表明其作为 SLC6A4 抑制剂靶标的潜力较低。然后我们检查了所有神经元亚型中基因模块 2 的活性。Ex_9 在所有神经元亚型中模块 2 的活性一直最高(图 5E)。此外,在 Ex_9 中模块 2 的活性在对照组和 MDD 组之间显著增加(图 5E)。通过比较对照组和 MDD 组中 Ex_9 的 DEGs,相关于治疗性 SSRI 反应的基因(SLC6A4、MAOA、HTR2A)的网络权重和表达倍增显著上调,使我们能够表征 Ex_9 表现出异常上调的 5- 羟色胺转运活性(图 5F 和 5G)。这些结果证明 Ex_9 对 MDD 的发展有贡献,并且由于 SLC6A4 相关子网络的强激活作用,成为药物靶标细胞类型。
最近的研究表明,SLC6A4 基因主要在前额叶皮层中的一部分深层神经元中表达,层 5 和层 6 中的神经元可能是 SSRIs 的靶标,而深层神经元表现出异常的神经元大小。因此,我们询问了 Ex_9 的空间分布是否与这些发现相对应。我们结合 MDD 的 scRNA-seq 数据与小鼠和人脑的空间转录组数据,使用 CellTrek 恢复了组织切片中 Ex_9 的空间坐标(图 5H 和 5I)。与先前的研究一致,大多数 Ex_9 神经元分布在脑的深层,表现出区别于这些层中其他神经元的网络特征(图 S10)。因此,scRank 预测的排名最高的 Ex_9 可能代表响应 SSRIs 的靶细胞类型。进一步分析这种神经元亚型可能对 MDD 药物开发有益。
展示对心肌梗死中丹参酮 IIA 响应的巨噬细胞亚群及其另一潜在靶标¶
巨噬细胞在导致心肌梗死的病理生理过程中发挥多种作用,表现出广泛的促炎和抗炎效应,这由其异质性表型决定。这些不同的巨噬细胞亚群也可能对药物表现出不同的反应。因此,识别关键的药物响应巨噬细胞亚群对于加深对药理机制的理解和发现细胞水平的新治疗靶标至关重要。因此,我们将 scRank 应用于我们之前研究中的小鼠心脏巨噬细胞的 scRNA-seq 数据集,包含三种条件(假手术、心肌梗死和丹参酮 IIA 治疗)(图 6A)。我们使用心肌梗死的 scRNA-seq 数据和丹参酮 IIA 的直接靶基因 Ctsk 作为输入数据。如图 6B 所示,巨噬细胞 5(M∅-5)是对丹参酮 IIA 响应的最高排名细胞类型,这与我们的原始结论一致,即该化合物可以显著减少 M∅-5 的比例和炎症效应(图 6C)。然后,我们放大了基于治疗前条件数据构建的最高排名和最低排名巨噬细胞亚型的 Ctsk 为中心的子网络。我们发现子网络被模块化为三个基因模块,药物靶基因 Ctsk 位于模块 1 中(图 6D 和 6E)。与 M∅-11 相比,M∅-5 表现出更高的模块 1 活性,主要富集在炎症相关功能中,通过基因本体分析确定(图 6D 和 6F)。此外,我们观察到丹参酮 IIA 显著降低了 M∅-5 中模块 1 的活性,但未在 M∅-11 中观察到,表明 M∅-5 亚群是治疗靶标。
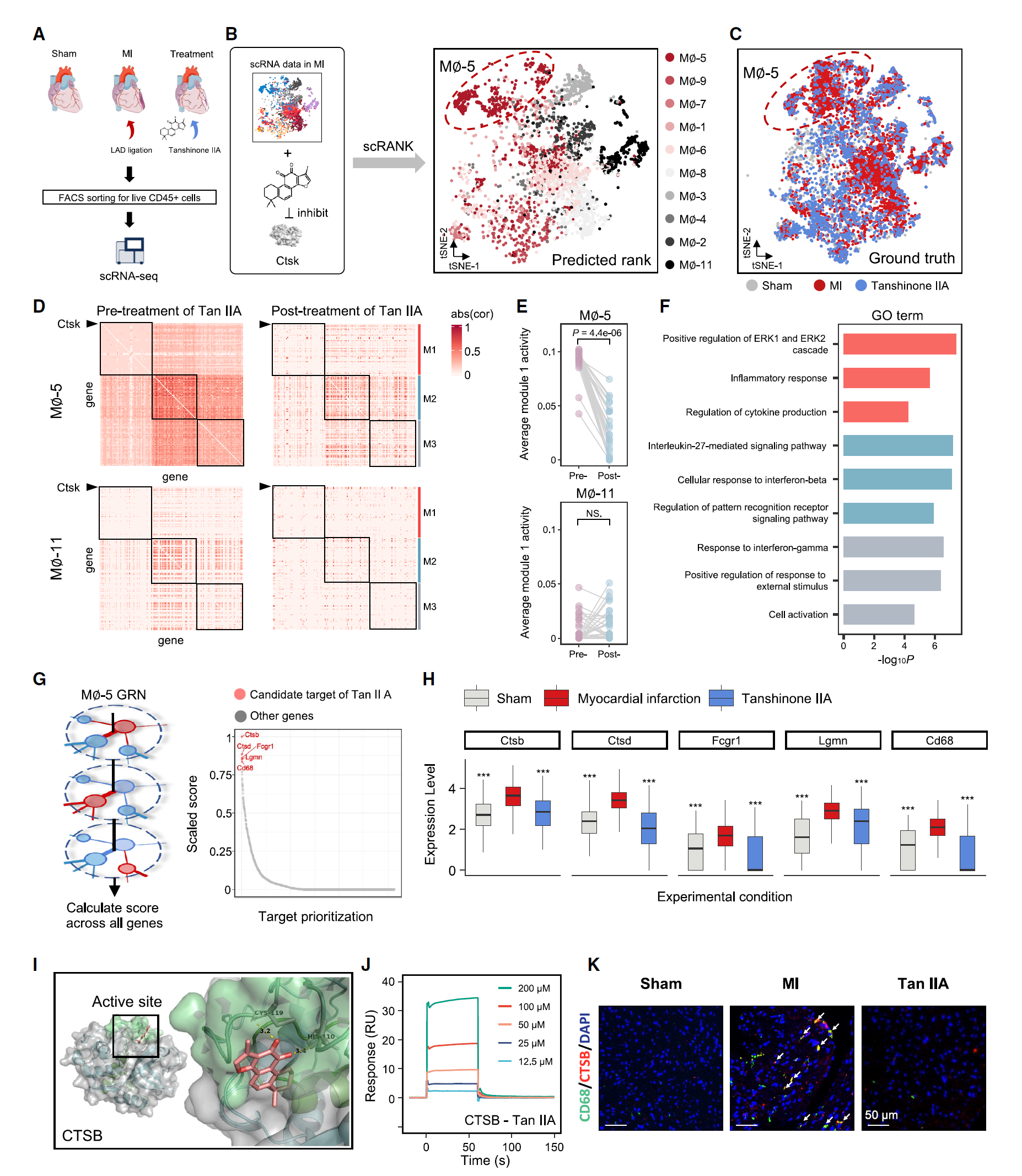
图 6:利用 scRank 识别对丹参酮 IIA 响应的巨噬细胞亚群及其潜在靶标
(A) 来自 C57BL/6 小鼠心脏左心室的免疫细胞 scRNA 数据,包括三种条件(假手术,左前降支结扎 3 天后导致心肌梗死(MI),以及结扎和丹参酮 IIA 治疗 3 天后)。 (B) MI 和丹参酮 IIA 靶基因 Ctsk 的 scRNA 数据是 scRank 的输入。t-SNE 可视化的 MI 数据中,细胞类型根据通过 scRank 预测的优先级着色,从红色(最高排名)到黑色(最低排名)。用虚线圈出最高排名的巨噬细胞亚型 M∅-5。 (C) t-SNE 可视化数据,结合组根据实验条件着色。先前验证的巨噬细胞亚型 M∅-5 作为丹参酮 IIA 的真实靶细胞类型,用虚线圈出。 (D) 在不同条件下(治疗前和丹参酮 IIA 治疗后)最高排名和最低排名巨噬细胞亚型(M∅-5 和 M∅-11)的 GRN 热图,包括 131 个 Ctsk 相关基因。 (E) 在治疗前和治疗后条件下,M∅-5(顶部)和 M∅-11(底部)之间的药物靶相关模块的比较,根据 47 个模块 1 基因的平均边权重通过配对双侧 Wilcoxon 检验评估。 (F) 不同模块基因的 GO 分析。 (G) scRank 用于基因排名的示意图。scRank 计算的 M∅-5 的 GRN 中每个基因的扰动评分,输入是靶细胞类型的 GRN,输出是基因的排名。 (H) 不同实验条件下预测靶基因在 M∅-5 中的表达变化。预测靶基因在 M∅-5 中的表达变化以箱线图表示(最小值,第 25 百分位数,中位数,第 75 百分位数和最大值)。每种条件下的数据点数量分别为 93, 896 和 140。星号表示表达值显著上调(MI 对比假手术)或下调(MI 对比丹参酮 IIA) (∗∗∗p < 0.0001)。 (I) 显示 CTSB 活性位点与丹参酮 IIA 之间形成氢键的 3D 结构和结合模式。 (J) 显示丹参酮 IIA 与 CTSB 相互作用的传感器图。 (K) 巨噬细胞标记物 CD68(绿色)、CTSB(红色)和 DAPI(4′,6- 二脒基 -2- 苯基吲哚;蓝色)的免疫荧光染色,显示丹参酮 IIA 治疗抑制巨噬细胞 CTSB 水平。
由于丹参酮 IIA 通过多种途径保护心血管系统,它可能具有多个靶标。因此,在确定其靶细胞类型后,我们试图确定 M∅-5 的 GRN 中基因的扰动评分是否可以指示其作为丹参酮 IIA 潜在靶标的可能性。换句话说,给定靶细胞类型,我们可以基于通过 scRank 计算的扰动评分对基因进行排序,以识别新的药物靶标。实际上,我们计算了 M∅-5 的 GRN 中所有基因的扰动评分(图 6G),并将前五个基因(Ctsb、Ctsd、Fcgr1、Lgmn、Cd68)视为潜在的丹参酮 IIA 靶标(图 6G)。值得注意的是,这些靶标通过简单的相关分析是无法识别的(图 S11)。我们还使用模拟数据验证了 scRank 在靶基因排序方面的准确性(图 S12)。基于丹参酮 IIA 对基因表达的抑制效应假设,我们调查了 scRank 预测的前五个候选基因是否确实被处理所抑制。实际上,所有这些基因都被丹参酮 IIA 显著下调(图 6H 和 S12),我们通过实验验证了这一点(图 S13)。这些基因在心肌梗死条件下也异常上调(图 6H 和 S13),这表明丹参酮 IIA 通过缓解心肌缺血下调了这些基因。事实上,这些基因在蛋白质 - 蛋白质相互作用网络中被分为一个模块,涉及溶酶体和免疫系统过程中的水解酶活性调节,从而表明了丹参酮 IIA 的作用机制(图 S12)。我们还通过分子对接分析检查了丹参酮 IIA 是否作为抑制剂直接结合这些基因编码的蛋白质(图 S12)。由于 Ctsb 在扰动评分中的主导作用,我们专注于它。分子对接结果显示丹参酮 IIA 通过与 His110 和 Cys119 形成氢键与 CTSB 活性位点的推定结合(图 6I)。为了确认丹参酮 IIA 与 CTSB 的结合,我们进行了表面等离子共振测定,发现丹参酮 IIA 可以直接结合 CTSB 蛋白,并且这种结合发生在浓度依赖的方式(图 6J)。此外,随后的免疫荧光染色实验确认丹参酮 IIA 显著减少了巨噬细胞中的 CTSB 表达(图 6K)。这些结果与最近的研究相一致,表明 CTSB 可能是丹参酮 IIA 的潜在靶标。
讨论¶
准确识别对药物治疗负责的关键细胞类型仍然是一个挑战。大多数推断治疗反应的方法集中于已知药物敏感性标志物的基因表达,可能错过药物靶标与下游信号基因之间的关键关联。这些关联对于理解异质药物反应非常重要,因为药物扰动不仅对单一靶标产生影响,还会影响信号转导和 GRNs。
在这项研究中,我们提出了 scRank,这是一种使用 tpGRN 对药物响应细胞类型进行排序和推断的新方法。其基本假设是,不同细胞类型的治疗反应取决于其与药物靶标相关的内在基因网络。scRank 的主要目标是模拟 GRN 中的计算机模拟药物扰动,并量化其在不同细胞类型中的效果。为此,scRank 创建了 tpGRNs 以模拟计算机模拟药物扰动,并将 tpGRN 与原始 GRN 进行比较,以测量药物引入的网络变化,从而对细胞类型进行排序。比较网络的常用方法是使用流形对齐算法将不同网络的节点投射到共享的流形空间,在那里计算节点相似性以评估网络结构的差异。然而,这种方法无法捕捉 tpGRN 中的动态变化,这在准确测量药物引起的网络变化方面可能是一个挑战。为了解决这个问题,我们将药物扰动在 tpGRN 中建模为具有全局和局部效应。具体而言,扰动的靶基因可以在每个基因节点上引起全局变形,结果变形可以沿着靶点为中心的子网络扩散。在实践中,距离与药物靶标及其相关的子网络信号相结合,以量化药物效应。虽然 tpGRN 的创建可能简化了响应药物扰动的复杂网络重组,但其设计旨在通过网络中的基因 - 基因相关性反映潜在药物靶标的遗传扰动。我们对 GRNs 和模拟的 tpGRNs 与来自处理细胞的 GRNs 进行比较,验证了这种方法可以准确模拟药物效应。我们所描述的方法,结合药物和生物网络以模拟和评估计算机模拟药物扰动,是基于未处理的 scRNA-seq 数据推断药物响应细胞类型的首例。
单细胞药物响应预测的一个重大挑战是准确识别不同的细胞状态,因为这些状态可能对同一药物表现出显著不同的响应,并且在治疗后可能转变为不同的命运。尽管 scRank 专门为预先标记的细胞类型设计,但在我们的补充基准测试中,它展示了跨各种聚类方法的适应性和鲁棒性,有效识别不同的细胞状态。另一个需要考虑的关键方面是药物耐药性,它对治疗结果有显著影响。我们结合了一个综合评分,在未处理样本中也考虑了内在药物耐药机制。此外,我们已将 scRank 的应用扩展到具有多个靶标的药物,初步结果表明 scRank 具有潜在的有效性。然而,需要承认的是,多靶标扰动的固有复杂性,例如协同效应。这些复杂的靶标间相互作用需要专门定制的方法,我们计划在未来的研究中开发这些方法。
总的来说,scRank 在以下几个方面取得了概念上的进步。我们的方法克服了实验药物治疗的限制,可以在不需要昂贵且耗时的实验的情况下提供药物响应的宝贵见解。此外,scRank 使用基于靶标相关网络的可解释模型来说明药物响应的基本机制,支持靶标为中心的密集连接子网络可能因更高的基因相互作用而受到更强药物扰动的假设。此外,我们的基于网络的方法超越了传统的对直接和下游药物靶标表达谱的依赖,这通常不足以准确预测药物响应。scRank 强调靶标相关网络,提供了药物响应的更全面视角。这种方法在药物靶基因不是特定细胞类型标志基因的情况下特别有效。考虑到药物的治疗效果取决于它如何影响对疾病最重要的细胞类型,我们也在我们的三个案例研究中确认了最高排名细胞类型的疾病相关性。此外,我们使用基因集合富集分析评估这些药物对最高排名细胞类型的功能影响,从而增强了我们工具的临床实用性。此外,scRank 还适用于处理作用机制未知的天然产物药物,例如丹参酮 IIA,因为它有助于识别和优先考虑潜在药物靶标。
在 scRank、beyondcell 和 scDEAL 的比较中,我们观察到预测性能的差异。例如,scDEAL 和 beyondcell 在索拉非尼上的最低性能在一定程度上受其使用的总体数据集的质量和不平衡的影响。同时,scRank 在处理具有复杂机制的药物时遇到挑战,例如阿糖胞苷。阿糖胞苷的作用机制不仅涉及抑制,还涉及直接的 DNA 损伤和整合到 DNA 中。这在生物网络中引入了复杂的扰动,使 scRank 难以预测。
我们预见到我们方法的若干未来应用。首先,通过了解不同细胞类型之间的相对药物敏感性,scRank 可以帮助优化治疗,通过更好的药物组合和递送策略最小化副作用并针对关键疾病细胞。其次,scRank 可以增强我们对 FDA 批准药物在细胞水平上的理解。尽管治疗效果已经确定,但对药物响应的细胞类型的精确知识有限。使用 scRank,我们可以识别这些重要的细胞类型进行进一步研究,从而增强我们对药物相关治疗机制的知识。在我们的研究中,我们选择 scRNA-seq 数据而不是蛋白质组数据,因为它们的可用性更广泛。展望未来,随着单细胞蛋白质组数据可用性的增加,我们预见到将类似方法应用于这些蛋白质测量所得的蛋白质 - 蛋白质相互作用网络的潜力。这种方法将使在蛋白质水平上对药物反应进行更直接的研究。
研究的局限性¶
scRank 的一个局限性是其主要集中于作为抑制剂作用的药物,这部分是由于可用数据的性质。我们目前访问的大多数数据都涉及抑制性药物,这指导了 scRank 的开发和测试主要在这种背景下。然而,认识到扩大 scRank 范围的需求,我们还对激动剂进行了初步测试,调整 scRank 的方法以模拟其效果。具体而言,为了在网络中反映激动剂的激活效应,我们将从靶基因节点出发的边缘权重设置为其最大可能值,从而生成 tpGRN。随着更多激动剂药物的单细胞数据变得可用,我们计划进一步完善和扩展 scRank 的能力。
方法细节¶
scRank 算法¶
我们设计了 scRank,以基于与抑制剂靶基因相关的基因网络活动对细胞类型进行排序。我们认为药物的效果会体现在网络连接性上。我们通过比较未处理的 GRNs 与 tpGRNs 来尝试量化药物的效力。scRank 量化了网络结构的差异和网络扩散的效果作为细胞类型排序的基础。该方法包括三个步骤:网络构建、计算机模拟药物扰动和扰动评估。第一步是从表达谱重建基因调控网络。第二步是在 GRN 中模拟计算机模拟药物扰动的效果。第三步是估计扰动。scRank 的资源使用情况已使用模拟数据集进行了评估,如表 S2 和 S3 所述。
网络构建¶
显然,构建网络是准确估计 tpGRN 中药物扰动的核心步骤。最近,已经开发了几种从 scRNA-seq 转录组数据构建基因调控网络的方法。scTenifoldNet 是一种先进的方法,在准确性和效率方面优于现有工具,如 GENIE3。这是通过使用主成分回归结合集成和去噪策略实现的,这有助于稳健地构建网络。鉴于其显著的效果,我们采用了类似的方法来构建网络。在实践中,经过深思熟虑,我们实施了进一步的修改,以定制适应我们特定研究目标的方法。为了确保药物扰动估计在所有细胞类型中具有可比性,我们使用逐群策略构建细胞类型特异性 GRNs,而不是为整个样本构建 GRN,在这种策略中,网络节点是一致的。构建 GRNs 时选择的基因特征是有目的的,重点是保持直接的调控关系、药物靶标信息和功能生物过程。为了尽量减少噪音和简化输入基因特征,我们进一步排除了线粒体和核糖体基因。子采样方法已被定制以解决稀有细胞类型及其不同丰度的问题。具体来说,选择固定比例的细胞进行子采样,而属于少于 25 个细胞类型的细胞则被排除。已经优化了几个参数,包括边权重阈值和去噪精度,从而增强了药物响应细胞类型识别的性能。详细步骤如下:
给定一个 scRNA-seq 表达矩阵 \(M\),其在细胞类型 \(i\) 中表示为 \(M_i[n \times c]\)(\(n\) 个基因和 \(c\) 个细胞,\(i \in\{1,2, \ldots, C\}\),其中 \(C\) 表示数据集中的细胞类型总数),我们利用 \(M_i\) 为每种细胞类型构建一个特异性基因调控网络。为了克服细胞类型之间的异质性并构建稳健的 GRNs,我们首先从 \(M_i\) 中随机选择 \(\delta c\) 个细胞(\(\delta \in(0,1)\) 表示选择比例,默认值为 0.5),重复 \(t\) 次,得到一组包含 \(\delta c\) 个细胞的基因表达矩阵 \(T_i=\left[X_1, \ldots, X_t\right]\)。这些基因表达矩阵代表了细胞类型 \(i\) 中更全面的细胞状态,用于构建 GRNs。
为了在保留最有意义的生物数据的同时降低计算成本,我们只选择了表达基因的一个子集,而不是整个转录组。实际上,我们整合了通过 Seurat 识别的整个 scRNA-seq 数据集中的前 2000 个高变异基因(HVGs)、来自 AnimalTFDB 的转录因子(TFs)和来自 DGIdb 的药物(抑制剂)靶基因来构建 GRN。我们还过滤了线粒体、核糖体和冗余基因,以保留有意义的细胞间变异和相关的生物过程。用于构建 GRN 的最终基因集 \(G=[HVG+TF+Tar]\) 的基因数量表示为 \(g\)。
为了以较低的计算成本从细胞类型 \(i\) 的基因表达矩阵 \(X\) 中恢复基因 - 基因相关性关系,我们采用了类似于 scTenifoldNet 的方法。为了构建细胞类型特异性的 GRN,我们对表达谱集 \(T_i\) 中的每个子集矩阵 \(X_t[g \times \delta c]\) 使用主成分分析(PCA)。这使我们能够通过将相关的独立变量组合成几个主成分来解决多重共线性问题,并保留细胞类型特异性信息。然后,我们应用回归模型来估计目标基因与所有其他基因之间的回归系数,这些系数用作 GRN 中的边权重。实际上,给定一个基因表达矩阵 \(X[n \times p]\),\(Y_{n \times 1}=\left(y_1, \ldots, y_n\right)^T\) 表示目标基因表达的向量,\(X_{n \times(p-1)}=\left(x_1, \ldots x_n\right)^T\) 表示其余 \(p-1\) 调节基因的表达向量。假设 \(Y\) 的值依赖于 \(X\) 的值,目标基因与其他 \(p-1\) 基因之间的关系可以建模为:
其中 \(\beta \in \mathbb{R}^{p-1}\) 表示回归系数向量,\(\varepsilon\) 表示随机误差向量。为了减少计算成本和过拟合,我们对通过 PCA 获得的 \(k\) 个潜在协变量而不是 \(p-1\) 个基因进行回归(\(k \ll p-1\))。令 \(X_{n \times k}^{\prime}=X_{n \times(p-1)}V_{(p-1) \times k}\) 表示选择的前 \(k\) 个主成分,其中 \(V_{(p-1) \times k}\) 是通过奇异值分解获得的 \(X\) 的主成分载荷。然后回归问题可以转化为:
这里,\(\beta^{\prime} \in \mathbb{R}^k\) 表示在变换数据矩阵 \(X^{\prime}\) 中的回归系数向量,可以使用普通最小二乘法进行估计。然后,表示目标基因与 \(p-1\) 基因之间相关性的 \(\beta\) 的估计值为:\(\widehat{\beta}=V_k \widehat{\beta}^{\prime} \in \mathbb{R}^{p-1}\)。通过对每个随机选择中的每个基因在 \(G\) 中进行迭代回归,我们最终获得了 \(t\) 个基因邻接矩阵,包括每对基因之间的关系和强度,以表示细胞类型 \(i\)。为了在 GRN 中保留显著的生物信号,我们只保留了绝对权重值排名前 \(\theta \%\) 的边(默认值为 \(\theta=5\%\))。由于主成分回归考虑了基因关系的不对称性,从基因 A 到基因 B 和反之亦然分配了不同的权重,这导致了双向关系,特别是在 TFs 及其靶基因之间。为了放大 TFs 的调控影响,我们在双向边之间进行了权衡,保留较低权重的边为其强度的 25\%。最后,我们通过将对角线值设置为 0 来消除自相关,从而获得最终的基因邻接矩阵,表示为 \(A\)。
为了提高 GRN 的稳健性并保留异质性,我们提取了每个 GRN \(A_t\) 中的主要信号,这些 GRN 来自 \(T_i\) 中的每个子集矩阵 \(X_t\),并使用张量成分分析(TCA)将它们整合到一个矩阵中。我们首先将 \(t\) 个 GRN 组合成一个 \(S \times T \times R\) 的三阶张量,表示为 \(\chi\),其中 \(S, T\) 和 \(R\) 分别对应于从随机选择的数据、目标基因和调节基因中获得的 GRN 数量。然后,在 \(\chi\) 上进行命名为 CANDECOMP/PARAFAC 的 TCA 方法,导致 \(\chi\) 的分解和近似表示为 \(R\) 个秩为 1 的张量的和:
其中符号 \(\otimes\) 表示外积,而 \(s^r, t^r\) 和 \(r^r\) 是包含张量中每个维度的元素载荷的因子 \(r\) 的向量。我们选择了前五个因子来表示 \(\chi\) 的关键组成部分,并将它们求和为一个新的张量 \(\chi^{\prime}\),近似表示原始张量 \(\chi\)。然后,我们在 \(\chi^{\prime}\) 中对 \(S\) 进行平均,并通过将每个权重除以其最大绝对值来进一步归一化,以获得给定细胞类型 \(i\) 的最终 GRN,表示为 \(A^{\prime}\)。最终的 GRN 包含所有输入基因的相互作用,并构成了后续创建目标扰动 GRN 的基础。
计算机模拟药物扰动¶
为了模拟抑制剂扰动对基因调控网络的影响,我们通过将药物直接靶基因所在行的所有值设置为 0,创建了目标扰动 GRN \(A_{tp}^{\prime}\)。相反,为了模拟激动剂扰动的效果,我们通过将靶基因节点发出的出边权重调整为其最大可能值(权重 \(=1\))来创建 tpGRN。
扰动评估¶
为了在 tpGRN 中建模计算机模拟药物扰动的效果,我们考虑了网络中的全局和局部效应,并使用流形空间中相同节点之间的距离和网络扩散效应进行公式化。
GRNs 之间基因节点距离的计算¶
为了从网络角度估计药物扰动,我们首先尝试使用流形对齐将两个高维邻接矩阵共同投影到共享的低维潜在空间中,并将每个匹配基因节点之间的距离解释为药物的扰动效果。流形对齐将从相似过程中生成的两个数据集投影到新的低维空间中,同时保持匹配点之间的最小距离并保留其原始结构。匹配点的更大距离表示该点在两个数据集中的拓扑变化更大。为了执行流形对齐,我们给定两个邻接矩阵,\(A^{\prime} \in \mathbb{R}^{p \times p}\) 和 \(A_{PB}^{\prime} \in \mathbb{R}^{p \times p}\),其中 \(p \in G\) 是用于构建 GRN 的基因集。然后,我们可以将 \(A^{\prime}\) 和 \(A_{PB}^{\prime}\) 组合成一个联合矩阵 \(W\):
其中 \(I\) 是描述两个 GRN 中基因节点之间对应关系的单位矩阵,\(\lambda\) 是一个调整参数。然后,我们利用从拉普拉斯特征映射问题中解决的 \(d\) 个最小非零特征值,因为它们共享低维投影。令 \(E_{2p \times d}=\left(e_1, \ldots, e_{2p}\right)\) 表示特征向量。然后,通过 \(D(p)=\left\|e_p-e_{2p}\right\|=\left(d_1, \ldots, d_p\right)\) 计算两个基因节点之间的欧氏距离。
网络扩散¶
为了更准确地推断药物扰动,我们考虑了网络中的扩散效应。给定一个靶基因 Tar,我们认为扰动的效果将沿着 GRN 中的边从靶基因扩散到下游基因。我们假设这种效应与边权重和节点距离有关。因此,扰动得分计算如下:
其中 \(D_{Tar}\) 是靶基因的距离,\(D_n\) 是下游基因的距离,\(\mathrm{Deg}\) 是相应基因节点的度,\(W_{in}\) 是入边的权重,\(W_{out}\) 是出边的权重。第一项是靶基因中的归一化药物扰动效应,第二项是 1 跳节点中的扩散效应,第三项是 2 跳节点中的归一化扩散效应。
数据处理¶
对于所有 ST 和 scRNA-seq 数据集,细胞类型注释由作者提供或根据其代码手动获得。所有具有超过 25 个细胞的细胞类型都经过 scRank 管道处理。对于小鼠大脑 ST 数据,我们使用 biomaRt (2.48.3) \({ }^{90}\) 将小鼠基因转换为人类基因,以便使用 CellTrek 映射人类 DLPFC 数据。
对于所有 ST 和 scRNA-seq 数据集,使用全局缩放归一化方法 LogNormalize 对原始计数进行归一化,以准备下游分析,并使用原始计数运行 scRank 管道。
模拟¶
使用“SERGIO” Python 包生成模拟数据。细胞类型表达谱根据预定义的 GRN 生成,这是 SERGIO 的输入文件,包含 GRN 结构及其参数。我们定义了可分为四个基因模块的基因,每个模块包含 25 个基因,表现出共调控。接下来,我们通过调整生产率和相互作用强度的参数来生成细胞类型特异性的基因模块活动。所有模拟数据的 GRNs 均使用从模拟的单细胞表达数据中 scRank 重建。通过参数调优,我们评估了 scRank 在多种情况下的性能。
第一个场景旨在生成模块活动有序增加的细胞类型。实际上,我们模拟了包含不同细胞数量的四个数据集(每个细胞类型 1000、3000、5000、10000 个细胞)。每个数据集由四种细胞类型组成,每种细胞类型具有 100 个基因。每个细胞类型有三个基因模块(基因模块 2、3 和 4)活动相似,而一个基因模块(基因模块 1)在细胞类型中活动梯度增加。我们假设药物靶基因在基因模块 1 中。我们将基因模块 1 的基因和单细胞基因表达谱逐次输入到 scRank 模型中。
第二个场景旨在生成分为两组、具有高或低基因模块活动的六种细胞类型。细胞类型 A、B 和 C 被分配到基因模块 1 活动高于细胞类型 D、E 和 F 的组中,后者在模块 1 中的活动较低。我们将基因模块 1 的基因和单细胞基因表达谱逐次输入到 scRank 模型中。
最后一个场景旨在生成具有特异性基因模块的细胞类型。具体而言,基因模块 1/2/3/4 分别对应于细胞类型 A/B/C/D。对于每个模块,我们将模块内的基因和单细胞基因表达谱逐次输入到 scRank 模型中。
为了模拟疾病 - 治疗配对数据集,我们首先模拟了具有异常高共表达基因模块 1 的疾病样数据集,其活动在细胞类型中梯度增加,如场景 1 所示。相应地,我们还模拟了具有非常低模块 1 活动的治疗样数据集,代表靶基因被药物抑制的潜在状态。
在第一个模拟中,我们通过比较细胞类型间的扰动得分来评估 scRank 在识别连续细胞状态方面的性能。考虑到基因模块 1 中的药物靶标,我们对每种细胞类型 GRN 中的基因 1-25 进行逐次扰动,以获得由 scRank 计算的最终扰动得分。性能通过细胞类型 A 的最高排名百分比和 Spearman 秩相关系数(SRCC)进行评估。在第二个模拟中,我们基于统计显著性评估了 scRank 在识别离散细胞状态方面的性能。在第三个模拟中,我们考虑了每个具有特定基因模块的细胞类型对特定药物的响应,表明当药物靶基因在基因模块 1 中时,具有特定高模块 1 活动的细胞类型 A 应在其他细胞类型中对药物最敏感。对于每个模块,我们逐次扰动模块内的基因,并基于统计显著性评估 scRank 在识别细胞类型特异性药物响应方面的性能。
在第四个模拟中,基于疾病 - 治疗配对数据集,我们将 scRank 与其他细胞类型排序方法进行了比较。对于 scRank,我们仅使用疾病样数据集来排序细胞类型。经典的差异表达分析(通过 Seurat 中的 Wilcoxon 秩和检验)和 Augur 都将疾病和治疗数据集作为输入数据进行分析,以优先排序细胞类型。在运行这两种方法之前,基因表达数据已根据条件进行归一化和整合,随后按照 Seurat 中的标准整合工作流程进行处理。
与其他方法的比较¶
使用模拟数据和五个真实数据集将 scRank 的性能与其他现有细胞类型排序方法进行比较。对于配对条件下的每种模拟和真实数据集,使用每种细胞类型在治疗前后条件之间的差异表达基因(DEGs)数量来量化药物扰动的程度并排序细胞类型。通过 Augur 量化的表示扰动程度的 AUC 得分用于排序细胞类型。计算目标细胞类型的最高排名百分比以评估方法的性能。
在与其他扰动预测方法(scGen、scVIDR 和 CellOracle)的比较研究中,我们使用估计的扰动向量的长度作为扰动的大小,并进一步使用它来排序细胞类型。鉴于 CellOracle 专门设计用于支持转录因子(TFs)的扰动,我们选择了在药物靶细胞类型内治疗前后状态之间具有最高折叠变化的 TF 基因作为输入到 CellOracle 中的扰动基因。
在与其他药物响应预测方法的比较研究中,我们采用不同的策略来评估 beyondcell 和 scDEAL 的性能。这涉及利用不同的度量标准根据其对治疗的响应性来排序细胞群体。对于 beyondcell,它为每个细胞生成 BSC(Beyondcell 评分),测量每个细胞对给定药物的易感性。为了使用 beyondcell 对细胞类型进行排序,我们计算了细胞亚群内所有细胞的平均 BSC 评分。这种方法称为“beyondcell”,侧重于群体级别的优先排序。此外,鉴于 beyondcell 提供了单细胞分辨率,我们实施了一种替代方法“bc_BCS”,其中我们根据每个细胞的 BSC 评分分别对每个细胞进行排序,然后计算每种细胞类型的平均排名。此外,beyondcell 定义了 BSC 评分的切换点,以将细胞分类为敏感或抗性。根据这一概念,我们根据二进制优先排序(0 表示抗性,1 表示敏感)的平均值来排序细胞类型,考虑切换点标准。这种方法称为“bc_SwitchBinary”。对于 scDEAL,它为每个细胞分配一个相对响应概率,称为敏感评分。使用 scDEAL 对细胞类型进行排序,我们计算了每种细胞类型内所有细胞的平均敏感评分。这种方法称为“scDEAL”,同样评估群体级别的响应性。像 beyondcell 一样,scDEAL 还为每个细胞提供二进制标签(敏感或抗性)。我们结合这一方面,通过计算每种细胞类型的二进制优先排序的平均值(0 表示抗性,1 表示敏感)来排序细胞类型。这种方法称为“scDEAL_binary”。
在与基于表达的方法的比较中,我们使用未处理条件下各细胞类型中直接和下游靶基因的平均表达水平来排序细胞类型。下游靶标是指模块化的靶基因相关网络中的网络邻近基因。
对于所有工具,我们遵循其原始库中的指导,并为所有参数设置默认值。
使用不同聚类方法对 scRank 的基准测试¶
对 scRank 在三种聚类方法(Leiden、Louvain 和 scSHC91)中的性能进行了基准测试。这些方法在三个现实世界的数据集上进行了基准测试,展示了细胞状态复杂性的增加,从不同的细胞类型到更复杂的细胞状态。实际上,通过 Seurat 包中的“FindClusters”函数执行 Leiden 和 Louvain,分辨率为 0.1、0.5 和 1。另一方面,scSHC 的实现不需要这样的参数调整。已知的响应性细胞类型用作真实标签。基于每种算法识别的聚类,scRank 预测的最高排名细胞类型中的这些标签的比例表明了 scRank 在不同聚类方法上的性能。
GSEA 和 GO 富集分析¶
使用“Metascape web tool [https://metascape.org/]” 92 进行途径和生物过程的富集分析,其中根据平均基因表达的折叠变化选择前 100 个 DEGs。使用 clusterProfiler93 工具对排名基因列表进行 GSEA,以丰富显著激活的途径和生物过程,其特征从 Molecular Signatures Database v7.494(“MSigDB [http://www.gsea-msigdb.org/gsea/msigdb]”)中获得,包括来自 KEGG、Reactome 和 WikiPathways 途径数据库的 GO 和规范途径基因集。